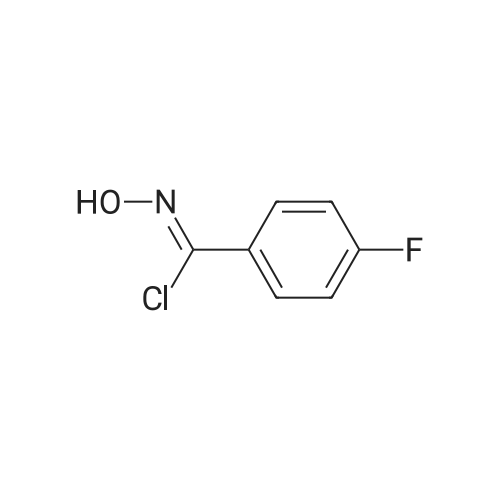
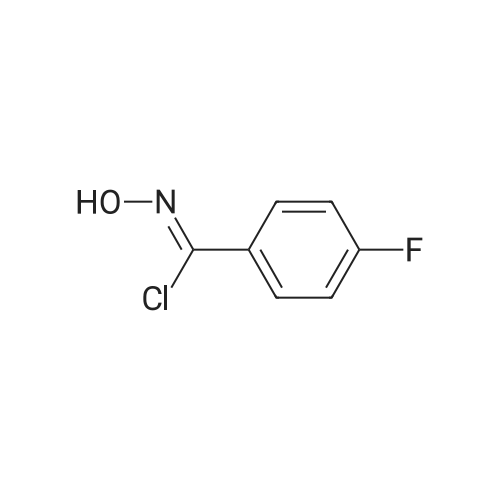
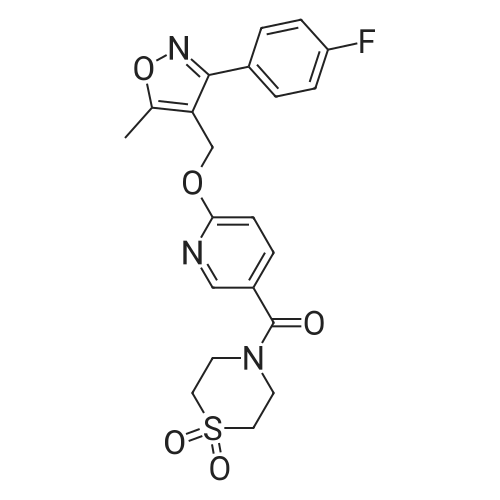
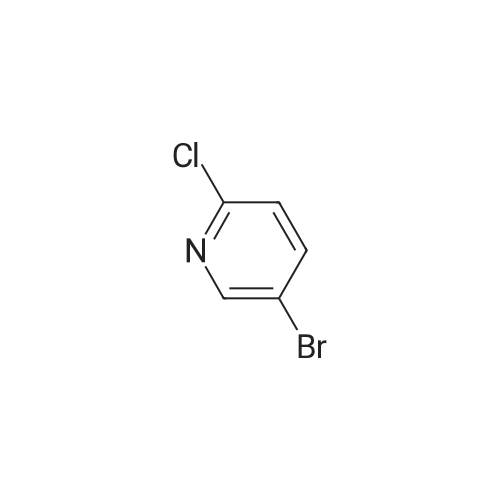
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With lithium aluminium tetrahydride In tetrahydrofuran at 0 - 20℃;
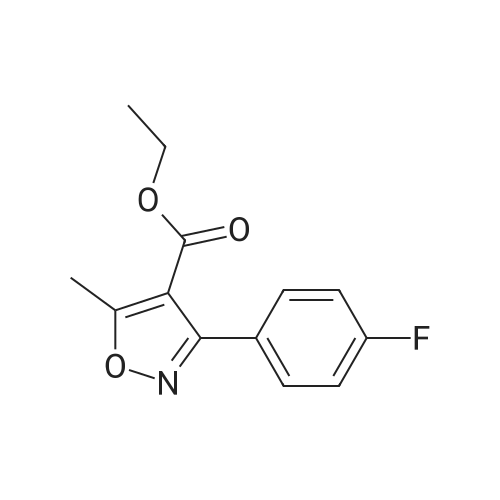
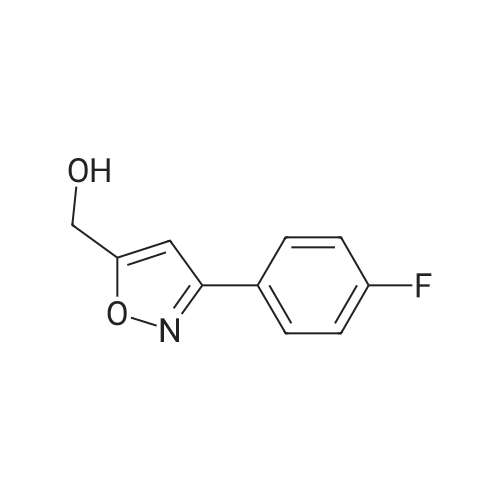
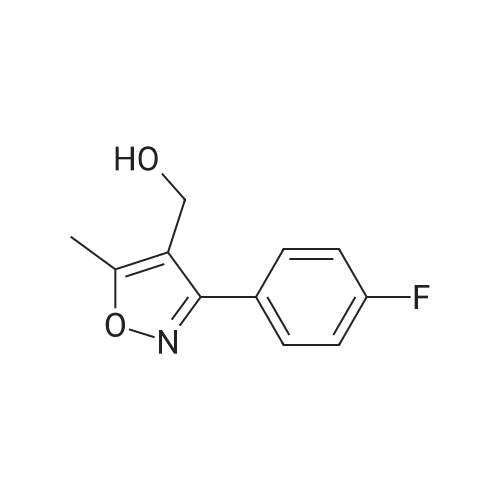
To a suspension of LiAlH4 (75.9 mg, 2.0 mmol) in THF (2 mL) was added at 0-10° C. within 15 minutes a solution of 3-(4-Fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid ethyl ester (0.50 g, 2.0 mmol) in THF (3 mL) and the resulting solution was allowed to warm to room temperature and subsequently stirred at this temperature for at least one hour. Water (15 mL) was added dropwise and the resulting suspension was then filtered and the filter cake washed with ethyl acetate (15 mL). From the biphasic filtrate the layers were separated and the organic layer was washed with water (1×15 mL). Both combined aqueous layers were back extracted with ethyl acetate (2×15 mL). The combined organic layers were dried over Na2SO4 and subsequently concentrated to dryness (45° C./25 mbar) to afford 0.375 g (90percent) of the title compound as a slightly yellow solid with a purity of 100percent (by HPLC).
71%
Stage #1: With lithium aluminium tetrahydride In tetrahydrofuran at 0 - 20℃; for 3 h; Stage #2: With water; sodium hydroxide In tetrahydrofuran at 0 - 20℃;
o a solution of 3-(4-fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid ethyl ester (3.0 g, 12 mmol) (6.18 g, 25 mmol) in THF (320 mL) was added portionwise lithiumaluminiumhydride (528 mg, 14 mmol) at 0 °C and the reaction mixture was stirred at room temperature for 3 h. The mixture was then cooled to 0 °C and water (518 μ) added followed by sodium hydroxide (15percent solution, 518 μ) and then again water (1.5 mL) and the mixture then stirred overnight at room temperature. The precipitate was then filtered off and washed with THF. The combined washings and filtrate were then evaporated. Purification by chromatography (Si02, heptane:ethyl acetate = 100:0 to 1: 1) afforded the title compound (1.8 g, 71percent) which was obtained as a white solid. MS: m/e = 208.1 [M+H]+.
71%
Stage #1: With lithium aluminium tetrahydride In tetrahydrofuran at 0 - 20℃; for 3 h; Stage #2: With water; sodium hydroxide In tetrahydrofuran at 0 - 20℃;
Step d: [3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol To a solution of 3-(4-fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid ethyl ester (3.0 g, 12 mmol) (6.18 g, 25 mmol) in THF (320 mL) was added portionwise lithiumaluminiumhydride (528 mg, 14 mmol) at 0° C. and the reaction mixture was stirred at room temperature for 3 h. The mixture was then cooled to 0° C. and water (518 μL) added followed by sodium hydroxide (15percent solution, 518 μL) and then again water (1.5 mL) and the mixture then stirred overnight at room temperature. The precipitate was then filtered off and washed with THF. The combined washings and filtrate were then evaporated. Purification by chromatography (SiO2, heptane:ethyl acetate=100:0 to 1:1) afforded the title compound (1.8 g, 71percent) which was obtained as a white solid. MS: m/e=208.1 [M+H]+.
71%
Stage #1: With lithium aluminium tetrahydride In tetrahydrofuran at 0 - 20℃; for 3 h; Stage #2: With water; sodium hydroxide In tetrahydrofuran at 0 - 20℃;
Step d: r3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-yll-methanol: To a solution of 3-(4-fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid ethyl ester (3.0 g, 12 mmol) (6.18 g, 25 mmol) in THF (320 mL) was added portionwise lithiumaluminiumhydride (528 mg, 14 mmol) at 0 °C and the reaction mixture was stirred at room temperature for 3 h. The mixture was then cooled to 0 °C and water (518 µl) added followed by sodium hydroxide (15percent solution, 518 µl) and then again water (1.5 mL) and the mixture then stirred overnight at room temperature. The precipitate was then filtered off and washed with THF. The combined washings and filtrate were then evaporated. Purification by chromatography (Si02, heptane:ethyl acetate = 100:0 to 1: 1) afforded the title compound (1.8 g, 71percent) which was obtained as a white solid. MS: m/e = 208.1 [M+H]+.
With sodium tetrahydroborate; zinc(II) chloride In tetrahydrofuran at 60 - 65℃; for 16 h;
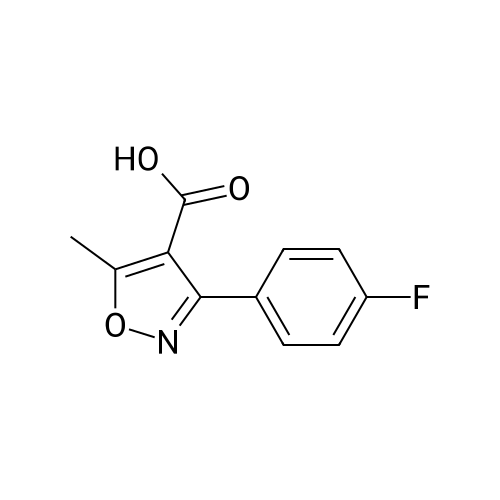
A suspension of 448 g of tetrahydrofuran and 95 g (0.70 mol) of zinc chloride was stirred at 20-30° C. for 1 h. 23.6 g (0.62 mol) of sodium borohydride were added in portions at 20-38° C. and the mixture subsequently stirred at 60-65° C. for 3 h. A solution of 69 g (0.31 mol) of 3-(4-Fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid in 220 g THF was added dropwise and the resulting mixture stirred at 60-65° C. for 16 h. The reaction was then quenched by the drop wise addition of a mixture of 93 g of HCl in 202 g of water at 5-10° C. The mixture was stirred at this temperature for 2 h to dissolve the solids completely. The solvent was removed under reduced pressure with a jacket temperature of 35-40° C. To the residue were added 510 g of water. The resulting suspension was cooled to 20-30° C. and the crystals were filtered off and washed with water. The crude wet product was stirred for 1 h in a mixture of 150 g of water, 31 g of HCl and 419 g of MTBE. The lower aqueous phase was removed and the organic phase was dried with anhydrous sodium sulfate, stirred for 0.5 h and filtered under nitrogen. The filtrate was almost completely concentrated under reduced pressure at 40-45° C. The residue was treated at 20-25° C. with 100 g of MTBE. The mixture was stirred at 55-60° C. for 2 h, cooled to 0° C. and subsequently stirred at this temperature for additional 2 h. The crystals were filtered off and dried at 45-50° C. in vacuum over night to afford 42 g (66percent yield) of the title alcohol as an off-white solid with a purity of 99.9percent (HPLC).
66%
With sodium tetrahydroborate; zinc(II) chloride In tetrahydrofuran at 60 - 65℃; for 16 h;
Step d) r3-(4-Fluorophenyl)-5-methyl-isoxazol-4-yll -methanol Alternative 1) Preparation by reduction of the acid A suspension of 448 g of tetrahydrofuran and 95 g (0.70 mol) of zinc chloride was stirred at 20- 30°C for 1 h. 23.6 g (0.62 mol) of sodium borohydride were added in portions at 20-38 °C and the mixture subsequently stirred at 60-65°C for 3 h. A solution of 69 g (0.31 mol) of 3-(4-Fluoro- phenyl)-5-methyl-isoxazole-4-carboxylic acid in 220 g THF was added dropwise and the resulting mixture stirred at 60-65 °C for 16 h. The reaction was then quenched by the drop wise addition of a mixture of 93 g of HC1 in 202 g of water at 5-10°C. The mixture was stirred at this temperature for 2 h to dissolve the solids completely. The solvent was removed under reduced pressure with a jacket temperature of 35-40°C. To the residue were added 510 g of water. The resulting suspension was cooled to 20-30°C and the crystals were filtered off and washed with water. The crude wet product was stirred for 1 h in a mixture of 150 g of water, 31 g of HC1 and 419 g of MTBE. The lower aqueous phase was removed and the organic phase was dried with anhydrous sodium sulfate, stirred for 0.5 h and filtered under nitrogen. The filtrate was almost completely concentrated under reduced pressure at 40-45 °C. The residue was treated at 20-25 °C with 100 g of MTBE. The mixture was stirred at 55-60°C for 2 h, cooled to 0°C and subsequently stirred at this temperature for additional 2 h. The crystals were filtered off and dried at 45-50°C in vacuum over night to afford 42 g (66 percent yield) of the title alcohol as an off- white solid with a purity of 99.9percent (HPLC).
66%
With sodium tetrahydroborate; zinc(II) chloride In tetrahydrofuran at 60 - 65℃; for 16 h;
A suspension of 448 g of tetrahydrofuran and 95 g (0.70 mol) of zinc chloride was stirred at 20- 30°C for 1 h. 23.6 g (0.62 mol) of sodium borohydride were added in portions at 20-38 °C and the mixture subsequently stirred at 60-65°C for 3 h. A solution of 69 g (0.31 mol) of 3-(4-Fluoro- phenyl)-5-methyl-isoxazole-4-carboxylic acid in 220 g THF was added dropwise and the resulting mixture stirred at 60-65 °C for 16 h. The reaction was then quenched by the drop wise addition of mixture of 93 g of HC1 in 202 g of water at 5-10°C. The mixture was stirred at this temperature for 2 h to dissolve the solids completely. The solvent was removed under reduced pressure with a jacket temperature of 35-40°C. To the residue were added 510 g of water. The resulting suspension was cooled to 20-30°C and the crystals were filtered off and washed with water. The crude wet product was stirred for 1 h in a mixture of 150 g of water, 31 g of HC1 and 419 g of MTBE. The lower aqueous phase was removed and organic phase was dried with 25 kg of anhydrous sodium sulfate, stirred for 0.5 h and filtered under nitrogen. The filtrate was almost completely concentrated under reduced pressure at 40-45 °C. The residue was treated at 20-25 °C with 100 g of MTBE. The mixture was stirred at 55-60°C for 2 h, cooled to 0°C andsubsequently stirred at this temperature for additional 2 h. The crystals were filtered off and dried at 45-50°C in vacuum over night to afford 42 g (66 percent yield) of the title alcohol as an off- white solid with a purity of 99.9percent (HPLC).
66%
With sodium tetrahydroborate; zinc(II) chloride In tetrahydrofuran at 60 - 65℃; for 16 h;
Step d) [3-(4-Fluorophenyl)-5-methyl-isoxazol-4-yl]-methanol A suspension of 448 g of tetrahydrofuran and 95 g (0.70 mol) of zinc chloride was stirred at 20-30° C. for 1 h. 23.6 g (0.62 mol) of sodium borohydride were added in portions at 20-38° C. and the mixture subsequently stirred at 60-65° C. for 3 h. A solution of 69 g (0.31 mol) of 3-(4-Fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid in 220 g THF was added dropwise and the resulting mixture stirred at 60-65° C. for 16 h. The reaction was then quenched by the drop wise addition of mixture of 93 g of HCl in 202 g of water at 5-10° C. The mixture was stirred at this temperature for 2 h to dissolve the solids completely. The solvent was removed under reduced pressure with a jacket temperature of 35-40° C. To the residue were added 510 g of water. The resulting suspension was cooled to 20-30° C. and the crystals were filtered off and washed with water. The crude wet product was stirred for 1 h in a mixture of 150 g of water, 31 g of HCl and 419 g of MTBE. The lower aqueous phase was removed and organic phase was dried with 25 kg of anhydrous sodium sulfate, stirred for 0.5 h and filtered under nitrogen. The filtrate was almost completely concentrated under reduced pressure at 40-45° C. The residue was treated at 20-25° C. with 100 g of MTBE. The mixture was stirred at 55-60° C. for 2 h, cooled to 0° C. and subsequently stirred at this temperature for additional 2 h. The crystals were filtered off and dried at 45-50° C. in vacuum over night to afford 42 g (66percent yield) of the title alcohol as an off-white solid with a purity of 99.9percent (HPLC).
66%
With sodium tetrahydroborate; zinc(II) chloride In tetrahydrofuran at 60 - 65℃; for 16 h;
Step d) r3-(4-Fluorophenyl)-5-methyl-isoxazol-4-yll -methanol: A suspension of 448 g of tetrahydrofuran and 95 g (0.70 mol) of zinc chloride was stirred at 20-30°C for 1 h. 23.6 g (0.62 mol) of sodium borohydride were added in portions at 20-38 °C and the mixture subsequently stirred at 60-65°C for 3 h. A solution of 69 g (0.31 mol) of 3-(4-Fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid in 220 g THF was added dropwise and the resulting mixture stirred at 60-65 °C for 16 h. The reaction was then quenched by the drop wise addition of mixture of 93 g of HCl in 202 g of water at 5-10°C. The mixture was stirred at this temperature for 2 h to dissolve the solids completely. The solvent was removed under reduced pressure with a jacket temperature of 35-40°C. To the residue were added 510 g of water. The resulting suspension was cooled to 20-30°C and the crystals were filtered off and washed with water. The crude wet product was stirred for 1 h in a mixture of 150 g of water, 31 g of HCl and 419 g of MTBE. The lower aqueous phase was removed and organic phase was dried with 25 kg of anhydrous sodium sulfate, stirred for 0.5 h and filtered under nitrogen. The filtrate was almost completely concentrated under reduced pressure at 40-45 °C. The residue was treated at 20-25 °C with 100 g of MTBE. The mixture was stirred at 55-60°C for 2 h, cooled to 0°C and subsequently stirred at this temperature for additional 2 h. The crystals were filtered off and dried at 45-50°C in vacuum over night to afford 42 g (66 percent yield) of the title alcohol as an off-white solid with a purity of 99.9percent (HPLC).
To a a solution of <strong>[1018297-63-6][3-(4-fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol</strong> (500 mg, 2.41 mmol) in THF (5 mL) at room temperature was added sodium hydride (55% dispersion in mineral oil, 137 mg, 3.1 mmol) and the reaction mixture stirred at room temperature for 1 h. Then a solution of 2-chloro-5-bromo-pyridine (484 mg, 2.41 mmol) in THF (5 mL) was added at room temperature and the reaction mixture was stirred at room temperature for 2 h. The reaction mixture was then diluted with methanol and water and the mixture was extracted with ethyl acetate. The combined organic layers were then washed with water and brine and then dried over sodium sulfate, filtered and evaporated. Purification by chromatography (SiO2, heptane:ethyl acetate=9:1 to 4:1) afforded the title compound (128 mg, 15%) which was obtained as a colourless oil. MS: m/e=363.1/365.1 [M+H]+.
With lithium aluminium tetrahydride; In tetrahydrofuran; at 0 - 20℃;
To a suspension of LiAlH4 (75.9 mg, 2.0 mmol) in THF (2 mL) was added at 0-10 C. within 15 minutes a solution of 3-(4-Fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid ethyl ester (0.50 g, 2.0 mmol) in THF (3 mL) and the resulting solution was allowed to warm to room temperature and subsequently stirred at this temperature for at least one hour. Water (15 mL) was added dropwise and the resulting suspension was then filtered and the filter cake washed with ethyl acetate (15 mL). From the biphasic filtrate the layers were separated and the organic layer was washed with water (1×15 mL). Both combined aqueous layers were back extracted with ethyl acetate (2×15 mL). The combined organic layers were dried over Na2SO4 and subsequently concentrated to dryness (45 C./25 mbar) to afford 0.375 g (90%) of the title compound as a slightly yellow solid with a purity of 100% (by HPLC).
71%
d) [3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol As described for example 48d, 3-(4-fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid ethyl ester (3.0 g, 12 mmol) was converted, instead of 3-(3-fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid ethyl ester, to the title compound (1.8 g, 71%) which was obtained as a white solid. MS: m/e=208.1 [M+H]+.
71%
o a solution of 3-(4-fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid ethyl ester (3.0 g, 12 mmol) (6.18 g, 25 mmol) in THF (320 mL) was added portionwise lithiumaluminiumhydride (528 mg, 14 mmol) at 0 C and the reaction mixture was stirred at room temperature for 3 h. The mixture was then cooled to 0 C and water (518 mu) added followed by sodium hydroxide (15% solution, 518 mu) and then again water (1.5 mL) and the mixture then stirred overnight at room temperature. The precipitate was then filtered off and washed with THF. The combined washings and filtrate were then evaporated. Purification by chromatography (Si02, heptane:ethyl acetate = 100:0 to 1: 1) afforded the title compound (1.8 g, 71%) which was obtained as a white solid. MS: m/e = 208.1 [M+H]+.
71%
Step d: [3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol To a solution of 3-(4-fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid ethyl ester (3.0 g, 12 mmol) (6.18 g, 25 mmol) in THF (320 mL) was added portionwise lithiumaluminiumhydride (528 mg, 14 mmol) at 0 C. and the reaction mixture was stirred at room temperature for 3 h. The mixture was then cooled to 0 C. and water (518 muL) added followed by sodium hydroxide (15% solution, 518 muL) and then again water (1.5 mL) and the mixture then stirred overnight at room temperature. The precipitate was then filtered off and washed with THF. The combined washings and filtrate were then evaporated. Purification by chromatography (SiO2, heptane:ethyl acetate=100:0 to 1:1) afforded the title compound (1.8 g, 71%) which was obtained as a white solid. MS: m/e=208.1 [M+H]+.
71%
Step d: r3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-yll-methanol: To a solution of 3-(4-fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid ethyl ester (3.0 g, 12 mmol) (6.18 g, 25 mmol) in THF (320 mL) was added portionwise lithiumaluminiumhydride (528 mg, 14 mmol) at 0 C and the reaction mixture was stirred at room temperature for 3 h. The mixture was then cooled to 0 C and water (518 mul) added followed by sodium hydroxide (15% solution, 518 mul) and then again water (1.5 mL) and the mixture then stirred overnight at room temperature. The precipitate was then filtered off and washed with THF. The combined washings and filtrate were then evaporated. Purification by chromatography (Si02, heptane:ethyl acetate = 100:0 to 1: 1) afforded the title compound (1.8 g, 71%) which was obtained as a white solid. MS: m/e = 208.1 [M+H]+.
With lithium aluminium tetrahydride; In tetrahydrofuran; at 20℃;
Step d) r3-(4-Fluorophenyl)-5-methyl-isoxazol-4-yll -methanol Alternative 2) Preparation by reduction of the ethyl ester (i) with L1AIH4 as reducing agent: To a suspension of LiAlH4 (75.9 mg, 2.0 mmol) in THF (2 mL) was added at 0-10C within 15 minutes a solution of 3-(4-Fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid ethyl ester (0.50 g, 2.0 mmol) in THF (3 mL) and the resulting solution was allowed to warm to room temperature and subsequently stirred at this temperature for at least one hour. Water (15 mL) was added dropwise and the resulting suspension was then filtered and the filter cake washed with ethyl acetate (15 mL). From the biphasic filtrate the layers were separated and the organic layer was washed with water (1x15 mL). Both combined aqueous layers were back extracted with ethyl acetate (2x15 mL). The combined organic layers were dried over Na2S04 and subsequently concentrated to dryness (45C/25 mbar) to afford 0.375 g (90%) of the title compound as a slightly yellow solid with a purity of 100% (by HPLC).
With triphenylphosphine; In tetrahydrofuran; toluene;
a) 2-[3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-ylmethyl]-isoindole-1,3-dione To a solution of <strong>[1018297-63-6][3-(4-fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol</strong> (5.0 g, 24 mmol) in THF (290 mL) was added phthalimide (4.7 g, 32 mmol) and triphenylphosphine (8.4 g, 32 mmol) at ambient temperature under an argon atmosphere. Then a solution of diethyl azodicarboxylate (40% in toluene, 12.5 mL, 32 mmol) was added and the reaction mixture was stirred for 1 h at room temperature. Concentration and repeated trituration and then purification by chromatography (SiO2, heptane:ethyl acetate=100:0 to 1:1) afforded the title compound (6.0 g, 74%) as a white solid. MS: m/e=337.1 [M+H]+.
e) 3-Chloro-6-[3-(4-fluoro-phenyl)-5-methyl-isoxazol-4-ylmethoxy]-pyridazine As described for example 48e, <strong>[1018297-63-6][3-(4-fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol</strong> (2.8 g, 14 mmol) was converted, instead of [3-(3-fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol, to the title compound (3.2 g, 74%) which was obtained as a white solid. MS: m/e=319.9 [M+H].
EXAMPLE 91 6-[3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-ylmethoxy]-[1,2,4]triazolo[4,3-b]pyridazine As described for example 90, <strong>[1018297-63-6][3-(4-fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol</strong> (100 mg, 0.53 mmol) was converted, instead of [3-(3-fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol, to the title compound (99 mg, 63%) which was obtained as a white solid. MS: m/e=326.1 [M+H]-.
With triphenylphosphine; diethylazodicarboxylate; In tetrahydrofuran; toluene; at 20℃; for 1h;
To a solution of [3-(3-fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol (5.0 g, 24 mmol) in THF (290 mL) was added phthalimide (4.7 g, 32 mmol) and triphenylphosphine (8.4 g, 32 mmol) at ambient temperature under an argon atmosphere. Then a solution of diethyl azodicarboxylate (40% in toluene, 12.5 mL, 32 mmol) was added and the reaction mixture was stirred for 1 h at room temperature. Concentration and repeated trituration and then purification by chromatography (SiO2, heptane:ethyl acetate=100:0 to 1:1) afforded the title compound (6.0 g, 74%) as a white solid. MS: m/e=337.1 [M+H]+.
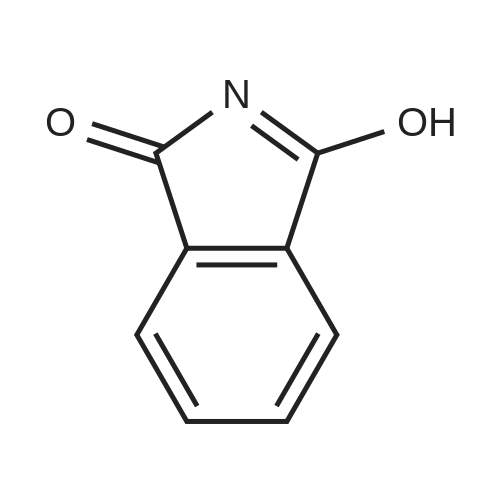
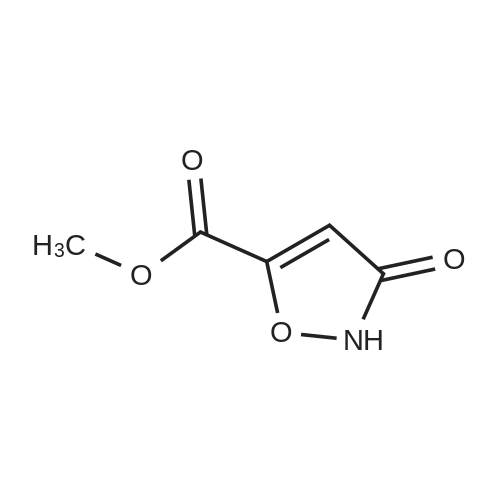
a) 3-[3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-ylmethoxy]-isoxazole-5-carboxylic acid methyl ester To a solution of <strong>[1018297-63-6][3-(4-fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol</strong> (1.0 g, 4.8 mmol) in THF (70 mL) was added 3-oxo-2,3-dihydro-isoxazole-5-carboxylic acid methyl ester (0.69 g, 4.8 mmol) and triphenylphosphine (1.65 g, 6.3 mmol) at ambient temperature under an argon atmosphere. Then diethyl azodicarboxylate (2.7 mL, 6.3 mmol) was added at 4 C. and the reaction mixture was stirred for 3 h at room temperature. Concentration and purification by chromatography (SiO2, heptane:ethyl acetate=100:0 to 7:3) afforded the title compound (958 mg, 60%) as a white solid. MS: m/e=333.2 [M+H]+.
With sodium hydride; In tetrahydrofuran; mineral oil; at 20℃; for 1h;
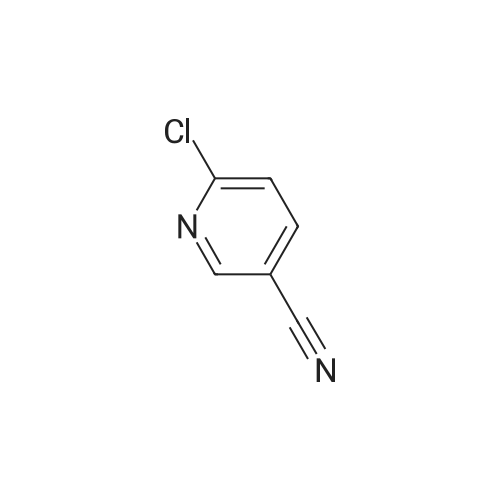
To a suspension of sodium hydride (60% in mineral oil, 7.9 g, 181 mmol, 1.5 eq.) in THF (65 mL) was added within 30 minutes at room temperature a solution of [3-(4-Fluorophenyl)-5-methyl-isoxazol-4-yl]-methanol (25.0 g, 121 mmol) and 6-chloronicotinonitrile (16.7 g, 121 mmol) in THF (120 mL) and the resulting mixture was stirred for one hour. A solution of citric acid (18.5 g, 96.5 mmol) in water (185 mL) was added to the reaction mixture within 30 minutes. From the resulting THF/water mixture THF was distilled off under reduced pressure at a jacket temperature of 60 C. and replaced by ethanol. In total 284 g of ethanol were added. The resulting suspension was stirred for one hour at room temperature. The crystals were filtered off, washed with a mixture of ethanol (60 mL) and water (60 mL) and subsequently dried at 50 C./<25 mbar to afford 36.5 g (91% corrected yield) of the title nitrile as an off-white solid with an assay of 93% (w/w).
91%
With sodium hydride; In tetrahydrofuran; mineral oil; at 20℃; for 1h;
Step 1) 6-r3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-ylmethoxyl-nicotinonitrile To a suspension of sodium hydride (60% in mineral oil, 7.9 g, 181 mmol, 1.5 eq.) in THF (65 mL) was added within 30 minutes at room temperature a solution of [3-(4-Fluorophenyl)-5- methyl-isoxazol-4-yl] -methanol (25.0 g, 121 mmol) and 6-chloronicotinonitrile (16.7 g, 121 mmol) in THF (120 mL) and the resulting mixture was stirred for one hour. A solution of citric acid (18.5 g, 96.5 mmol) in water (185 mL) was added to the reaction mixture within 30 minutes. From the resulting THF/water mixture THF was distilled off under reduced pressure at a jacket temperature of 60C and replaced by ethanol. In total 284 g of ethanol were added. The resulting suspension was stirred for one hour at room temperature. The crystals were filtered off, washed with a mixture of ethanol (60 mL) and water (60 mL) and subsequently dried at 50C/<25 mbar to afford 36.5 g (91% corrected yield) of the title nitrile as an off-white solid with an assay of 93 %(w/w).
91%
With sodium hydride; In tetrahydrofuran; mineral oil; for 1h;
To a suspension of sodium hydride (60% in mineral oil, 7.9 g, 181 mmol, 1.5 eq.) in THF (65 mL) was added within 30 minutes at room temperature a solution of [3-(4-Fluorophenyl)-5- methyl-isoxazol-4-yl] -methanol (25.0 g, 121 mmol) and 6-chloronicotinonitrile (16.7 g, 121 mmol) in THF (120 mL) and the resulting mixture was stirred for one hour. A solution of citric acid (18.5 g, 96.5 mmol) in water (185 mL) was added to the reaction mixture within 30 minutes. From the resulting THF/water mixture THF was distilled off under reduced pressure at a jacket temperature of 60C and replaced by ethanol. In total 284 g of ethanol were added. The resulting suspension was stirred for one hour at room temperature. The crystals were filtered off, washed with a mixture of ethanol (60 mL) and water (60 mL) and subsequently dried at 50C/<25 mbar to afford 36.5 g (91 corrected yield) of the title nitrile as an off-white solid with an assay of 93 %(w/w).
91%
With sodium hydride; In tetrahydrofuran; mineral oil; at 20℃; for 1.5h;
Step e) 6-[3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-ylmethoxy]-nicotinonitrile To a suspension of sodium hydride (60% in mineral oil, 7.9 g, 181 mmol, 1.5 eq.) in THF (65 mL) was added within 30 minutes at room temperature a solution of [3-(4-Fluorophenyl)-5-methyl-isoxazol-4-yl]-methanol (25.0 g, 121 mmol) and 6-chloronicotinonitrile (16.7 g, 121 mmol) in THF (120 mL) and the resulting mixture was stirred for one hour. A solution of citric acid (18.5 g, 96.5 mmol) in water (185 mL) was added to the reaction mixture within 30 minutes. From the resulting THF/water mixture THF was distilled off under reduced pressure at a jacket temperature of 60 C. and replaced by ethanol. In total 284 g of ethanol were added. The resulting suspension was stirred for one hour at room temperature. The crystals were filtered off, washed with a mixture of ethanol (60 mL) and water (60 mL) and subsequently dried at 50 C./<25 mbar to afford 36.5 g (91% corrected yield) of the title nitrile as an off-white solid with an assay of 93% (w/w).
91%
With sodium hydride; In tetrahydrofuran; mineral oil; at 20℃; for 1.5h;
Step e) 6-r3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-ylmethoxyl-nicotinonitrile: To a suspension of sodium hydride (60% in mineral oil, 7.9 g, 181 mmol, 1.5 eq.) in THF (65 mL) was added within 30 minutes at room temperature a solution of [3-(4-Fluorophenyl)-5-methyl-isoxazol-4-yl] -methanol (25.0 g, 121 mmol) and 6-chloronicotinonitrile (16.7 g, 121 mmol) in THF (120 mL) and the resulting mixture was stirred for one hour. A solution of citric acid (18.5 g, 96.5 mmol) in water (185 mL) was added to the reaction mixture within 30 minutes. From the resulting THF/water mixture THF was distilled off under reduced pressure at a jacket temperature of 60C and replaced by ethanol. In total 284 g of ethanol were added. The resulting suspension was stirred for one hour at room temperature. The crystals were filtered off, washed with a mixture of ethanol (60 mL) and water (60 mL) and subsequently dried at 50C/<25 mbar to afford 36.5 g (91 corrected yield) of the title nitrile as an off-white solid with an assay of 93 %(w/w).
With sodium tetrahydroborate; zinc(II) chloride; In tetrahydrofuran; at 60 - 65℃; for 16h;
A suspension of 448 g of tetrahydrofuran and 95 g (0.70 mol) of zinc chloride was stirred at 20-30 C. for 1 h. 23.6 g (0.62 mol) of sodium borohydride were added in portions at 20-38 C. and the mixture subsequently stirred at 60-65 C. for 3 h. A solution of 69 g (0.31 mol) of 3-(4-Fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid in 220 g THF was added dropwise and the resulting mixture stirred at 60-65 C. for 16 h. The reaction was then quenched by the drop wise addition of a mixture of 93 g of HCl in 202 g of water at 5-10 C. The mixture was stirred at this temperature for 2 h to dissolve the solids completely. The solvent was removed under reduced pressure with a jacket temperature of 35-40 C. To the residue were added 510 g of water. The resulting suspension was cooled to 20-30 C. and the crystals were filtered off and washed with water. The crude wet product was stirred for 1 h in a mixture of 150 g of water, 31 g of HCl and 419 g of MTBE. The lower aqueous phase was removed and the organic phase was dried with anhydrous sodium sulfate, stirred for 0.5 h and filtered under nitrogen. The filtrate was almost completely concentrated under reduced pressure at 40-45 C. The residue was treated at 20-25 C. with 100 g of MTBE. The mixture was stirred at 55-60 C. for 2 h, cooled to 0 C. and subsequently stirred at this temperature for additional 2 h. The crystals were filtered off and dried at 45-50 C. in vacuum over night to afford 42 g (66% yield) of the title alcohol as an off-white solid with a purity of 99.9% (HPLC).
66%
With sodium tetrahydroborate; zinc(II) chloride; In tetrahydrofuran; at 60 - 65℃; for 16h;
Step d) r3-(4-Fluorophenyl)-5-methyl-isoxazol-4-yll -methanol Alternative 1) Preparation by reduction of the acid A suspension of 448 g of tetrahydrofuran and 95 g (0.70 mol) of zinc chloride was stirred at 20- 30C for 1 h. 23.6 g (0.62 mol) of sodium borohydride were added in portions at 20-38 C and the mixture subsequently stirred at 60-65C for 3 h. A solution of 69 g (0.31 mol) of 3-(4-Fluoro- phenyl)-5-methyl-isoxazole-4-carboxylic acid in 220 g THF was added dropwise and the resulting mixture stirred at 60-65 C for 16 h. The reaction was then quenched by the drop wise addition of a mixture of 93 g of HC1 in 202 g of water at 5-10C. The mixture was stirred at this temperature for 2 h to dissolve the solids completely. The solvent was removed under reduced pressure with a jacket temperature of 35-40C. To the residue were added 510 g of water. The resulting suspension was cooled to 20-30C and the crystals were filtered off and washed with water. The crude wet product was stirred for 1 h in a mixture of 150 g of water, 31 g of HC1 and 419 g of MTBE. The lower aqueous phase was removed and the organic phase was dried with anhydrous sodium sulfate, stirred for 0.5 h and filtered under nitrogen. The filtrate was almost completely concentrated under reduced pressure at 40-45 C. The residue was treated at 20-25 C with 100 g of MTBE. The mixture was stirred at 55-60C for 2 h, cooled to 0C and subsequently stirred at this temperature for additional 2 h. The crystals were filtered off and dried at 45-50C in vacuum over night to afford 42 g (66 % yield) of the title alcohol as an off- white solid with a purity of 99.9% (HPLC).
66%
With sodium tetrahydroborate; zinc(II) chloride; In tetrahydrofuran; at 60 - 65℃; for 16h;
A suspension of 448 g of tetrahydrofuran and 95 g (0.70 mol) of zinc chloride was stirred at 20- 30C for 1 h. 23.6 g (0.62 mol) of sodium borohydride were added in portions at 20-38 C and the mixture subsequently stirred at 60-65C for 3 h. A solution of 69 g (0.31 mol) of 3-(4-Fluoro- phenyl)-5-methyl-isoxazole-4-carboxylic acid in 220 g THF was added dropwise and the resulting mixture stirred at 60-65 C for 16 h. The reaction was then quenched by the drop wise addition of mixture of 93 g of HC1 in 202 g of water at 5-10C. The mixture was stirred at this temperature for 2 h to dissolve the solids completely. The solvent was removed under reduced pressure with a jacket temperature of 35-40C. To the residue were added 510 g of water. The resulting suspension was cooled to 20-30C and the crystals were filtered off and washed with water. The crude wet product was stirred for 1 h in a mixture of 150 g of water, 31 g of HC1 and 419 g of MTBE. The lower aqueous phase was removed and organic phase was dried with 25 kg of anhydrous sodium sulfate, stirred for 0.5 h and filtered under nitrogen. The filtrate was almost completely concentrated under reduced pressure at 40-45 C. The residue was treated at 20-25 C with 100 g of MTBE. The mixture was stirred at 55-60C for 2 h, cooled to 0C andsubsequently stirred at this temperature for additional 2 h. The crystals were filtered off and dried at 45-50C in vacuum over night to afford 42 g (66 % yield) of the title alcohol as an off- white solid with a purity of 99.9% (HPLC).
66%
With sodium tetrahydroborate; zinc(II) chloride; In tetrahydrofuran; at 60 - 65℃; for 16h;
Step d) [3-(4-Fluorophenyl)-5-methyl-isoxazol-4-yl]-methanol A suspension of 448 g of tetrahydrofuran and 95 g (0.70 mol) of zinc chloride was stirred at 20-30 C. for 1 h. 23.6 g (0.62 mol) of sodium borohydride were added in portions at 20-38 C. and the mixture subsequently stirred at 60-65 C. for 3 h. A solution of 69 g (0.31 mol) of 3-(4-Fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid in 220 g THF was added dropwise and the resulting mixture stirred at 60-65 C. for 16 h. The reaction was then quenched by the drop wise addition of mixture of 93 g of HCl in 202 g of water at 5-10 C. The mixture was stirred at this temperature for 2 h to dissolve the solids completely. The solvent was removed under reduced pressure with a jacket temperature of 35-40 C. To the residue were added 510 g of water. The resulting suspension was cooled to 20-30 C. and the crystals were filtered off and washed with water. The crude wet product was stirred for 1 h in a mixture of 150 g of water, 31 g of HCl and 419 g of MTBE. The lower aqueous phase was removed and organic phase was dried with 25 kg of anhydrous sodium sulfate, stirred for 0.5 h and filtered under nitrogen. The filtrate was almost completely concentrated under reduced pressure at 40-45 C. The residue was treated at 20-25 C. with 100 g of MTBE. The mixture was stirred at 55-60 C. for 2 h, cooled to 0 C. and subsequently stirred at this temperature for additional 2 h. The crystals were filtered off and dried at 45-50 C. in vacuum over night to afford 42 g (66% yield) of the title alcohol as an off-white solid with a purity of 99.9% (HPLC).
66%
With sodium tetrahydroborate; zinc(II) chloride; In tetrahydrofuran; at 60 - 65℃; for 16h;
Step d) r3-(4-Fluorophenyl)-5-methyl-isoxazol-4-yll -methanol: A suspension of 448 g of tetrahydrofuran and 95 g (0.70 mol) of zinc chloride was stirred at 20-30C for 1 h. 23.6 g (0.62 mol) of sodium borohydride were added in portions at 20-38 C and the mixture subsequently stirred at 60-65C for 3 h. A solution of 69 g (0.31 mol) of 3-(4-Fluoro-phenyl)-5-methyl-isoxazole-4-carboxylic acid in 220 g THF was added dropwise and the resulting mixture stirred at 60-65 C for 16 h. The reaction was then quenched by the drop wise addition of mixture of 93 g of HCl in 202 g of water at 5-10C. The mixture was stirred at this temperature for 2 h to dissolve the solids completely. The solvent was removed under reduced pressure with a jacket temperature of 35-40C. To the residue were added 510 g of water. The resulting suspension was cooled to 20-30C and the crystals were filtered off and washed with water. The crude wet product was stirred for 1 h in a mixture of 150 g of water, 31 g of HCl and 419 g of MTBE. The lower aqueous phase was removed and organic phase was dried with 25 kg of anhydrous sodium sulfate, stirred for 0.5 h and filtered under nitrogen. The filtrate was almost completely concentrated under reduced pressure at 40-45 C. The residue was treated at 20-25 C with 100 g of MTBE. The mixture was stirred at 55-60C for 2 h, cooled to 0C and subsequently stirred at this temperature for additional 2 h. The crystals were filtered off and dried at 45-50C in vacuum over night to afford 42 g (66 % yield) of the title alcohol as an off-white solid with a purity of 99.9% (HPLC).
Alternative 2: Telescoped process To as suspension of sodium hydride (60% in mineral oil, 3.95 g, 99 mmol, 1.6 eq.) in THF (120 mL) was added within 120 minutes at 25-32C a solution of [3-(4-Fluorophenyl)-5-methyl- isoxazol-4-yl] -methanol (12.50 g, 60 mmol) and 6-chloronicotinonitrile (8.36 g, 60 mmol) in THF (60 mL) and the resulting mixture was stirred for one hour at approx. 30C. The mixture was then treated drop wise at room temperature with water (100 mL). THF was distilled off under reduced pressure (200-70 mbar) with a jacket temperature of 50C. The residue was diluted with ethanol (90 mL) and subsequently treated at 20 to 35C with 28% sodium hydroxide solution (69.6 g, 487 mmol). The mixture was heated to 50-55C and subsequently stirred at this temperature for 15 hour. The reaction mixture was treated with toluene (150 mL) and the resulting biphasic mixture was stirred for 15 minutes and the layers were then allowed to separate for 30 minutes. The lower product-containing aqueous layer was separated and the toluene layer was extracted at 30C with water (1x50 mL). The combined aqueous layers were acidified with 20% sulfuric acid (approx. 150 g) until a pH of 3.0-3.3 was obtained. The suspension was treated with THF (120 mL) and the resulting biphasic mixture was stirred for 15 minutes and the layers were then allowed to separate for 30 minutes. The lower aqueous layer was removed and the product-containing organic layer was diluted with toluene (150 mL) to afford a biphasic mixture from which the lower aqueous layer was separated. The aqueous layer was removed and the organic layer was washed with water (2x30 mL). From the organic layer THF, Ethanol and water were then completely distilled off under reduced pressure and at a jacket temperature of 40-80C and continuously replaced by toluene (250 mL in total). At the end of the distillation a volume of approx. 300 mL was adjusted in the reactor. The partly precipitated product was completely re-dissolved by heating the suspension to 100-105C. The clear solution was cooled to 15-20C within 5-10 hours whereupon crystallization occurred. The crystals were filtered off, washed with toluene (100 mL) and subsequently dried at 55C/
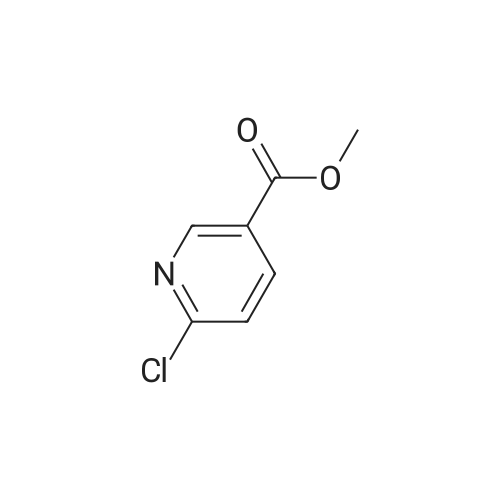
Step e: 6-[3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-ylmethoxy]-nicotinic acid methyl ester To a suspension of sodium hydride (55percent dispersion in mineral oil, 852 mg, 20 mmol) in THF (27 mL) was added a solution of [3-(4-fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol (103 mg, 0.55 mmol) (3.68 g, 18 mmol) in THF (54 mL) at 0° C. and the reaction mixture warmed to room temperature over 30 min. Then a solution of methyl 6-chloronicotinate (3.35 g, 20 mmol) in THF (1.5 mL) was added dropwise at 0° C. and the reaction mixture was stirred at room temperature overnight. The reaction mixture was then poured into aqueous sodium chloride (saturated) and the mixture was extracted with ethyl acetate. The combined organic layers were then washed with water and brine and then dried over sodium sulfate, filtered and evaporated. Purification by chromatography (SiO2, heptane:ethyl acetate=7:3) afforded the title compound (81 mg, 47percent) which was obtained as a light yellow solid. MS: m/e=343.3 [M+H]+.
47%
To a suspension of sodium hydride (55percent dispersion in mineral oil, 852 mg, 20 mmol) in THF (27 mL) was added a solution of [3-(4-fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol (103 mg, 0.55 mmol) (3.68 g, 18 mmol) in THF (54 mL) at 0 °C and the reaction mixture warmed to room temperature over 30 min. Then a solution of methyl 6-chloronicotinate (3.35 g, 20 mmol) in THF (1.5 mL) was added dropwise at 0 °C and the reaction mixture was stirred at room temperature overnight. The reaction mixture was then poured into aqueous sodium chloride (saturated) and the mixture was extracted with ethyl acetate. The combined organic layers were then washed with water and brine and then dried over sodium sulfate, filtered and evaporated. Purification by chromatography (Si02, heptane:ethyl acetate = 7:3) afforded the title compound (81 mg, 47percent) which was obtained as a light yellow solid. MS: m/e = 343.3 [M+H]+.
47%
Step e: 6- r3-(4-Fluoro-phenyl)-5-methyl-isoxazol-4-ylmethoxyl -nicotinic acid methyl ester: To a suspension of sodium hydride (55percent dispersion in mineral oil, 852 mg, 20 mmol) in THF (27 mL) was added a solution of [3-(4-fluoro-phenyl)-5-methyl-isoxazol-4-yl]-methanol (103 mg, 0.55 mmol) (3.68 g, 18 mmol) in THF (54 mL) at 0 °C and the reaction mixture warmed to room temperature over 30 min. Then a solution of methyl 6-chloronicotinate (3.35 g, 20 mmol) in THF (1.5 mL) was added dropwise at 0 °C and the reaction mixture was stirred at room temperature overnight. The reaction mixture was then poured into aqueous sodium chloride (saturated) and the mixture was extracted with ethyl acetate. The combined organic layers were then washed with water and brine and then dried over sodium sulfate, filtered and evaporated. Purification by chromatography (Si02, heptane:ethyl acetate = 7:3) afforded the title compound (81 mg, 47percent) which was obtained as a light yellow solid. MS: m/e = 343.3 [M+H]+.
(3-(4-fluorophenyl)-5-(methyl-d3)isoxazol-4-yl)methanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
99%
With [D]-sodium hydroxide; In d(4)-methanol; water-d2; at 85℃; for 2h;Microwave irradiation; Sealed tube;
(3 -(4-Fluorophenyl)-5 -(methyl-d3)isoxazol-4-yl)methanol (500 mg, 2.4 mmol) was placed in a microwave vial with a stir bar. To this was added 2.0 mL of deuterium oxide, 8.0 ml of CD3OD and 1.0 ml of 30% NaOD in in D20. The vial was sealed and heated to 85 C for two hours with microwave irradiation. The vial then was cooled, and quenched by addition to aqueous ammonium chloride. The compound was extracted into ethyl acetate, and the solvent was removed by rotary evaporation to give the title product as a white solid in 99% yield (504 mg) without further purification. ?H NMR analysis indicated about 80% deuterium incorporation. ?H NMR (400 MHz, cdcl3) oe 7.91 - 7.68 (m, 2H), 7.18 - 6.98 (m, 2H), 4.48 (s, 2H), 2.41 - 2.36 (m, 0.6 H, 80% deuterium incorporation). ?3C NMR (101 MHz, cdcl3) oe 168.7, 163.7 (d, JF=25l.5 Hz, 1C) 161.7, 130.4, 130.3, 125.2, 116.1, 115.9, 113.0, 53.6, 10.7 (m, C-D splitting). MS: Expected: 211.2 (M+H). Observed: 211.1.
4-(bromomethyl)-3-(4-fluorophenyl)-5-methyl-1,2-oxazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
97%
With phosphorus tribromide; In dichloromethane; at 20℃; for 1h;
4.98 g (24.0 mmol) of [3-(4-fluorophenyl)-5-methyl-1 ,2-oxazol-4-yl]methanol (WO 2013/057123 A1 , Hoffmann-La Roche) was dissolved in 80 ml_ of anhydrous dichloromethane, and 9.76 g (3.39 ml_, 36.1 mmol) of phosphorus tribromide was added dropwise to the stirred solution. The reaction mixture was stirred for 1 hour at room temeprature, and poured into 50 ml_ of saturated sodium bicarbonate solution. The mixture was stirred for another 10 minutes, and the phases were separated. The organic phase was washed with water, dried over anhydrous sodium sulfate, and evaporated to afford 5.89 g (97%) of the title compound as a yellow-brownish solid. MS (ESI) m/z: 269.9 [M+H]+.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping