* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Alternatively, not previously purified crude reaction mixture can be dissolved in methanol (375 mL) in the presence of triethylamine (60 mL) and stirred at 65°C for 2 hours. The solvents were removed under reduced pressure and the residue treated with water / ethyl acetate. Organic phase was dried over sodium sulfate and evaporated to dryness. Purification of the crude by chromatography over silica gel (DCM/EtOH/NH3 5N in MeOH = 1000/50/5) and crystallisation of the so obtained compound from EtOAc / hexane afforded 8.4 g of the title compound as a white solid (78percent yield). IH-NMR (400 MHz), δ (ppm, DMSO-d6): 1.26 - 1.43 (m, 2H) 1.86 - 2.02 (m, 2H) 2.23 (s, 3H) 2.42 - 2.46 (m, 4H) 3.23 - 3.29 (m, 4H) 3.45 - 3.54 (m, 2H) 3.62 - 3.75 (m, IH) 3.82 (dt, J=I 1.61, 3.83 Hz, 2H) 4.05 (s, 2H) 6.14 (d, J=2.07 Hz, IH) 6.24 (dd, J=8.90, 2.19 Hz, IH) 6.94 - 7.06 (m, 3H) 7.26 (dd, J=8.66, 1.46 Hz, IH) 7.41 (d, J=8.66 Hz, IH) 7.50 (d, IH) 7.80 (d, J=9.15 Hz, IH) 8.29 (d, J=7.68 Hz, IH) 10.08 (s, IH) 12.63 (s, IH).
71%
With potassium carbonate In methanol; water at 20℃; for 3 h; Inert atmosphere
Compound 40 (0.66 g, 1.0 mmol) with magnetic stirring Potassium carbonate (0.42 g, 3.0 mmol) was added to a methanol/water solution (11 mL, 10/1). The reaction was stirred at room temperature for 3 hours under a nitrogen atmosphere. Water (30 mL) was added, a large amount of a gray solid was precipitated, filtered and washed with water (10mL). Dissolved in dichloromethane (20 mL), dried and concentrated. The residue was passed through a silica gel column to give a white solid, 0.4 g. The yield was 71.0percent.
2.88 kg
With water; potassium carbonate In methanol at 10℃; Large scale
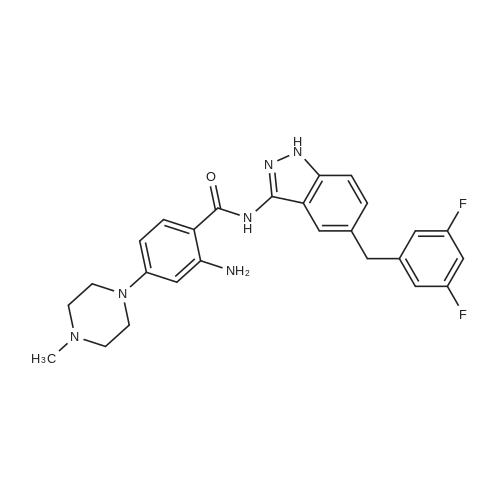
The so obtained crude N-[5-(3,5-difluorobenzyl)-1 H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-[(tetrahydro-pyran-4-yl)- 2,2,2-trifluoro-acetyl)-amino]-benzamide, with HPLC purity > 95percent, is dissolved in methanol and added with a solution of K2CO3 in water/methanol at 10°C. The solution is filtered and dropped into water; the precipitate amorphous N-[5-(3,5- difluorobenzyl)-1 H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-(tetrahydro-pyran-4-ylamino)-benzamide is filtered, washed with water and dried under vacuum (2.88 Kg).
2.88 kg
With water; potassium carbonate In methanol at 10℃; Large scale
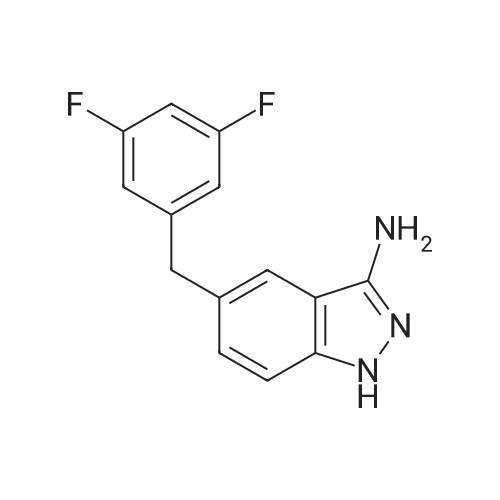
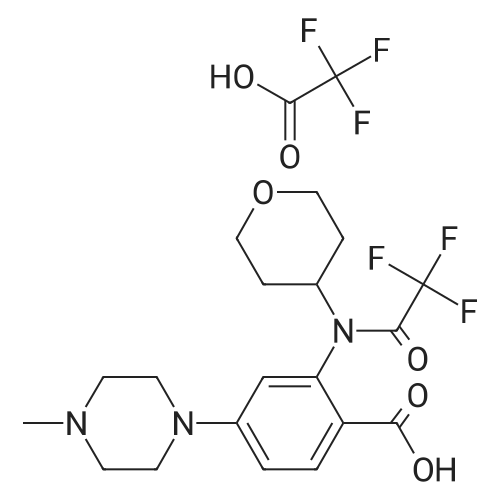
To a suspension of 4-(4-methylpiperazin-1-yl)-2-[tetrahydro-2H-piran-4-yl(trifluoroacetyl)-amino]-benzoic acid trifluoroacetate (3.7 Kg, 7 mol) in dry DCM (36 L) and N,N-dimethylformamide (14 mL), oxalyl chloride (1.78 L, 21 mol) is added. The mixture is stirred for about 1.5 hours and evaporated to oily residue; dry DCM is then added and evaporated twice. [0117] The acyl chloride of formula (II) is suspended in dry DCM and the suspension is added slowly and gradually to a solution of 5-(3,5-difluoro-benzyl)-1H-indazol-3-ylamine (1.6 Kg, 6.1 mol) in dry pyridine (16 L) at −40/-30° C. The addition is blocked when the 5-(3,5-difluoro-benzyl)-1H-indazol-3-ylamine is completely reacted. After about 1 hour the solvent is evaporated and DCM (55 L), methanol (6.5 L), and MTBE (55 L) are sequentially added. The purified protected compound of formula (IV) is filtered, washed with a mixture 10/10/1 of DCM/MTBE/MeOH and dried under vacuum (3.8 Kg). [0118] The so obtained crude N-[5-(3,5-difluorobenzyl)-1H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-[(tetrahydro-pyran-4-yl)-2,2,2-trifluoro-acetyl)-amino]-benzamide, with HPLC purity >95percent, is dissolved in methanol and added with a solution of K2CO3 in water/methanol at 10° C. The solution is filtered and dropped into water; the precipitate amorphous N-[5-(3,5-difluorobenzyl)-1H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-(tetrahydro-pyran-4-ylamino)-benzamide is filtered, washed with water and dried under vacuum (2.88 Kg). [0119] 5.5 g of the dried amorphous N-[5-(3,5-difluorobenzyl)-1H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-(tetrahydro-pyran-4-ylamino)-benzamide are suspended in 130 mL of ethanol and heated to reflux for 10 minutes; about 70 mL of ethanol are distilled before cooling to room temperature. 110 mL of water are added and the suspension is seeded with 55 mg of crystalline form 1. The suspension is stirred for about 72 hours sampling to monitor conversion into crystalline crystalline form 1 by DSC. The suspension is then filtered and dried to give 4.3 g of the desired crystalline form 1. [0120] The dried amorphous N-[5-(3,5-difluorobenzyl)-1H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-(tetrahydro-pyran-4-ylamino)-benzamide (2.88 Kg) is slurred in about 10 volumes of ethanol to allow conversion to the desired crystalline form 2; 20 volumes of water are then added and the suspension is filtered. The product is finally dried under vacuum so giving about 2.6 Kg of N-[5-(3,5-difluorobenzyl)-1H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-(tetrahydro-pyran-4-ylamino)-benzamide (4.6 mol) in the desired crystalline form 2.
Reference:
[1] Patent: WO2009/13126, 2009, A1, . Location in patent: Page/Page column 76
[2] Patent: CN108623576, 2018, A, . Location in patent: Paragraph 0202; 0205; 0219; 0220
[3] Patent: WO2013/174876, 2013, A1, . Location in patent: Page/Page column 11
[4] Patent: US2015/51222, 2015, A1, . Location in patent: Paragraph 0115-0120
2
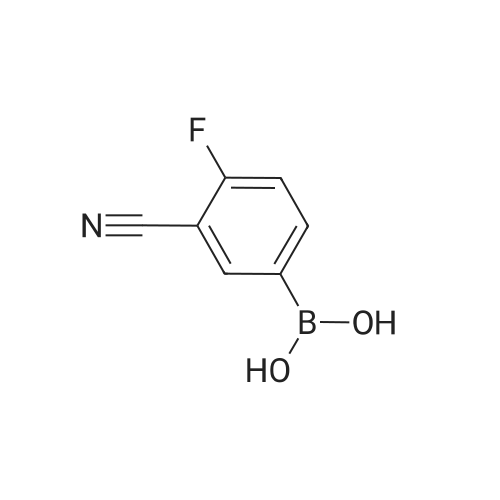
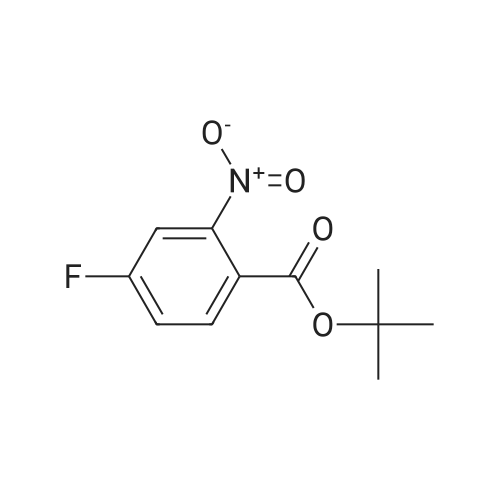
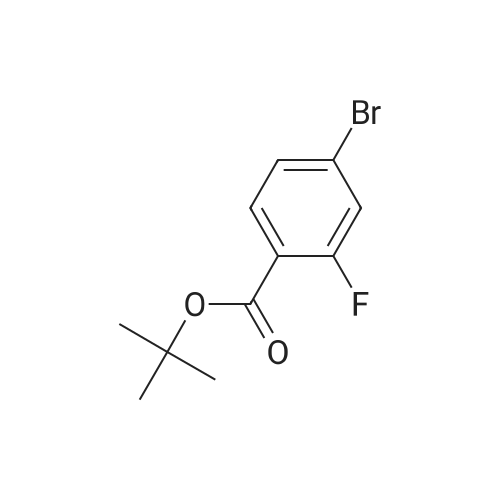
[ 29943-42-8 ]
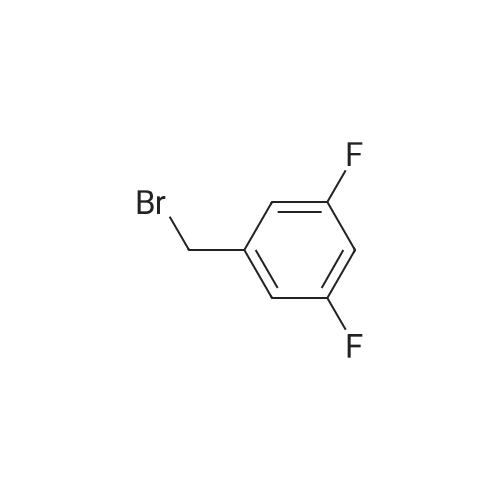
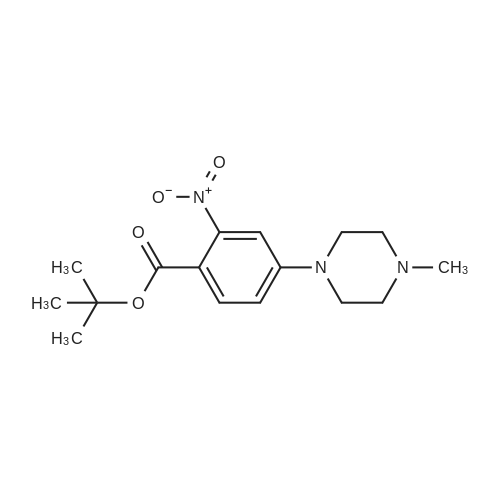
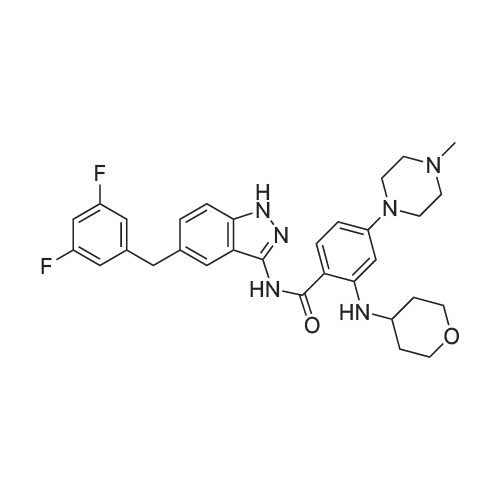
[ 1108743-96-9 ]
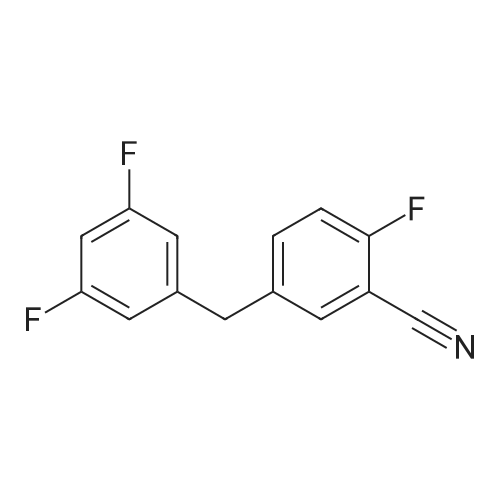
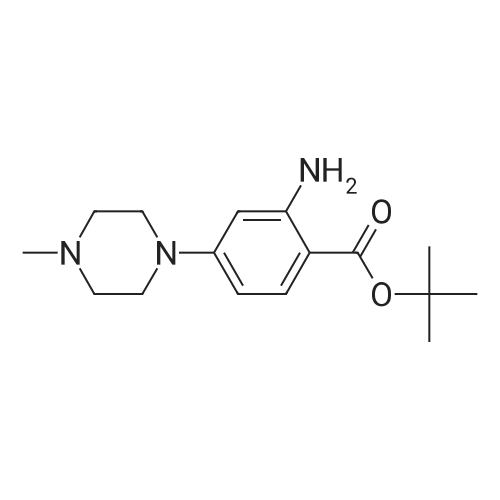
[ 1108743-60-7 ]
Yield
Reaction Conditions
Operation in experiment
72%
Stage #1: With trifluoroacetic acid; tetramethylammonium triacetoxyborohydride In dichloromethane at 20℃; for 3 h; Stage #2: With sodium hydroxide; water In dichloromethane
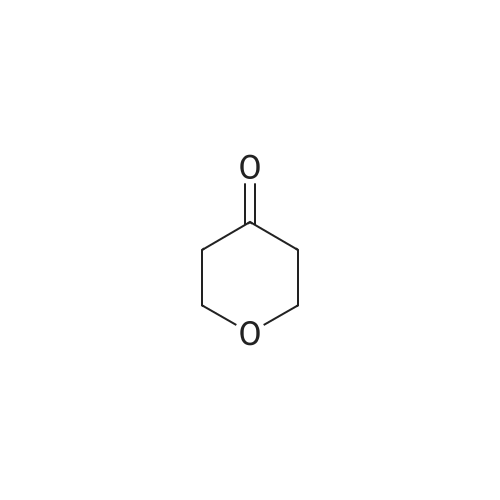
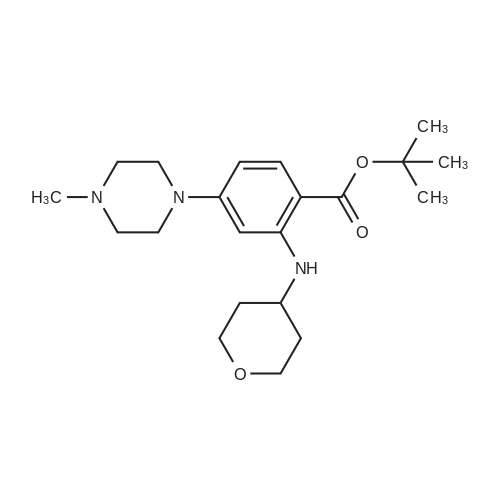
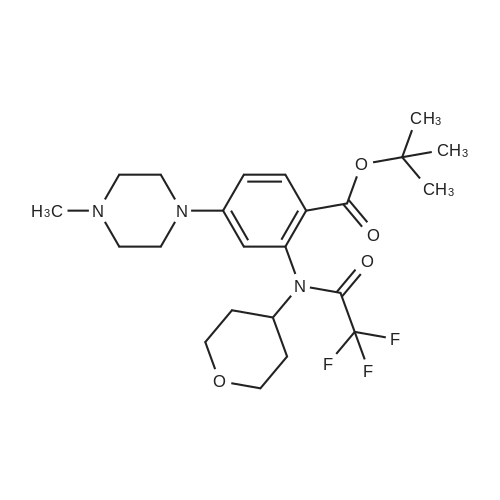
Example 7 Conversion 6; N-[5-(3,5-Difluoro-benzyl)-lH-indazol-3-yl]-4-(4-methyl-piperazin-l-yl)-2-(tetrahydro-pyran-4-ylamino)-benzamide [(IA)? R1=R2=R3=H, R=3,5- difluorophenyl, Ar=4-(4-methyl-piperazin-l-yl)-2-(tetrahydro-pyran-4-ylamino)- phenyl] cpd. 11To a solution of 2-amino-N-[5-(3,5-difluoro-benzyl)-lH-indazol-3-yl]-4-(4-methyl- piperazin-l-yl)-benzamide (1.9 g, 3.98 mmol) in dichloromethane (80 mL) were added <n="101"/>tetrahydro-pyran-4-one (0.55 mL, 5.98 mmol), trifluoroacetic acid (4 niL) and tetramethylammonium triacetoxyborohydride (1.57 g, 5.98 mmol). The mixture was stirred at room temperature overnight, and then more tetramethylammonium triacetoxyborohydride (1.57 g) was added. After stirring for additional 3 hours at room temperature the mixture was diluted with dichloromethane, washed with 2N sodium hydroxide and brine, dried over sodium sulfate and evaporated to dryness. The crude was purified by flash chromatography on silica gel using dichloromethane/methanol/ NH3 5N in MeOH 96:4:0.5 as the eluant, affording 1.61 g of the title compound (72percent yield). IH-NMR (400 MHz), δ (ppm, DMSO-d6): 1.26 - 1.43 (m, 2H) 1.86 - 2.02 (m, 2H) 2.23 (s, 3H) 2.42 - 2.46 (m, 4H) 3.23 - 3.29 (m, 4H) 3.45 - 3.54 (m, 2H) 3.62 - 3.75 (m, IH) 3.82 (dt, J=I 1.61, 3.83 Hz, 2H) 4.05 (s, 2H) 6.14 (d, J=2.07 Hz, IH) 6.24 (dd, J=8.90, 2.19 Hz, IH) 6.94 - 7.06 (m, 3H) 7.26 (dd, J=8.66, 1.46 Hz, IH) 7.41 (d, J=8.66 Hz, IH) 7.50 (d, IH) 7.80 (d, J=9.15 Hz, IH) 8.29 (d, J=7.68 Hz, IH) 10.08 (s, IH) 12.63 (s, IH)
With triethylamine; In methanol; at 65℃; for 2h;Product distribution / selectivity;
Alternatively, not previously purified crude reaction mixture can be dissolved in methanol (375 mL) in the presence of triethylamine (60 mL) and stirred at 65C for 2 hours. The solvents were removed under reduced pressure and the residue treated with water / ethyl acetate. Organic phase was dried over sodium sulfate and evaporated to dryness. Purification of the crude by chromatography over silica gel (DCM/EtOH/NH3 5N in MeOH = 1000/50/5) and crystallisation of the so obtained compound from EtOAc / hexane afforded 8.4 g of the title compound as a white solid (78% yield). IH-NMR (400 MHz), delta (ppm, DMSO-d6): 1.26 - 1.43 (m, 2H) 1.86 - 2.02 (m, 2H) 2.23 (s, 3H) 2.42 - 2.46 (m, 4H) 3.23 - 3.29 (m, 4H) 3.45 - 3.54 (m, 2H) 3.62 - 3.75 (m, IH) 3.82 (dt, J=I 1.61, 3.83 Hz, 2H) 4.05 (s, 2H) 6.14 (d, J=2.07 Hz, IH) 6.24 (dd, J=8.90, 2.19 Hz, IH) 6.94 - 7.06 (m, 3H) 7.26 (dd, J=8.66, 1.46 Hz, IH) 7.41 (d, J=8.66 Hz, IH) 7.50 (d, IH) 7.80 (d, J=9.15 Hz, IH) 8.29 (d, J=7.68 Hz, IH) 10.08 (s, IH) 12.63 (s, IH).
71%
With potassium carbonate; In methanol; water; at 20℃; for 3h;Inert atmosphere;
Compound 40 (0.66 g, 1.0 mmol) with magnetic stirring Potassium carbonate (0.42 g, 3.0 mmol) was added to a methanol/water solution (11 mL, 10/1). The reaction was stirred at room temperature for 3 hours under a nitrogen atmosphere. Water (30 mL) was added, a large amount of a gray solid was precipitated, filtered and washed with water (10mL). Dissolved in dichloromethane (20 mL), dried and concentrated. The residue was passed through a silica gel column to give a white solid, 0.4 g. The yield was 71.0%.
2.88 kg
With water; potassium carbonate; In methanol; at 10℃;Large scale;
The so obtained crude N-[5-(3,5-difluorobenzyl)-1 H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-[(tetrahydro-pyran-4-yl)- 2,2,2-trifluoro-acetyl)-amino]-benzamide, with HPLC purity > 95%, is dissolved in methanol and added with a solution of K2CO3 in water/methanol at 10C. The solution is filtered and dropped into water; the precipitate amorphous N-[5-(3,5- difluorobenzyl)-1 H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-(tetrahydro-pyran-4-ylamino)-benzamide is filtered, washed with water and dried under vacuum (2.88 Kg).
2.88 kg
With water; potassium carbonate; In methanol; at 10℃;Large scale;
To a suspension of 4-(4-methylpiperazin-1-yl)-2-[tetrahydro-2H-piran-4-yl(trifluoroacetyl)-amino]-benzoic acid trifluoroacetate (3.7 Kg, 7 mol) in dry DCM (36 L) and N,N-dimethylformamide (14 mL), oxalyl chloride (1.78 L, 21 mol) is added. The mixture is stirred for about 1.5 hours and evaporated to oily residue; dry DCM is then added and evaporated twice. [0117] The acyl chloride of formula (II) is suspended in dry DCM and the suspension is added slowly and gradually to a solution of 5-(3,5-difluoro-benzyl)-1H-indazol-3-ylamine (1.6 Kg, 6.1 mol) in dry pyridine (16 L) at -40/-30 C. The addition is blocked when the 5-(3,5-difluoro-benzyl)-1H-indazol-3-ylamine is completely reacted. After about 1 hour the solvent is evaporated and DCM (55 L), methanol (6.5 L), and MTBE (55 L) are sequentially added. The purified protected compound of formula (IV) is filtered, washed with a mixture 10/10/1 of DCM/MTBE/MeOH and dried under vacuum (3.8 Kg). [0118] The so obtained crude N-[5-(3,5-difluorobenzyl)-1H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-[(tetrahydro-pyran-4-yl)-2,2,2-trifluoro-acetyl)-amino]-benzamide, with HPLC purity >95%, is dissolved in methanol and added with a solution of K2CO3 in water/methanol at 10 C. The solution is filtered and dropped into water; the precipitate amorphous N-[5-(3,5-difluorobenzyl)-1H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-(tetrahydro-pyran-4-ylamino)-benzamide is filtered, washed with water and dried under vacuum (2.88 Kg). [0119] 5.5 g of the dried amorphous N-[5-(3,5-difluorobenzyl)-1H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-(tetrahydro-pyran-4-ylamino)-benzamide are suspended in 130 mL of ethanol and heated to reflux for 10 minutes; about 70 mL of ethanol are distilled before cooling to room temperature. 110 mL of water are added and the suspension is seeded with 55 mg of crystalline form 1. The suspension is stirred for about 72 hours sampling to monitor conversion into crystalline crystalline form 1 by DSC. The suspension is then filtered and dried to give 4.3 g of the desired crystalline form 1. [0120] The dried amorphous N-[5-(3,5-difluorobenzyl)-1H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-(tetrahydro-pyran-4-ylamino)-benzamide (2.88 Kg) is slurred in about 10 volumes of ethanol to allow conversion to the desired crystalline form 2; 20 volumes of water are then added and the suspension is filtered. The product is finally dried under vacuum so giving about 2.6 Kg of N-[5-(3,5-difluorobenzyl)-1H-indazol-3-yl]-4-(4-methyl-piperazin-1-yl)-2-(tetrahydro-pyran-4-ylamino)-benzamide (4.6 mol) in the desired crystalline form 2.
With NaO2(H); water-d2; In dimethylsulfoxide-d6; water; at 70℃;Inert atmosphere;
For magnetic stirring and condensing tubes Compound 41 (200 mg, 0.35 mmol) and D2O (5 mL) were sequentially added to a 50 mL single-necked flask.DMSO-D6 (5 mL) and 40% aqueous NaOD (180 mg, 0.178 mL),The mixture was warmed to 70 C, and the reaction was stirred overnight under a nitrogen atmosphere. Cool to room temperature,Ethyl acetate extraction (20 mL x 3), combined organics, washed with water (40 mL x 3)Dry over anhydrous sodium sulfate, filter,Concentrate and pass through a silica gel column to obtain 120 mg of a white solid.The yield was 60%.
N-(5-(3,5-difluorobenzyl)-1H-indazol-3-yl)-4-(4-methylpiperazin-1-yl)-2-((tetrahydro-2H-pyran-4-yl)amino)benzamide-3,5-d[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
71.4%
With deuteroacetic acid; zinc; at 70℃; for 2h;Inert atmosphere;
50mL with magnetic stirring and condenser Compound 41 (112 mg, 0.2 mmol) was added in succession to a single-necked flask. CH3COOD (8mL), add zinc powder (56 mg, 1 mmol) with stirring, vacuum and replace N3 three times. The temperature was raised to 70 C and the reaction was stirred for 2 hours with stirring. Cool to room temperature and add ethyl acetate (20 mL).The unreacted zinc powder was filtered off, and the filtrate was concentrated to dryness. Add saturated NaHCO3 dissolved in (10 mL), Extracted with ethyl acetate (15 mL x 3),The organic phase is dried over anhydrous sodium sulfate and filtered.Concentrated and passed through a silica gel column to give a white solid, 80mg.The yield was 71.4%.
47-53 Example 47 - Form G (monohydrate)
Entrectinib (38.6 mg) was dissolved in 1 -propanol (200 pi, 5 vol) and left to stir for 5 minutes at 50 °C. After 5 minutes of stirring, the sample was treated with water (400 pi). The resulting suspension was then left at 50 °C for 1 hour before cooling to 5 °C at 0.1 °C/min and left overnight. The resulting suspension was filtered and dried under suction before analysis by XRPD.
54-56 Example 54 - Form H
Entrectinib (39.6 mg) was dissolved in 2-methyl THF (200 pi, 5 vol) at 25 °C and the solution was left uncapped to evaporate. Once dry, the solid was analysed by XRPD.
57-58 Example 57 - Form J
Entrectinib (39.5 mg) was treated with methanol (4 ml, 100 vol) and heated to 50 °C before being placed into maturation (RT/50 °C, cycles of 4 hours at each temperature) for 6 days. The resulting suspension was filtered and dried under suction before analysis by XRPD.
11; 12 Example 11 - Form B (THF solvate)
Entrectinib (39.2 mg) was dissolved in THF (200 mI, 5 vol) at 50 °C. The solution was left at 50 °C for 1 hour before cooling to 5 °C at 0.1 °C/min and left overnight. The resulting suspension was aliquoted for XRPD.
31-38 Example 31 - Form D (nitromethane solvate)
Entrectinib (39.1 mg) was treated with nitromethane (4 ml, 100 vol) and heated to 50 °C before being placed into maturation (RT/50 °C, cycles of 4 hours at each temperature) for 6 days. The resulting suspension was filtered and dried under suction before analysis by XRPD.
39-44 Example 39 - Form E (acetonitrile solvate)
Entrectinib (39.0 mg) was dissolved in THF (200 mI, 5 vol) and left to stir for 5 minutes at 50 °C. After 5 minutes of stirring, the sample was treated with acetonitrile (1000 mI). The resulting solution was then left at 50 °C for 1 hour before cooling to 5 °C at 0.1 °C/min and left overnight. The resulting suspension was filtered and dried under suction before analysis by XRPD.
45-46 Example 45 - Form F (isopropyl acetate solvate)
Entrectinib (38.3 mg) was dissolved in 1 -propanol (200 pi, 5 vol) and left to stir for 5 minutes at 50 °C. After 5 minutes of stirring, the sample was treated with isopropyl acetate (1000 pi). The resulting solution was then left at 50 °C for 1 hour before cooling to 5 °C at 0.1 °C/min and left overnight. The resulting suspension was filtered and dried under suction before analysis by XRPD.
With 10% Pd/C; hydrogen In methanol at 20℃; for 1h; Inert atmosphere;
13-15 Example 13 Synthesis of the compound represented by formula I
Compound 7 (1.5 g, 2.30 mmol) was dissolved in a MeOH solution (15 mL) and 10% Pd/C (100 mg) was added, vacuumed and replaced with nitrogen, replaced with H2 for3 times, and hydrogenated at room temperature (1 atm) for 1 h.The reaction solution was filtered and concentrated, and the residue was subjected to silica gel column chromatography (column chromatography using a mixture of petroleum ether and ethyl acetate with a volume ratio of 2:1) to obtain the compound represented by formula I, entritinib, as a white solid 1.05 g, the yield is 81.2%, and the HPLC purity is 99.7%.
64.7%
With trimethylsilyl iodide; triethylamine In acetonitrile at 20℃; for 2.25h;
13; 14; 15 Example 13 Synthesis of the compound represented by formula I
At room temperature, compound 6 (1.8 g, 3.31 mmol) was dissolved in an acetonitrile solution (18 mL), trimethylsilyl iodide (Me3SiI) (1.46 g, 7.28 mmol) was added, and the reaction was stirred for 2 h. Triethylamine (1.38mL, 9.93mmol) was added to the reaction solution and stirred for 15min.The reaction solution was concentrated under reduced pressure, and the residue was dissolved in ethyl acetate and washed with saturated sodium bicarbonate solution.The organic phase is dried and concentrated, and passed through a silica gel column (column chromatography uses a mixture solvent of petroleum ether and ethyl acetate with a volume ratio of 2:1) to obtain the compound of formula I, entritinib, as a white solid. The yield is 1.20 g, the yield is 64.7%, and the HPLC purity is 99.8%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping