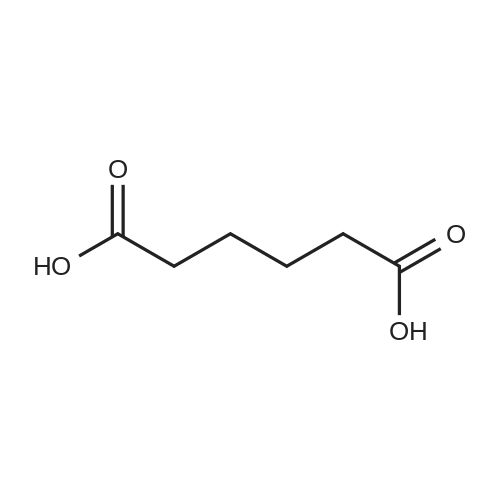
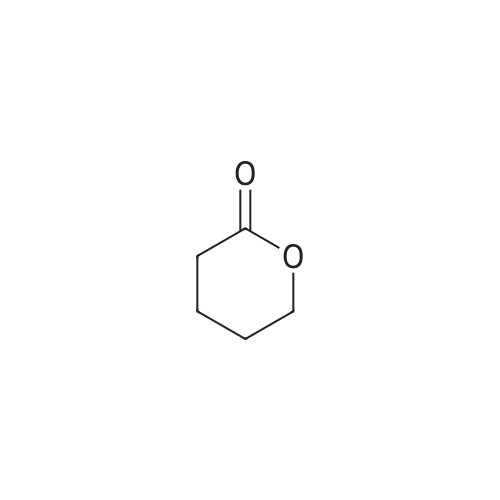
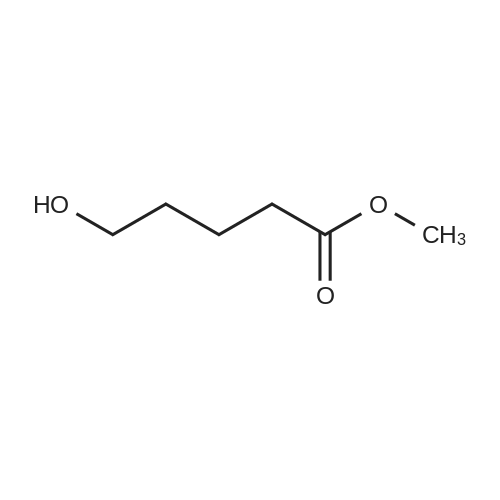
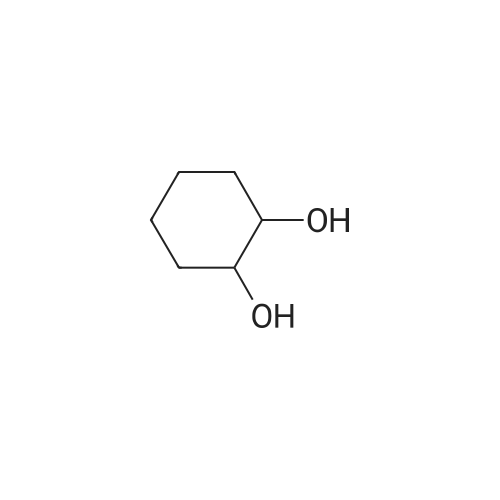
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: With potassium hydroxide In methanol at 20℃; for 4 h; Stage #2: With hydrogenchloride In water
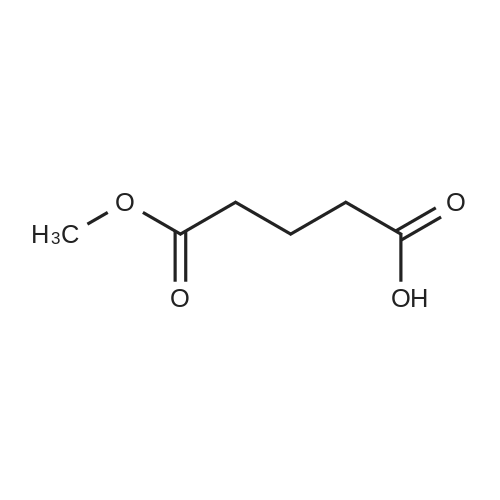
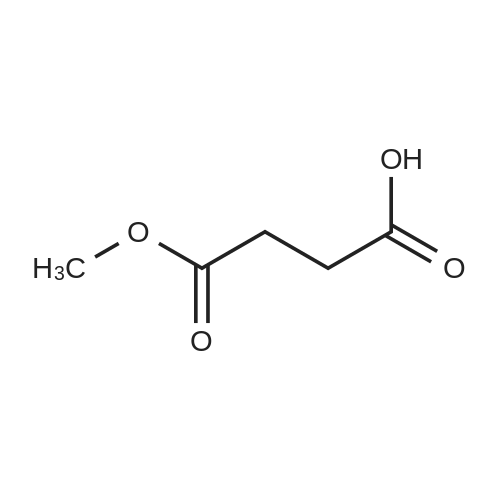
General procedure: A solution of KOH (5.87 g, 104.65 mmol) in MeOH (150 ml) was added to dimethyl glutarate (13.15 g, 90 mmol), and the mixture was stirred for 4 h at rt. The solvent was then removed, and Et2O (100 ml) and H2O (200 ml) were added. The organic layer was separated, washed with brine, dried (MgSO4), and concentrated under reduced pressure to afford 3a as a yellow oil (4.61 g, 32percent). The aqueous layer was acidified with concentrated HCl to pH 3, and extracted with Et2O (3 .x. 100 ml). The combined organic phase was washed with brine (3 .x. 100 ml) and dried (MgSO4). The solvent was removed to give a mixture of a white solid and an oil. Filtration and concentration in vacuum and purification with silica gel column chromatography gave 5.79 g (44percent) of 4a as a colorless oil.
Reference:
[1] Bioorganic and Medicinal Chemistry, 2012, vol. 20, # 12, p. 3865 - 3872
[2] Chemistry Letters, 1995, # 7, p. 539 - 540
2
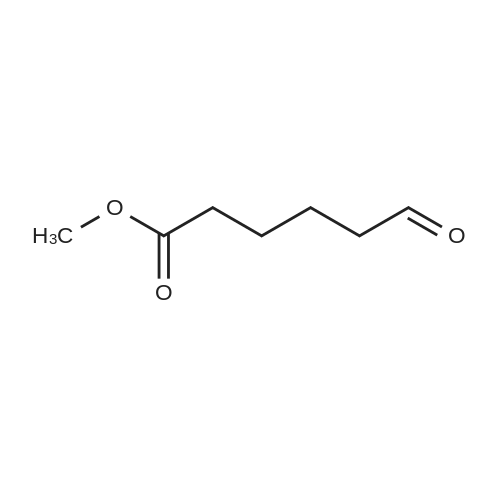
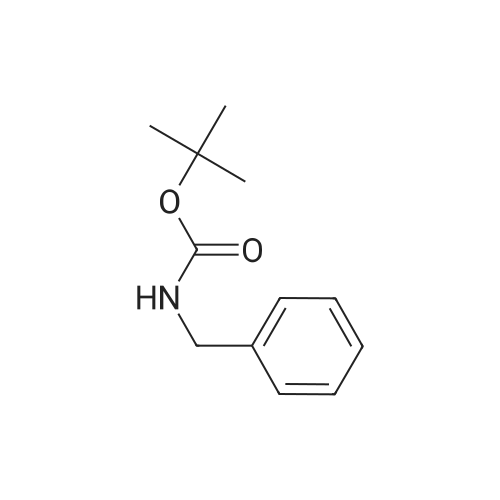
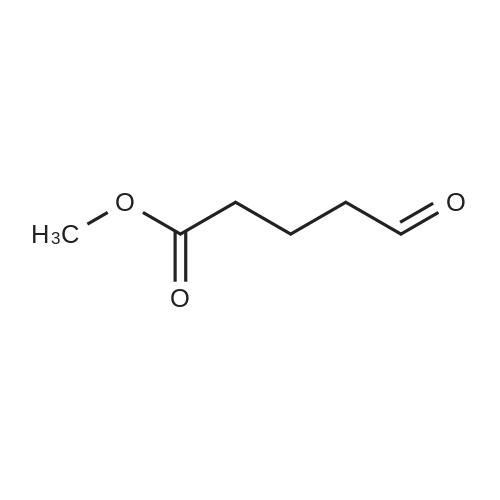
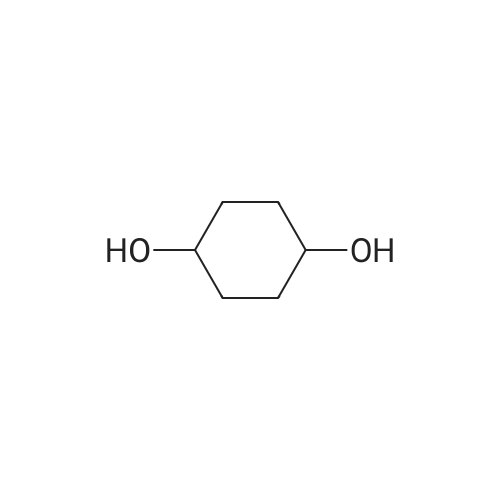
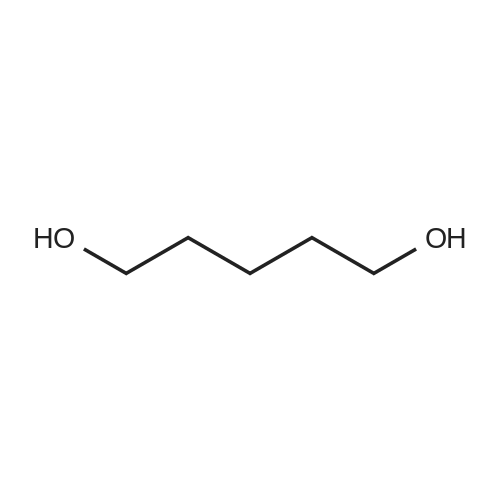
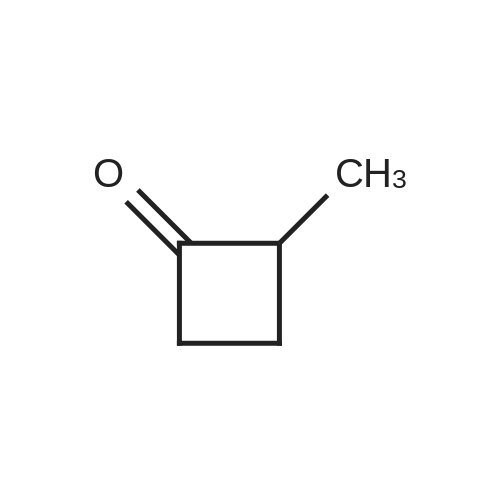
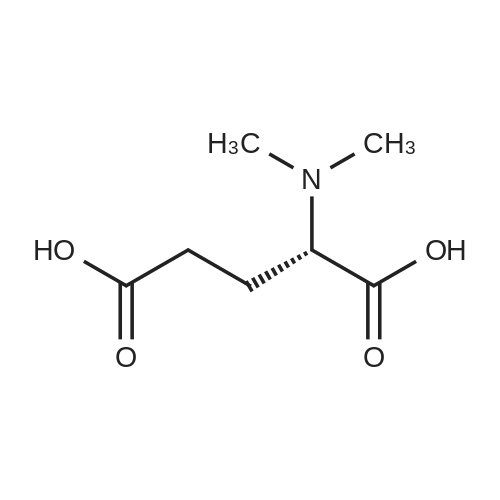
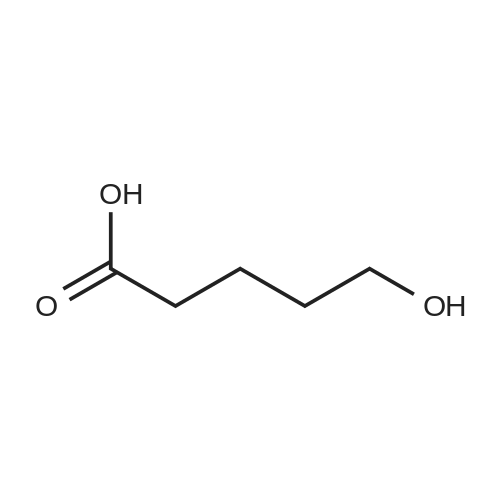
[ 1119-40-0 ]
[ 110-94-1 ]
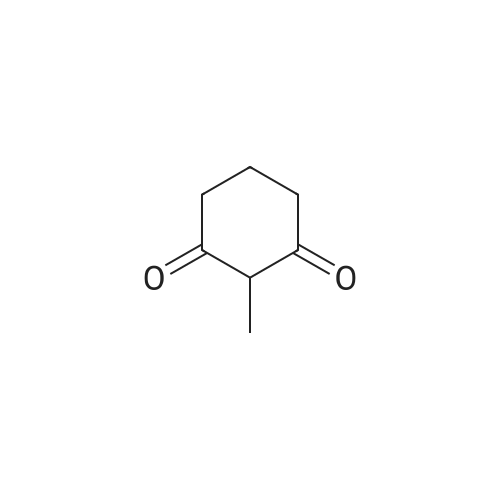
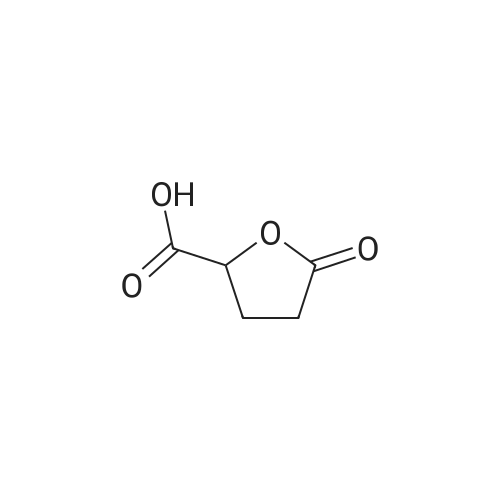
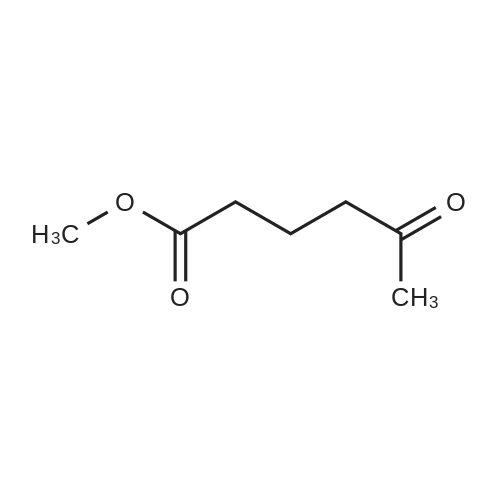
[ 1501-27-5 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 2005, vol. 78, # 3, p. 498 - 500
[2] Patent: WO2008/150487, 2008, A2, . Location in patent: Page/Page column 27-28
[3] Patent: WO2008/150487, 2008, A2, . Location in patent: Page/Page column 27-28
[4] Patent: WO2008/150487, 2008, A2, . Location in patent: Page/Page column 27-28
[5] Patent: WO2008/150487, 2008, A2, . Location in patent: Page/Page column 27-28
3
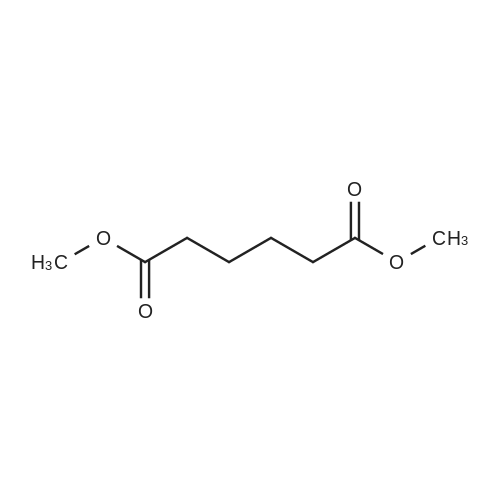
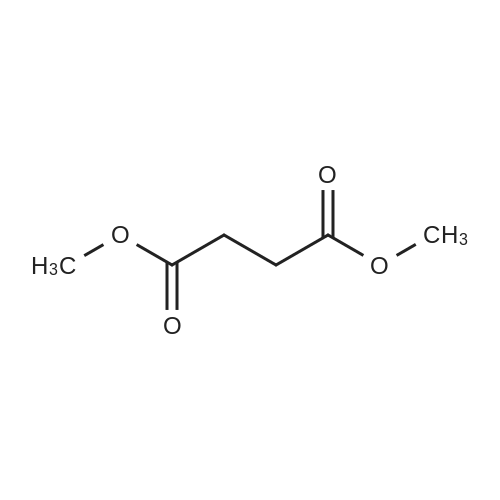
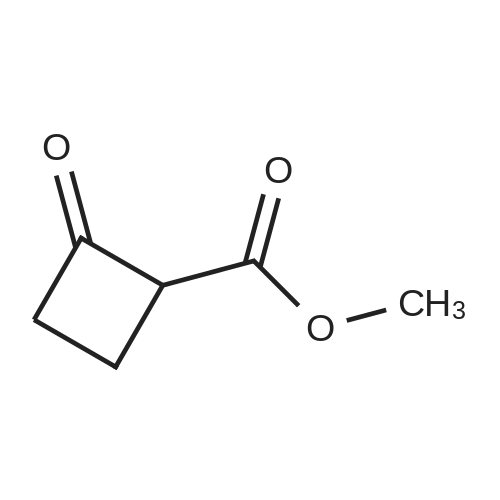
[ 110-94-1 ]
[ 1119-40-0 ]
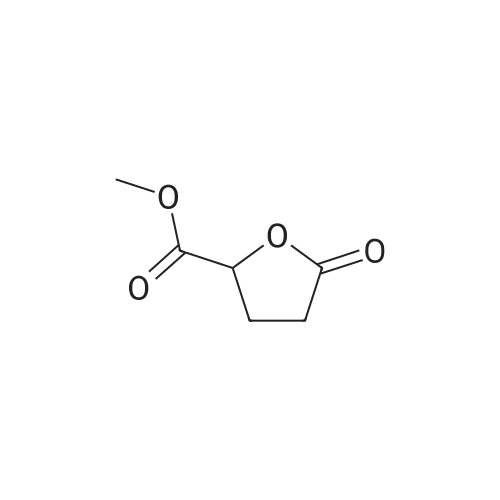
[ 1501-27-5 ]
Reference:
[1] Bulletin de la Societe Chimique de France, 1929, vol. <4> 45, p. 841
4
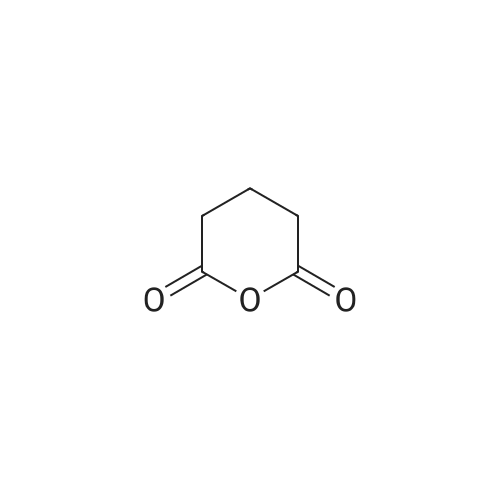
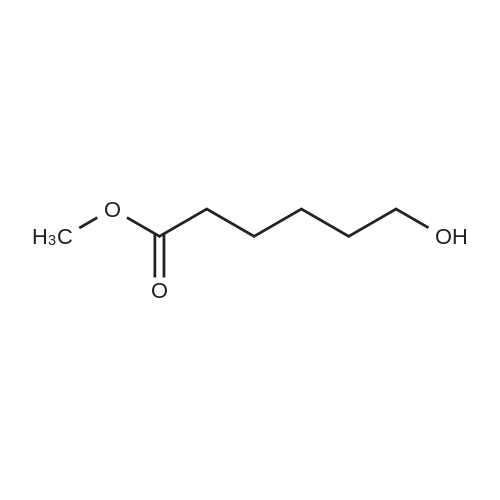
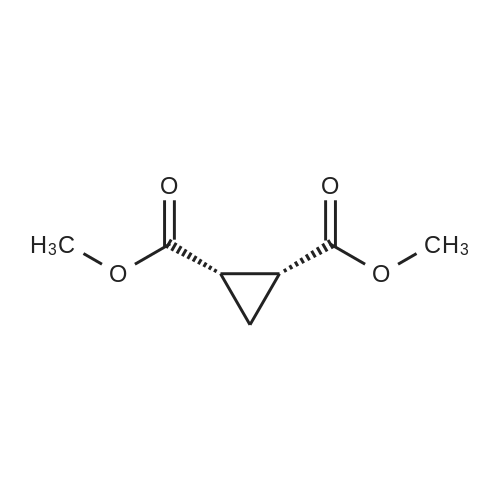
[ 1119-40-0 ]
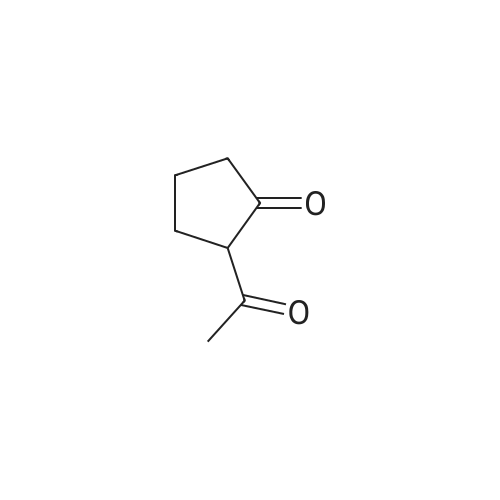
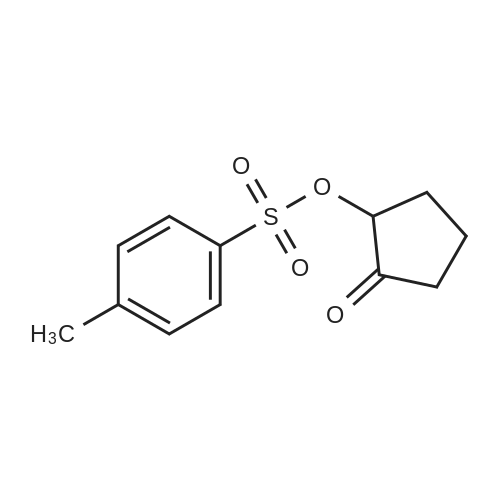
[ 1501-26-4 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 2012, vol. 20, # 12, p. 3865 - 3872
5
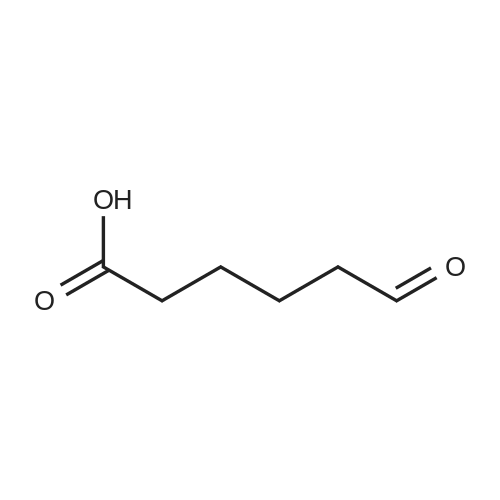
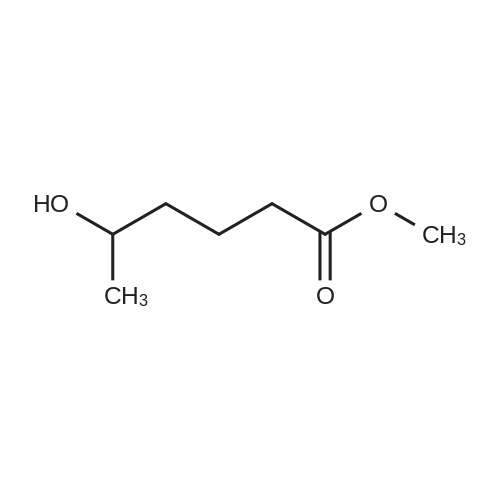
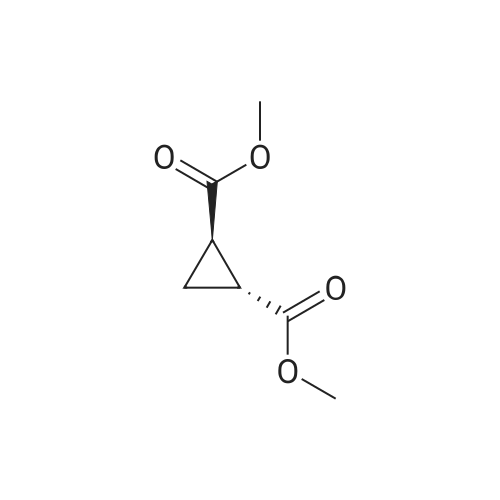
[ 1119-40-0 ]
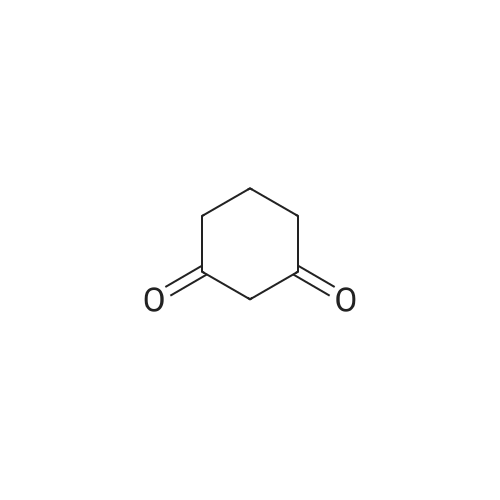
[ 1070-62-8 ]
Reference:
[1] Journal of the American Chemical Society, 1946, vol. 68, p. 99
[2] Bulletin of the Chemical Society of Japan, 1949, vol. 22, p. 125[3] Chem.Abstr., 1950, p. 6478
[4] Arch. neerl. Physiol., 1930, vol. 15, p. 532[5] Chem. Zentralbl., 1931, vol. 102, # I, p. 1120
6
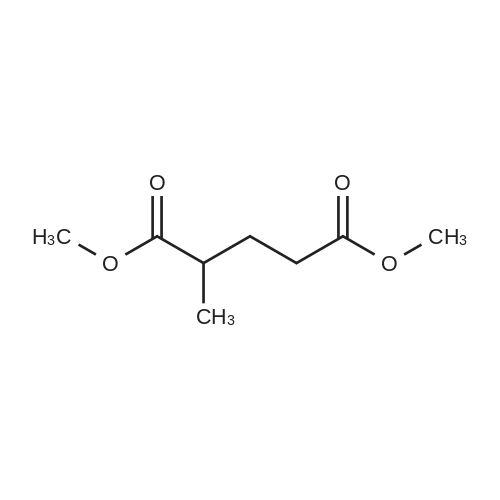
[ 67-56-1 ]
[ 110-94-1 ]
[ 1119-40-0 ]
Yield
Reaction Conditions
Operation in experiment
92%
Cooling
PCl3 (8 ml) was added batchwise to a solution of glutaric acid (13.2 g; 0.10 mol) in methanol (50 ml) under cooling and stirring. The solvent was removed from the reaction mixture under vacuum. The resulting residue was distilled off under vacuum. The amount of the resulting glutaric acid dimethyl ester with a boiling point of 110-112° C. was 14.7 (92percent).
Reference:
[1] Analytical Chemistry, 2004, vol. 76, # 16, p. 4765 - 4778
[2] Patent: US2016/31858, 2016, A1, . Location in patent: Paragraph 0143
[3] Recueil des Travaux Chimiques des Pays-Bas, 1899, vol. 18, p. 373
[4] Chem. Zentralbl., 1918, vol. 89, # I, p. 1144
[5] Recueil des Travaux Chimiques des Pays-Bas, 1926, vol. 45, p. 586
[6] Zhurnal Obshchei Khimii, 1953, vol. 23, p. 212,214; engl.Ausg.S.219
[7] Recueil des Travaux Chimiques des Pays-Bas, 1899, vol. 18, p. 373
[8] Journal of the Chemical Society, 1934, p. 339[9] Journal of the Chemical Society, 1948, p. 640
[10] Journal of Organic Chemistry, 1988, vol. 53, # 15, p. 3587 - 3593
[11] Patent: US2009/264674, 2009, A1, . Location in patent: Page/Page column 5-6
[12] Patent: US2013/303796, 2013, A1, . Location in patent: Paragraph 0027; 0028; 0034
[13] Green Chemistry, 2016, vol. 18, # 7, p. 2193 - 2200
[14] New Journal of Chemistry, 2018, vol. 42, # 15, p. 12745 - 12753
7
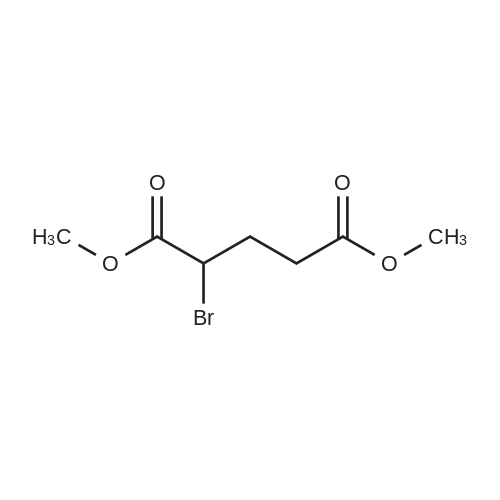
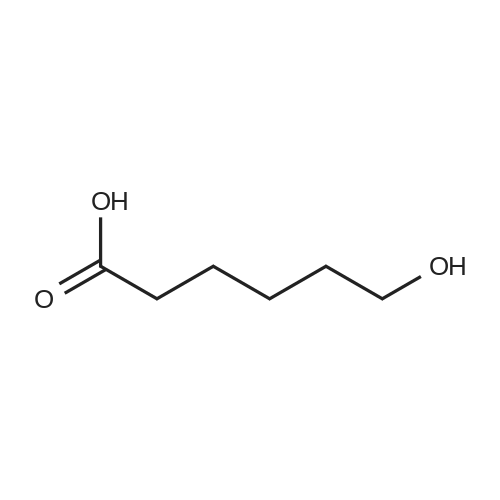
[ 6654-36-0 ]
[ 627-93-0 ]
[ 1119-40-0 ]
Yield
Reaction Conditions
Operation in experiment
56 %Chromat.
With 5H(1+)*[PMo10V2O40](5-)*H2O; oxygen In water; acetic acid at 70℃; Schlenk technique; Green chemistry
General procedure: All catalytic tests were carried out using Schlenk flasks (20mL), which were attached to a vacuum line with a manometer and a gas inlet. In a typical experiment, the Schlenk was charged with 5.0mL of the corresponding solvent (CH3CN, H2O, CH3OH, CH3COOH or a mixture of CH3COOH/H2O) and HPA-n (0.02mmol). The substrate (5.0mmol) was then added and the vessel was immersed in an oil bath preheated at 70°C. O2 was introduced at atmospheric pressure and the mixture was stirred magnetically for the time indicated in the tables. Three parallel catalytic experiments were carried out for each test.
Glutaric anhydride (29.4 mmol, 3.35 g) was dissolved in anhydrous MeOH (100 mL) and conc. H2SO4 (5.9 mmol, 0.31 mL) was added. After being stirred at room temperature for 18 h, the mixture was concentrated to a half volume and quenched with water (40 mL) afterwards. The aqueous phase was extracted with CH2Cl2 (3 x 50 mL), the combined organic phases were washed with brine (30 mL), dried over Na2SO4 and concentrated to give dimethyl glutarate 1l (4.53 g, 96percent) as a colourless clear oil; 1H NMR (CDCl3): δ =1.96 (quint, J = 7.2 Hz, 2H), 2.39 (t, J = 7.2 Hz, 4H).
General procedure: Into a stainless steel pressure microreactor of capacity 17 mL was charged 5 wt percent of zeolite NaY-Bf, 100 mmol of carboxylic acid, and 300–400 mmol of dimethyl carbonate, the reactor was hermetically closed, and the reaction mixture was heated at 180–200°C for 5 h. On completion of the reaction the reactor was cooled to room temperature, opened, the reaction mixture was filtered through a bed of Al2O3. Unreacted dimethyl carbonate was distilled off, the residue was distilled at atmospheric pressure or in a vacuum, or it was crystallized from ethanol.
Reference:
[1] Russian Journal of Organic Chemistry, 2017, vol. 53, # 2, p. 163 - 168[2] Zh. Org. Khim., 2017, vol. 53, # 2, p. 177 - 181,5
10
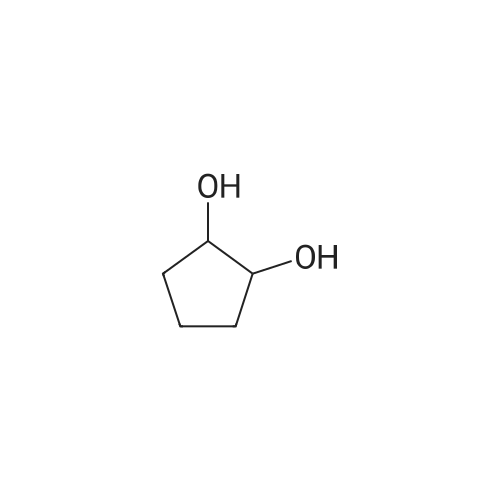
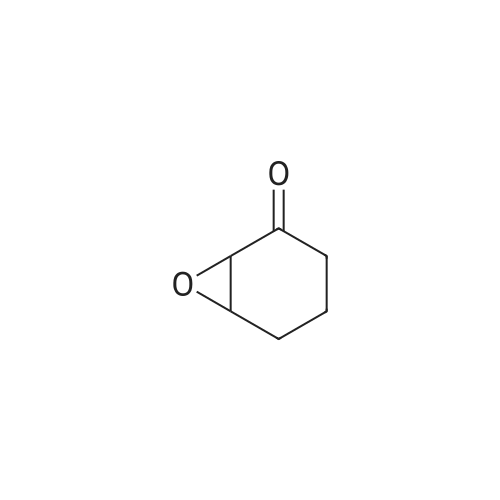
[ 186581-53-3 ]
[ 928-81-4 ]
[ 627-93-0 ]
[ 1119-40-0 ]
Yield
Reaction Conditions
Operation in experiment
60 %Chromat.
Stage #1: With 5H(1+)*[PMo10V2O40](5-)*H2O; oxygen; nickel(II) acetylacetonate In water; acetic acid at 70℃; for 8 h; Schlenk technique; Green chemistry Stage #2: Schlenk technique; Green chemistry
General procedure: All catalytic tests were carried out using Schlenk flasks (20mL), which were attached to a vacuum line with a manometer and a gas inlet. In a typical experiment, the Schlenk was charged with 5.0mL of the corresponding solvent (CH3CN, H2O, CH3OH, CH3COOH or a mixture of CH3COOH/H2O) and HPA-n (0.02mmol). The substrate (5.0mmol) was then added and the vessel was immersed in an oil bath preheated at 70°C. O2 was introduced at atmospheric pressure and the mixture was stirred magnetically for the time indicated in the tables. Three parallel catalytic experiments were carried out for each test.
Reference:
[1] Patent: EP1544190, 2005, A1, . Location in patent: Page/Page column 8
[2] Journal of the American Chemical Society, 2009, vol. 131, # 4, p. 1382 - 1383
24
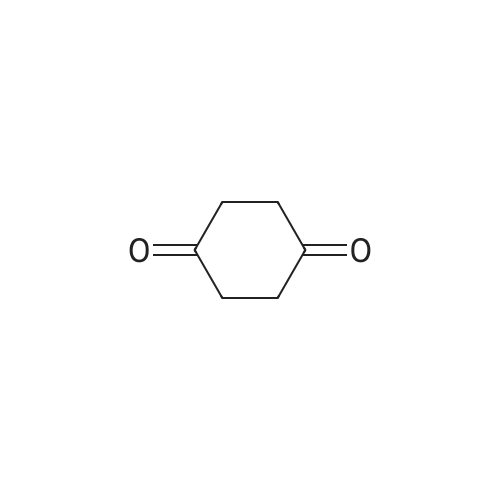
[ 67-56-1 ]
[ 1119-40-0 ]
Reference:
[1] Synlett, 2002, # 10, p. 1679 - 1680
25
[ 67-56-1 ]
[ 930-29-0 ]
[ 1119-40-0 ]
Reference:
[1] Canadian Journal of Chemistry, 1980, vol. 58, # 19, p. 2049 - 2054
With sodium hydride In 1,4-dioxane; methanol at 80℃; for 24h;
40%
With sodium hydride In 1,4-dioxane; methanol; mineral oil at 80℃; for 22h; Inert atmosphere;
Synthesis of meso-6,7,8,9-tetrahydro-5H-benzocycloheptene-5,9-diol
To a 100-mL three-necked flask were added NaH (60% oil suspension, 1.22 g, 30.5 mmol),dimethyl phthalate (2.0 mL, 12.3 mmol), 1,4-dioxane (20 mL), 20 drops of CH3OH under Ar. Thesolution was allowed to warm to 80 °C, and 1,4-dioxane solution (20 mL) of dimethyl glutarate (1.9mL, 12.9 mmol) was added. The reaction mixture was stirred for 22 h and then cooled to 0 °C followed by addition of 10% H2SO4 33 mL and excess amount of water. The organic layer was concentrated and filtered off. The residue was recrystallized from hot CH3OH to give5H-benzocycloheptene-6,8-dicarboxylic acid-6,7,8,9-tetrahydro-5,9-dioxo-6,8-dimethyl ester in 40 %yield (1.43 g, 4.93 mmol). A mixture of the obtained diester (1.43 g, 4.93 mmol), 1,4-dioxane (50 mL) and 6 N HCl (50 mL)in a 100-mL flask was refluxed at 120 °C for 3 h. Then, the solution was extracted with AcOEt (3 ×20 mL), and the organic phase was washed with a saturated aqueous solution of NaHCO3 (2 × 10mL) and brine (20 mL). The combined organic layer was dried over MgSO4 and volatiles wereremoved under reduced pressure to afford 5,9-dioxo-6,7,8,9-tetrahydro-5H-benzocycloheptene aspale yellow crystals in 78% yield (673 mg, 3.87 mmol).The resulted diketone (673 mg, 3.87 mmol) was diastereoselectively reducted by a method similar to diol 2e to give corresponding meso-diol 2f as a white solid in 42% yield (281 mg, 1.58 mmol).
With di(n-butyl)tin oxide at 5 - 10℃; Heating / reflux;
7.i
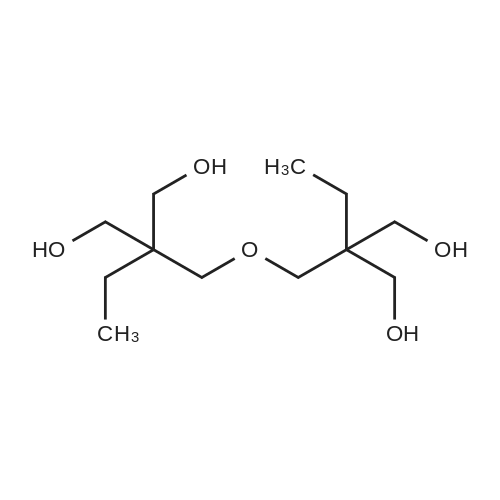
EXAMPLE 7; [0075] Step (i): 800 g of Estasol (mixed dibasic ester comprising 21%-w/w of dimethyladipate, 59%-w/w of dimethylglutarate and 20%-w/w of dimethylsuccinate), 309.5 g of di(trimethylolpropane), 3.3 g dibutyltinoxide and 2.2 g of trisnonylphenylphosphite were charged in a reaction flask equipped with electrical heating, a Dean-Starkseparator, a vertical cooler, mechanical stirrer and nitrogen inlet. The temperature was under stirring and nitrogen blanket raised until all di(trimethylolpropane) was dissolved and formed methanol progressively distilled off. The temperature was allowed to decrease 5-10[deg.] C. during the alcoholysis. The alcoholysis was considered completed when 160 ml of methanol was collected and unreacted Estasol was distilled off from formed di(trimethylolpropane) ester. [0076] Step (ii): 200 g of the di(trimethylolpropane) ester obtained in Step (i), 111 g of diethanolamine and 0.3 g dibutyltinoxide were charged in a reaction flask equipped as in Step (i). The temperature slowly raised to 220[deg.] C. and methanol removed from the reaction mixture. The aminolysis was considered completed when 34 ml of methanol was collected. Vacuum was now applied to remove unreacted diethanolamine and obtained [beta]-hydroxyamide was recovered. [0077] Obtained [beta]-hydroxyamide had a hydroxyl value of 7.62 mequiv/g and a viscosity of 428 mPas at 100[deg.] C.
With di(n-butyl)tin oxide; at 165 - 185℃;Product distribution / selectivity;
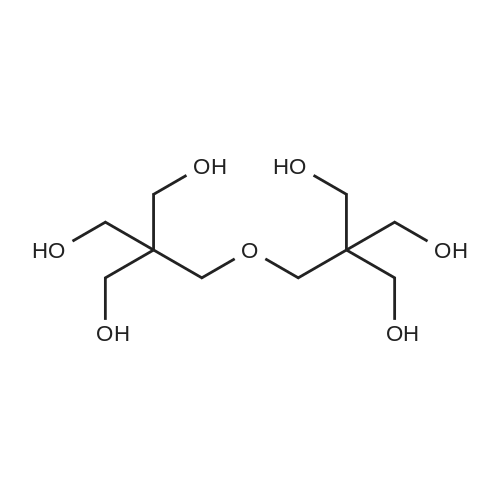
EXAMPLE 2; [0060] Step (i): 800 g of Estasol (mixed dibasic ester comprising 21%-w/w of dimethyladipate, 59%-w/w of dimethylglutarate and 20%-w/w of dimethylsuccinate), 193 g of dipentaerythritol, 3 g dibutyltinoxide and 2 g of trisnonylphenylphosphite were charged in a reaction flask equipped with electrical heating, a Dean-Stark separator, a vertical cooler, mechanical stirrer and nitrogen inlet. The temperature was under stirring and nitrogen blanket raised to 170[deg.] C. and formed methanol progressively distilled off. The temperature was maintained at 170[deg.] C. and the alcoholysis was considered completed when 140 ml of methanol was collected. Vacuum was applied and excess of Estasol was evaporated from formed dipentaerythritol ester. [0061] Step (ii): 200 g of the dipentaerythritol ester obtained in Step (i), 124 g of diethanolamine and 0.3 g dibutyltinoxide were charged in a reaction flask equipped as in Step (i). The temperature slowly raised to 220[deg.] C. and methanol removed from the reaction mixture. The aminolysis was considered completed when 37 ml of methanol was collected. Vacuum was now applied to remove unreacted diethanolamine and obtained [beta]-hydroxyamide was recovered.[0062] Obtained [beta]-hydroxyamide had a hydroxyl value of 8.23 mequiv/g and a viscosity of 740 mPas at 100[deg.] C.; EXAMPLE 3; [0063] Step (i): 500 g of Estasol (mixed dibasic ester comprising 21%-w/w of dimethyladipate, 59%-w/w of dimethylglutarate and 20%-w/w of dimethylsuccinate), 106.5 g of pentaerythritol, 1.8 g dibutyltinoxide and 1.2 g of trisnonylphenylphosphite were charged in a reaction flask equipped with electrical heating, a Dean-Stark separator, a vertical cooler, mechanical stirrer and nitrogen inlet. The temperature was under stirring and nitrogen blanket raised to 185[deg.] C. and formed methanol progressively distilled off. The temperature was allowed to decrease to 165[deg.] C. during the alcoholysis and the alcoholysis was considered completed when 100 ml of methanol was collected and unreacted Estasol was distilled off from formed pentaerythritol ester. [0064] Step (ii): 200 g of the pentaerythritol ester obtained in Step (i), 130 g of diethanolamine and 0.3 g dibutyltinoxide were charged in a reaction flask equipped as in Step (i). The temperature slowly raised to 220[deg.] C. and methanol removed from the reaction mixture. The aminolysis was considered completed when 39 ml of methanol was collected. Vacuum was now applied to remove unreacted diethanolamine and obtained [beta]-hydroxyamide was recovered. [0065] Obtained [beta]-hydroxyamide had a hydroxyl value of 8.52 mequiv/g and a viscosity of 660 mPas at 100[deg.] C.
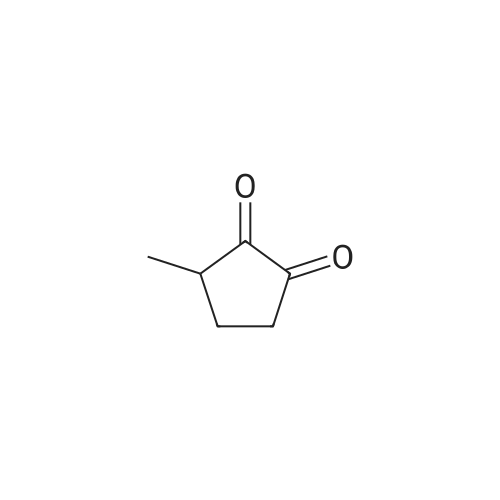
EXAMPLE 1 3-Methyl Cyclopentane-1,2-Dione (2-Hydroxy-3-methyl cyclopent-2-ene-1-one) A 5-liter three-necked round bottom flask is fitted with a thermometer and nitrogen inlet tube, a mechanical stirrer, and a ten-tray Oldershaw column with a condenser and take-off head. The flask is purged with dry nitrogen and charged with two liters of dry dimethylformamide, 292 grams (2 moles) diethyl oxalate, 320 grams (2 moles) dimethyl glutarate, and 240 grams (4.44 moles) of sodium methoxide. With the addition of sodium methoxide, the temperature rose to approximately 50 C. Continuing the nitrogen purge and while stirring, the pot temperature is taken to about 110 C. During this time the thick, light-brown solution is stirred vigorously and the methanol and ethanol formed during the reaction is allowed to distill. It was not necessary to remove all of the alcohol formed during the reaction. The reaction is complete at 110 C. pot temperature in about 30 minutes. The pot contains the condensation products:






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping