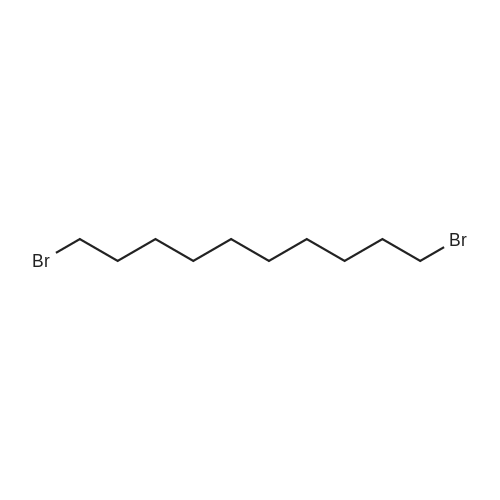
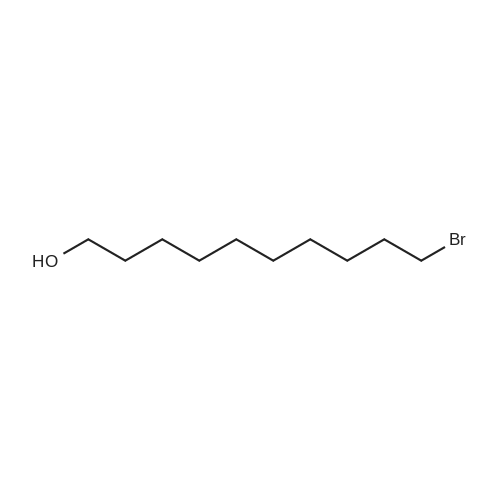
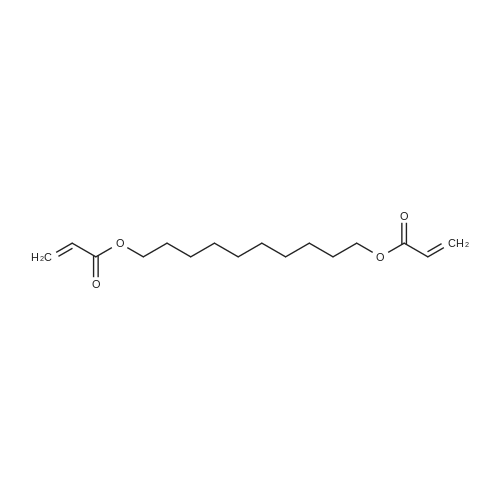
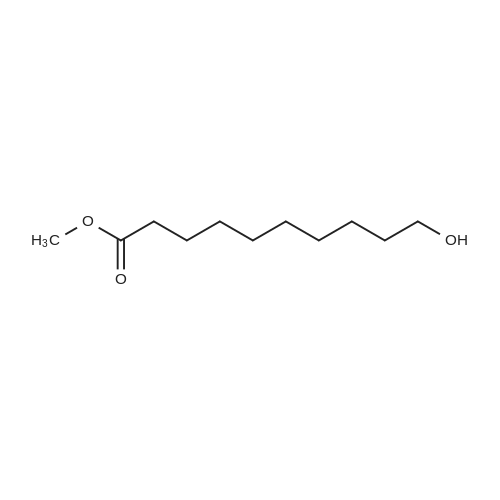
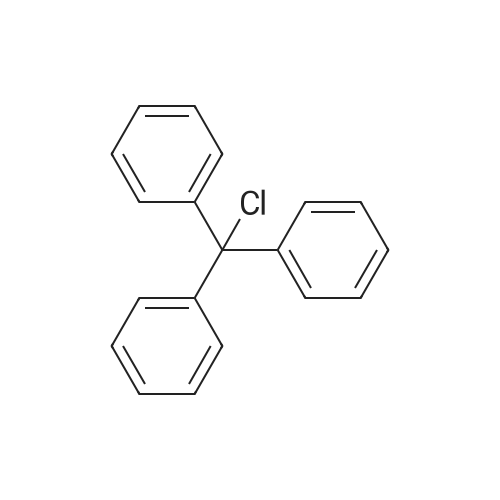
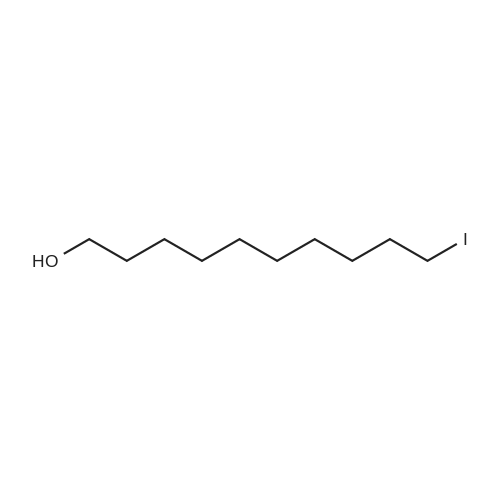
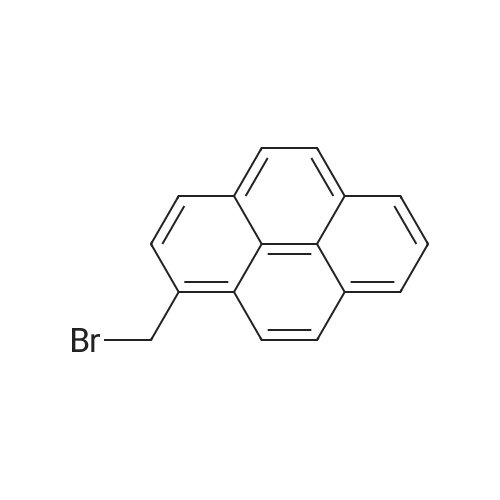
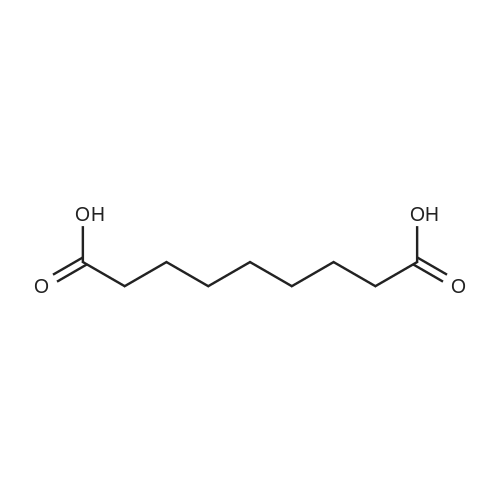
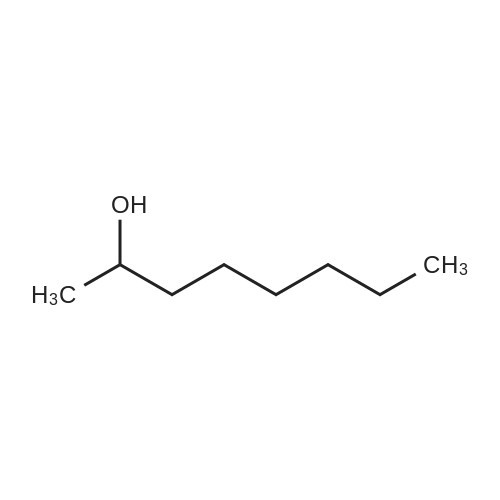
| 92% |
With hydrogen bromide In toluene at 180℃; for 24h; |
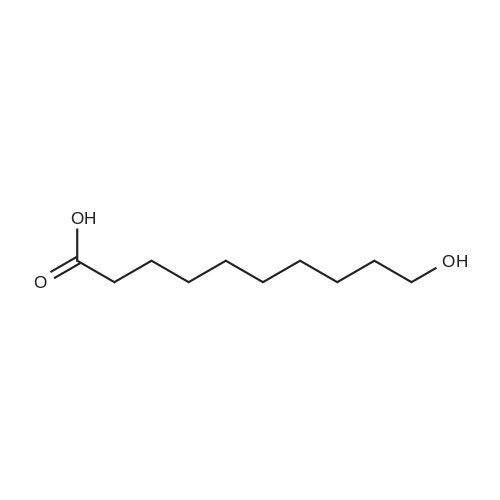
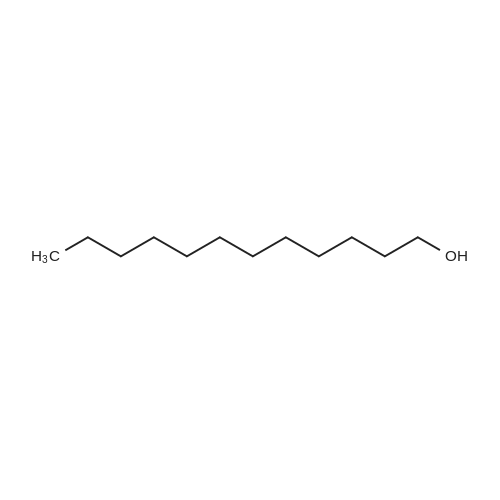
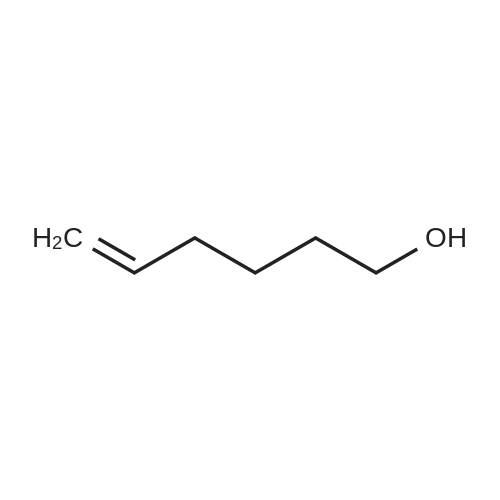
3.2.3. Synthesis of 10-bromodecanoic acid (4d)
To a solution of 1,10-decandiol (3) (34.8 g, 0.2 mol, 1 equiv) in toluene (400 mL) was added 48% HBr (22.6 mL, 0.2 mol, 1 equiv) dropwise with stirring and refluxed at 180 °C using Dean-Stark trap for 24 h. The mixture was cooled to room temperature and washed with 6 N NaOH (150 mL), 10% HCl (150 mL), H2O (2 x 250 mL) and brine (200 mL). The organic layer was dried over Na2SO4, concentrated and chromatographed on silica gel eluting with cyclohexane/ethylacetate (4:1) to give 43.5 g (92%) of 10-bromo-1-decanol as a colourless liquid. 1H NMR δ 3.65 (t, J = 6.7 Hz, 2H, H-1), 3.43 (t, J = 7.0 Hz, 2H, H-10), 1.87 (m, 2H, H-9), 1.57 (m, 2H, H-2), 1.43 (m, 2H, H-3), 1.26-1.41 (m, 10H, H-4-8). 13C NMR δ 63.0 (C1), 34.1 (C10), 32.8, 32.6, 29.5, 29.4, 29.3, 28.6, 28.2, 25.6.To a solution of 10-bromo-1-decanol (41 g, 0.17 mol, 1 equiv) in 130 mL of acetone at -5 °C was added slowly chromic acid solution prepared from CrO3 (25.7 g, 0.26 mol, 1.5 equiv), water (25 mL) and conc H2SO4 (22.5 mL, 0.34 mol, 2 equiv) at 0 °C, then stirred for 2 h and left over night at room temperature. The mixture was extracted with diethyl ether (3 x 250 mL), washed with water (250 mL) and brine (250 mL), dried over Na2SO4 and concentrated. The residue was chromatographed on silica gel eluting with CH2Cl2 afforded 31.0 g of 10-bromodecanoic acid (4d) (73%) as a white solid after recrystallization from petroleum ether. |
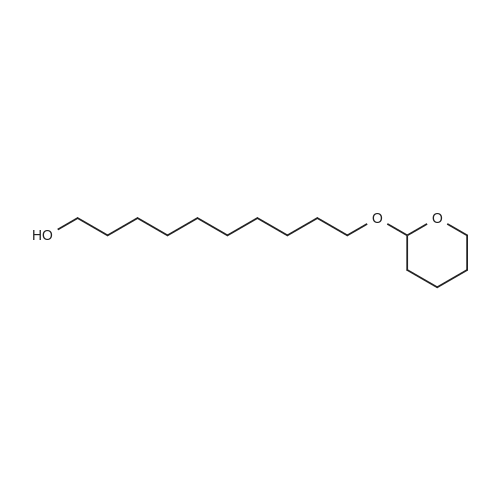
| 90% |
With hydrogen bromide In water; toluene for 72h; Heating / reflux; |
1
EXAMPLE 1 To a mixture of 1,10-decanediol (35.73g, 0.205 mol) and toluene (700 mL) was added concentrated HBr (29 mL of 47% aqueous solution, 0.24 mol). The heterogeneous mixture was stirred and heated at reflux for 36 hours. TLC analysis indicated substantial amounts of 1,10-decanediol still remained. Thus a further quantity of HBr (15 mL, 0.12 mol) was added and the mixture was heated at reflux for further 36 h, at which time TLC analysis showed no diol remaining. The reaction mixture was allowed to cool to room temperature and the phases were separated. The organic layer was concentrated by evaporating the toluene and diluted with ethyl acetate and washed with water, sodium bicarbonate and brine. Then the organic layer was dried over Na2SO4 and concentrated to yellow liquid and purification of this crude reaction mixture by column chromatography provided pure 10-bromodecanol (43.0 g) in 90% yield. |
| 90% |
With hydrogen bromide In water; toluene for 72h; Heating / reflux; |
1
EXAMPLE 1 To a mixture of 1,10-decanediol (35.73g, 0.205 mol) and toluene (700 mL) was added concentrated HBr (29 mL of 47% aqueous solution, 0.24 mol). The heterogeneous mixture was stirred and heated at reflux for 36 hours. TLC analysis indicated substantial amounts of 1,10-decanediol still remained. Thus a further quantity of HBr (15 mL, 0.12 mol) was added and the mixture was heated at reflux for further 36 h, at which time TLC analysis showed no diol remaining. The reaction mixture was allowed to cool to room temperature and the phases were separated. The organic layer was concentrated by evaporating the toluene and diluted with ethyl acetate and washed with water, sodium bicarbonate and brine. Then the organic layer was dried over Na2SO4 and concentrated to yellow liquid and purification of this crude reaction mixture by column chromatography provided pure 10-bromodecanol (43.0 g) in 90% yield. |
| 89% |
With hydrogen bromide In toluene for 16h; Heating; |
|
| 89% |
With hydrogen bromide In cyclohexane; water for 6h; Heating / reflux; |
A-1
Synthesis Example A-(1) 1,10-Decanediol (4 g) was dissolved in 100 ml of cyclohexane, and 57% aqueous hydrobromic acid solution (58 ml) was added to this solution. The reaction mixture was refluxed for six hours while stirring. After the reaction, the mixture was extracted three times with diethyl ether. The organic layer was neutralized with saturated sodium hydrogen carbonate solution, washed with saline solution, dried over magnesium sulfate, and filtered, and the solvent was distilled off under reduced pressure. Purification of the residue by silica gel flash chromatography (hexane: ethyl acetate = 7:3) gave 10-bromodecan-1-ol as white crystals at an 89% yield. Molecular weight: 237.18 (C10H21BrO) TLC: (hexane-ethyl acetate=7-3) Rf value: 0.53 1H-NMR: (300MHz, CDCl3)δ: 1.26 (s large, 12H, -(CH2)6-); 1.56 (qt, 2H, J=7.0Hz, -CH2-); 1.85 (qt, 2H, J=7.1Hz, -CH2-); 3.40 (t, 2H, J=6.9Hz, -CH2-Br); 3.64 (t, 2H, J=6.6Hz, -CH2-O-) 13C-NMR: (75MHz, CDCl3) δ: 25.70; 28.14-29.45; 32.77; 32.80; 34.03; 63.05 |
| 88.93% |
With hydrogen bromide In toluene for 24h; Reflux; |
Bromodecan-1-ol (8).
Decane-1,10-diol (5 g, 28.69 mmol) was dissolved in toluene, 48% aqueous HBr (3.73 mL, 32.99 mmol) was added, and the mixture was refluxed for 24 h. A yellow oily liquid was obtained. Yield 6.12 g (88.93%). 1H NMR spectrum, δ, ppm: 3.80-3.59 m (1H, CH), 1.71-1.54 m (1H, CH), 1.50-1.12 m (4H, CH2), 0.93 d.d (3H, CH2, J = 6.5,4.0 Hz). 13C NMR spectrum, δC, ppm: 61.25 (CH2), 40.01 (CH2), 36.83 (CH2), 29.51 (CH2), 29.20 (CH2), 22.97 (CH2), 19.66 (CH2), 14.11 (CH2). |
| 88.18% |
With hydrogen bromide In toluene Reflux; |
9 Step (9) Synthesis of 10-bromodecanol
Take a 250mL single-mouth bottle, weigh (5g, 28.69mmol, 1eq) 1,10-decanediol, add 125mL of toluene, add HBr (3.73mL, 32.99mmol, 1.15eq, W=48%), connect to a water separator With the condenser, heat to reflux overnight. Point plate detection, no raw material point, wash with saturated sodium bisulfite solution (50mL×3, that is, wash with 50mL saturated sodium bisulfite solution each time, wash 3 times in total), and extract with ethyl acetate (50mL×3, namely Extract with 50 mL of ethyl acetate each time, 3 times in total), combine the organic phases, dry with anhydrous magnesium sulfate, filter with suction, and spin-dried through the column. 6 g of light yellow oily liquid was obtained with a yield of 88.18%. |
| 87% |
With hydrogen bromide In water; Petroleum ether for 4h; Heating / reflux; |
|
| 85% |
With hydrogen bromide In benzene for 28h; Heating; |
|
| 85% |
With hydrogen bromide In toluene |
|
| 84% |
With hydrogen bromide In toluene for 72h; Reflux; Inert atmosphere; |
|
| 84% |
With hydrogen bromide In toluene at 110℃; for 24h; |
1 Example 1
ynthesis of 10-bromo-1-decanol[0025] Add 1,10-decanediol (321g, 1.712mol), toluene (1000ml), 48% hydrobromic acid in a 2L three-necked bottle(231ml, 2.054mol, 1.2eq), heated to 110C, after reflux for 24 hours, add 48% hydrobromic acid (84ml, 0.753mol,40.44eq), continue heating and refluxing for 24 hours, a small amount of raw material remains detected by gas chromatography. Cool to room temperature and add 500ml of petroleum etherDilute, separate hydrobromic acid, and wash the organic phase with saturated sodium bicarbonate (400ml X 2) and saturated brine (400ml X 2),Dry over anhydrous sodium sulfate. Spin-dry the column to obtain 10-bromo-1-decanol (yield 84%). |
| 82% |
With hydrogen bromide In water; toluene at 95 - 97℃; for 72h; |
|
| 82% |
With hydrogen bromide In water; toluene for 48h; Reflux; Inert atmosphere; |
|
| 81% |
With hydrogen bromide |
|
| 80% |
With hydrogen bromide In benzene for 30h; Heating; |
|
| 80% |
With hydrogen bromide In benzene for 28h; Heating; |
|
| 80% |
With hydrogen bromide In benzene Heating; |
|
| 80% |
With tetrabutylammomium bromide; hydrogen bromide In water for 0.0833333h; Microwave irradiation; |
|
| 80% |
With hydrogen bromide; tetra-(n-butyl)ammonium iodide In water for 0.0833333h; Microwave irradiation; |
10-Bromodecan-1-ol (3)
A mixture of 1, 10-decanediol (0.3 g, 1.72mmol), 48% aq. hydrogen bromide (0.29 g, 3.58 mmol) and tetrabutylammonium iodide (0.12 g, 0.34 mmol) was subjected to MWI at 355 W for 5 min. The reaction mixture was cooled, extracted with diethyl ether (3x10 mL) and washed with saturated sodium bicarbonate solution (2 x 5 mL), 10% aq. sodium thiosulfate (2 x 5 mL), water (2 x5 mL), and brine and dried. The evaporation of solvent under reduced pressure furnished the crude product, which was purified by column chromatography over silica gel using 5% ethyl acetate in n-hexane to afford pure product (3, 0.32 g, 80%) as a colourless oil. |
| 79% |
With hydrogen bromide In water; toluene for 43h; Inert atmosphere; Reflux; |
|
| 78% |
With hydrogen bromide at 85℃; for 3h; |
|
| 78% |
With hydrogen bromide |
|
| 78.5% |
With hydrogen bromide In cyclohexane; water at 80℃; for 5h; Inert atmosphere; |
|
| 77% |
With hydrogen bromide In water; toluene for 6h; Reflux; |
|
| 74% |
With hydrogen bromide at 70℃; for 40h; |
|
| 74.5% |
With hydrogen bromide In water; toluene for 36h; Reflux; |
|
| 74% |
With hydrogen bromide In water; toluene for 48h; Reflux; |
|
| 74% |
With hydrogen bromide In water; toluene for 16h; Dean-Stark; Reflux; |
2.2.2. Synthesis of 10-bromo-decan-1-ol 2
Hydrobromic acid (1.2 mL 48% aqueous solution, 9.5 mmol) was added to a suspension of 1,10-decanediol 1 (1.5 g, 8.6 mmol) in 30 mL of toluene in a round-bottomed flask equipped with a Dean-Stark trap and a cooler. The mixture was refluxed for 16 h. After cooling, the solvent was removed under reduced pressure. The residue obtained was chromatographed on silica gel using hexane/ethyl acetate (8:2) to yield the pure compound 2 (Scheme 1). Yield 74%; yellow oily product: IR (KBr): v = 3325, 2925, 2850,1450, 1250, 1050, 700, 625, 550 cm-1. 1H NMR (200 MHz, CDCl3): δ1.20-1.40 (m, 12H), 1.45-1.60 (m, 2H), 1.84 (qn, J=6.8 Hz, 2H), 3.39 (t, J=6.8 Hz, 2H), 3.62 (t, J=6.4 Hz, 2H) ppm. 13C NMR (50 MHz, CDCl3) δ: 25.67, 28.11, 28.69, 29, 32, 29.42, 32.73, 32.77, 33.98, 62.99 ppm. |
| 73% |
With hydrogen bromide In water; toluene for 16h; Reflux; |
10-Bromodecan-1-ol (3)
HBr (48% in H2O, w/w, 16.3 mL, 138 mmol) was added to a mixture ofdecane-1,10-diol (2; 20 g, 115 mmol) and toluene (250 mL). The resultingmixture was stirred at reflux temperature for 16 h. Afterreaching r.t. and careful addition of sat. aq Na2S2O3 (50 mL), the aqueouslayer was extracted with EtOAc (3 × 200 mL). The combined extractswere dried (MgSO4), filtered, and the solvent was evaporated.Purification of the residue by flash chromatography (silica gel, hexanes/EtOAc, 4:1 to 2:1) afforded the title compound as a colorless oil(19.8 g, 73%).IR (ATR): 3328, 2924, 2853, 1463, 1437, 1371, 1352, 1256, 1242, 1129,1055, 899, 756, 722, 644, 562, 505, 465, 445, 428, 417 cm-1.1H NMR (400 MHz, CDCl3): δ = 3.63 (t, J = 6.6 Hz, 2 H), 3.40 (t, J = 6.9Hz, 2 H), 1.88-1.80 (m, 2 H), 1.60-1.51 (m, 2 H), 1.46-1.24 (m, 13 H).13C NMR (100 MHz, CDCl3): δ = 63.2, 34.2, 32.92, 32.89, 29.6, 29.49,29.48, 28.9, 28.3, 25.8.HRMS-ESI: m/z calcd for [C10H21BrO + Na]+: 259.0669; found:259.0668. |
| 70% |
With hydrogen bromide In n-heptane |
|
| 70% |
With hydrogen bromide In n-heptane for 25h; Heating; |
|
| 69% |
With hydrogen bromide |
|
| 69% |
In hydrogen bromide |
XXXV.a 10-Bromodecanol
Example XXXVa 10-Bromodecanol A solution of 50 g of decane-1,10-diol in 300 ml of 48% aqueous hydrobromic acid was heated at 90° C. for 24 hours while the solution was extracted continuously with petroleum ether 100-140. After cooling to room temperature, the petroleum ether solution was evaporated and the residue was purified by distillation. Boiling point 121°-124° C. (0.1 mm Hg). Yield 69%. 1H-NMR (CDCl3): 0.98-2.20 ppm, m, 16.0 H (C-(CH2)8 COH); 3.43 ppm, t, J=6.9 Hz, 2.0 H (OCH2); 3.65 ppm, t, J=6.2 Hz, 1.9 H (BrCH2). |
| 68% |
With hydrogen bromide In toluene Heating; |
|
| 67.1% |
With hydrogen bromide In ligroin for 72h; |
|
| 65% |
With hydrogen bromide In Petroleum ether at 90 - 95℃; for 65h; |
|
| 63% |
With hydrogen bromide In benzene for 24h; Heating; |
|
| 59% |
With hydrogen bromide In toluene at 20℃; for 18h; |
1.1 1). Synthesis of 10-bromononanol (compound b)
adding toluene 500 ml in a 1 L three-necked flask, 1 175g of 10-decanediol, adding 200ml of 40% hydrobromic acid, stirring at room temperature for 18h, separating the liquid, washing the toluene phase with 200ml saturated laboratory, drying, concentrated crude product 240g,Column chromatography (EA: PE = 4:1) gave 140 g of 10-bromofurfuryl alcohol in a yield of 59% |
| 57% |
With hydrogen bromide In water; toluene for 48h; Reflux; Inert atmosphere; |
|
| 57% |
With hydrogen bromide In water; toluene Reflux; Dean-Stark; |
|
| 56% |
With hydrogen bromide In water; toluene at 90 - 95℃; for 48h; |
|
| 48% |
With hydrogen bromide |
|
|
With water; hydrogen bromide |
|
|
With hydrogen bromide In n-heptane Heating; |
|
|
With hydrogen bromide In Petroleum ether |
|
|
With hydrogen bromide In toluene |
|
|
With hydrogen bromide In water for 60h; Heating; |
|
|
With sulfuric acid; hydrogen bromide for 2h; Heating; |
|
|
With hydrogen bromide In n-heptane Heating; |
|
|
With hydrogen bromide In cyclohexane for 6h; Heating; |
|
|
With hydrogen bromide In toluene for 72h; Heating; |
|
|
With hydrogen bromide In cyclohexane for 6h; Heating; |
|
|
In hexane; hydrogen bromide |
3 10-Bromo-1-decanol
EXAMPLE 3 10-Bromo-1-decanol A solution of 1,10-dihydroxydecane (80 g) in 47% hydrobromic acid (650 ml) was stirred at 80° C. whilst being continuously extracted with petroleum ether (boiling range 80°-100°), for 24 hours. The organic extracts were neutralised with solid potassium carbonate, filtered, and the filtrates evaporated to dryness. The resultant dark oil was fractionally distilled (Vigreaux apparatus) to give a colourless oil, boiling point 102°-104° (0.01 mm Hg). Part of this oil (52 g) was purified by preparative HPLC (silica; eluents:diethyl ether/hexane 1:1) to remove a significant 1,10-dibromodecane impurity, finally yielding the title compound as a colourless oil. |
|
In hexane; hydrogen bromide |
3 10-Bromo-1-decanol
EXAMPLE 3 10-Bromo-1-decanol A solution of 1,10-dihydroxydecane (80 g) in 47% hydrobromic acid (650 ml) was stirred at 80°C whilst being continuously extracted with petroleum ether (boiling range 80-100°), for 24 hours. The organic extracts were neutralised with solid potassium carbonate, filtered, and the filtrates evaporated to dryness. The resultant dark oil was fractionally distilled (Vigreaux apparatus) to give a colourless oil, boiling point 102-104° (0.01 mm Hg). Part of this oil (52 g) was purified by preparative HPLC (silica; eluents:diethyl ether/hexane 1:1) to remove a significant 1,10-dibromodecane impurity, finally yielding the title compound as a colourless oil. |
|
With hydrogen bromide In toluene for 16h; Reflux; |
|
| 73.47 mg |
With hydrogen bromide In benzene for 2.5h; Reflux; |
|
|
With hydrogen bromide In toluene for 75h; Reflux; |
|
|
With hydrogen bromide In toluene at 120℃; for 16h; |
4.1.2. General Synthesis Procedure for Bromoalcohols 8a, 8b,and 8c.
To a stirred solution of diol 7 (300 mmol, 1.0 equiv)in toluene (600 mL) was added slowly 48% HBr (360 mmol,1.2 equiv) at RT. -e reaction mixture was reLuxed at 120°Cfor 16 h. After cooling to 0°C for 30 min, the organic layerwas separated and washed with saturated NaHCO3 solution.-e resulting aqueous layer was extracted with hexane(120 mL× 3). -e combined organic layer was washed withbrine (300 mL), dried over anhydrous MgSO4, :ltered, andconcentrated in vacuo to obtain the corresponding bromoalcohol8 as a light yellow oil. -e crude bromoalcohol 8was employed in the next step without further puri:cation.Bromoalcohols 8a, 8b, and 8c were synthesized independentlyaccording to the general procedure with 90%, 85%,and 91% yields, respectively. 1H NMR (200 MHz, CDCl3 ppm) for 8a: δ 3 3.63 (t, J 6.5 Hz, 2H), 3.40 (t, J 6.8 Hz, 2H),1.90-1.75 (m, 2H), 1.55-1.40 (m, 3H), 1.40-1.20 (m, 12H);13C NMR (100 MHz, CDCl3, ppm) for 8a: δ 3 62.54, 33.76,32.66, 32.53, 29.33, 29.24, 29.21, 28.58, 27.99, 25.59. |
|
With hydrogen bromide In water; toluene for 48h; Reflux; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping