|
With sodium carbonate;tetrakis(triphenylphosphine)palladium (0); In ethanol; hexane; dichloromethane; water; toluene; |
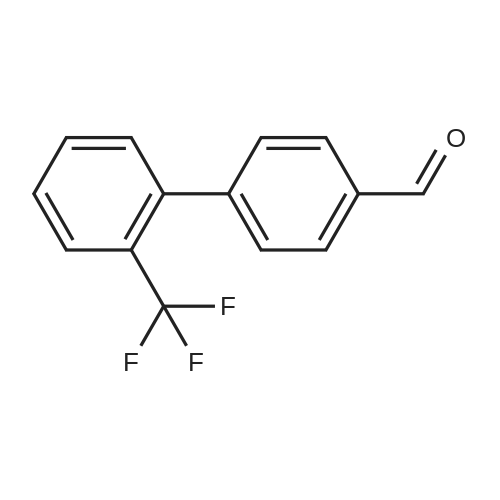
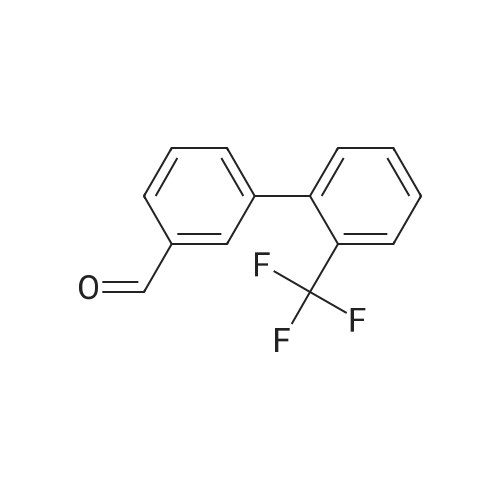
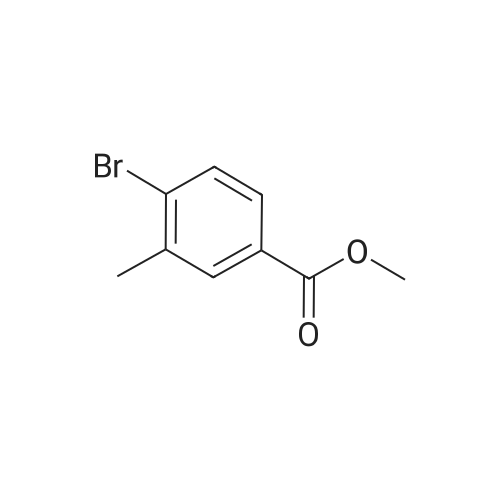
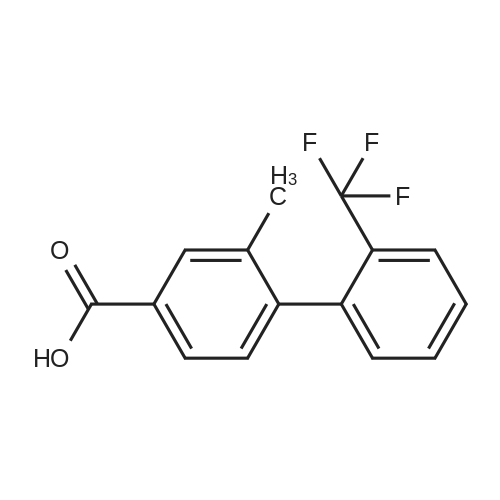
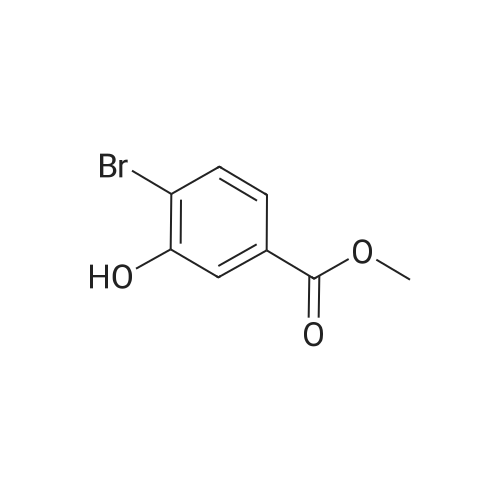
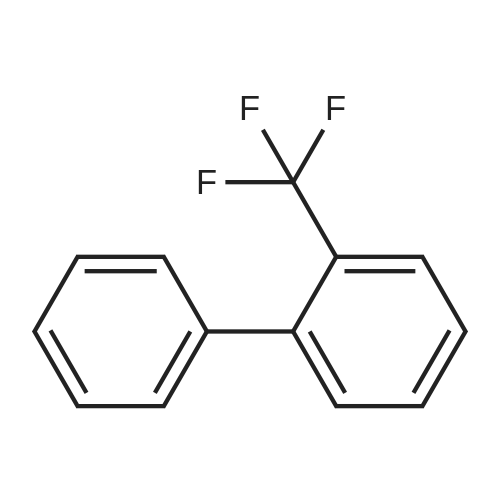
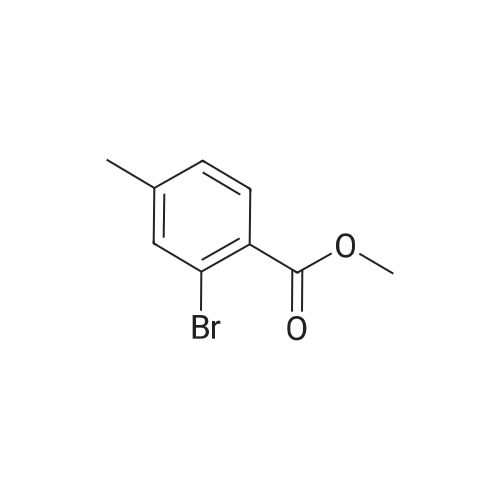
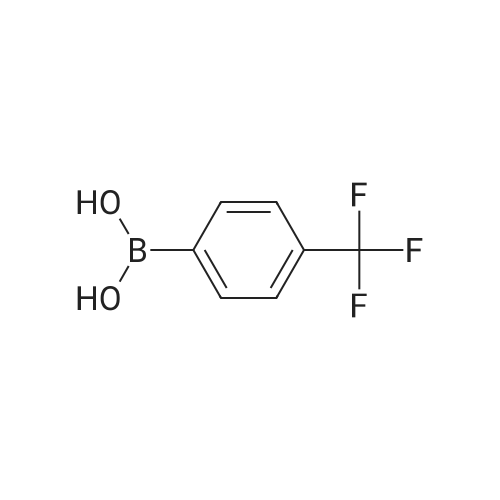
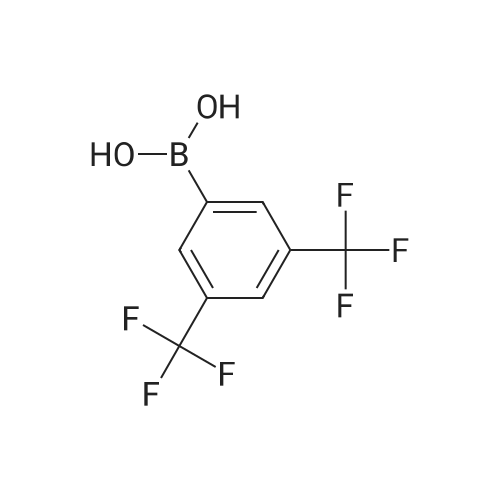
Step B. (2-Methyl-2'-trifluoromethyl-[1,1'-biphenyl]-4-yl)-carboxylic acid methyl ester A mixture of 4-bromo-3-methylbenzoic acid methyl ester of Step A (2.0 g, 8.7 mmol), 2-trifluoromethyl-phenyl boronic acid (1.65 g, 8.7 mmol) and sodium carbonate (4.1 g, 38.7 mmol) in toluene:ethanol:water (50 mL:25 mL: 25 mL) was purged with nitrogen for 1 hour. After addition of the tetrakis(triphenylphosphine) palladium(0) catalyst (0.50 g, 0.43 mmol) the reaction was heated at 100 C. overnight. The cooled reaction mixture was filtered through Celite and the cake washed with ethyl acetate. The organic layer was washed with water, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to give a brown oil. Purification by flash chromatography with a solvent gradient of 25% to 50% dichloromethane in hexane provided 2.0 g of the title compound as a colorless oil. 1H NMR (DMSO-d6, 400 MHz): delta2.03 (s, 3H), 3.88 (s, 3H), 7.26 (d, 1H), 7.34 (d, 1H), 7.66 (t, 1H), 7.75 (t, 1H), 7.81-7.83 (m, 1H), 7.86-7.88 (m, 1H), 7.90-7.91 (m, 1H) MS [ESI, m/z]: 312 [M+NH4]+. Anal. Calcd. for C16H13F3O2: C, 65.31; H, 4.45. Found: C, 64.92; H, 4.54. |
| 5.61 g |
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 24h; |
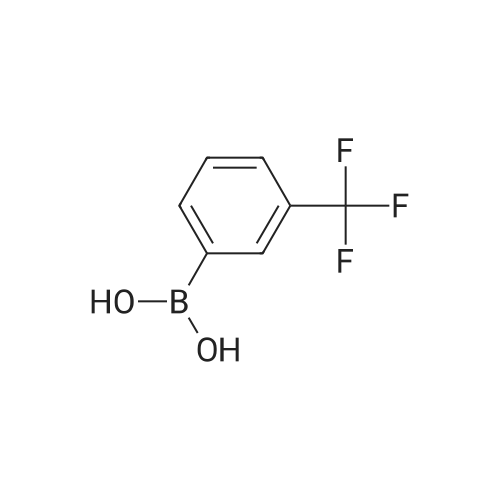
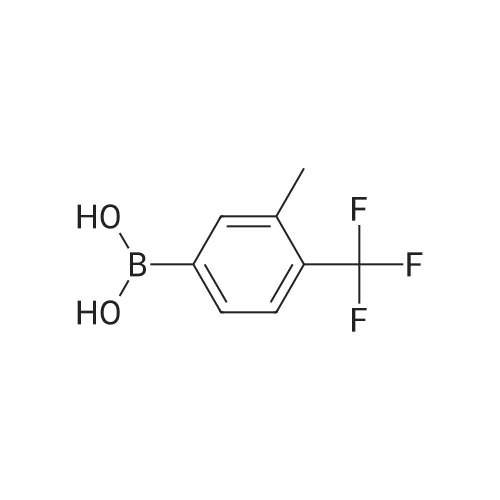
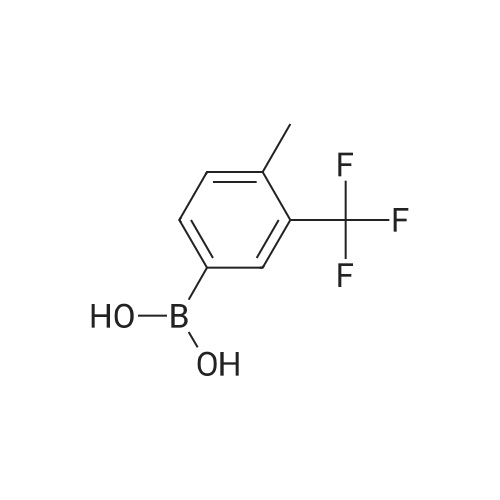
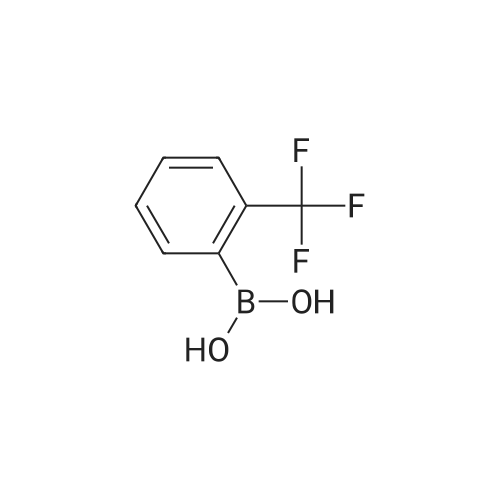
Reference Example 11 [0605] [0606] A mixture of methyl 4-bromo-3-methylbenzoate (4.58 g), [1,1?-bis(diphenylphosphino)-ferrocene]dichloropalladium (II) (1.46 g), potassium carbonate (11.0 g) and <strong>[1423-27-4]2-(trifluoromethyl)phenylboronic acid</strong> (7.60 g) was stirred in DMF (100 mL) at 80 C. for 24 hours. After cooling to room temperature, the reaction mixture was diluted with a mixed solvent of ethyl acetate-toluene (1:1) and filtered. Water was added to the filtrate and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine successively, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give a residue, which was purified with silica gel column chromatography (hexane/ethyl acetate) to give compound 11-1 (5.61 g). N-Bromosuccinimide (2.06 g) and benzoyl peroxide (133 mg) were added to a solution of compound 11-1 (3.07 g) in carbon tetrachloride (47 mL) and the mixture was heated to reflux for 6 hours. After cooling to room temperature, the reaction mixture was filtered. The filtrate was concentrated under reduced pressure and the resulting residue was purified with silica gel column chromatography (hexane/ethyl acetate) to give compound 11-2 (4.05 g) as a crude product. The crude product (3.78 g) of compound 11-2 was dissolved in acetic acid (73 mL), sodium acetate (4.17 g) was added thereto, and the mixture was heated to reflux for 16 hours. After cooling to room temperature, the reaction solution was concentrated under reduced pressure to give a residue, to which water was added and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine successively, dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure to give a residue, which was purified with silica gel column chromatography (hexane/ethyl acetate) to give compound 11-3 (2.39 g). Compound 11-3 (2.04 g) was dissolved in a mixed solvent of tetrahydrofuran-methanol (1:1) (40 mL), 1N aqueous solution of sodium hydroxide (23 mL) was added dropwise thereto, and the mixture was stirred at room temperature for 25 hours. The reaction solution was acidified by addition of 2N aqueous solution of hydrochloric acid, concentrated under reduced pressure and ethyl acetate and 1N aqueous solution of hydrochloric acid were added. The organic layer was extracted, washed with saturated brine, dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure to give a residue, which was azeotropically distilled with toluene to give compound 11-4 (1.69 g). Compound 11-5 (89 mg) was prepared from compound 11-4 (94 mg) in the same manner as the second step of Reference Example 9. Compound 11-1: 1H-NMR (CDCl3) delta 2.08 (s, 3H), 3.94 (s, 3H), 7.22 (d, J=8.1 Hz, 2H), 7.50 (t-like, J=7.6 Hz, 1H), 7.59 (t-like, J=7.1 Hz, 1H), 7.77 (d, J=7.7 Hz, 1H), 7.88 (md, J=7.9 Hz, 1H), 7.94 (m, 1H). Compound 11-2: 1H-NMR (CDCl3) delta 3.96 (s, 3H), 4.04 (d, J=10.4 Hz, 1H), 4.40 (d, J=10.4 Hz, 1H), 7.27 (d, J=8.1 Hz, 2H), 7.43 (d, J=7.3 Hz, 1H), 7.52-7.64 (m, 2H), 7.79 (d, J=7.3 Hz, 1H), 7.99 (dd, J=8.0, 1.7 Hz, 1H), 8.21 (d, J=1.7 Hz, 1H). Compound 11-3: 1H-NMR (CDCl3) delta 2.00 (s, 3H), 3.94 (s, 3H), 4.81 (AB q, JAB=22.8 Hz, 2H), 7.28 (d, J=7.9 Hz, 2H), 7.48-7.59 (m, 2H), 7.76 (d, J=7.2 Hz, 1H), 8.00 (dd, J=8.0, 1.7 Hz, 1H), 8.14 (d, J=1.3 Hz, 1H). Compound 11-4: 1H-NMR (CDCl3) delta 4.42 (AB q, JAB=23.0 Hz, 2H), 7.29 (d, J=8.1 Hz, 2H), 7.50-7.61 (m, 2H), 7.78 (d, J=7.5 Hz, 1H), 8.05 (dd, J=8.0, 1.6 Hz, 1H), 8.34 (s, 1H). [0607] Compound 11-5: 1H-NMR (CDCl3) delta 4.83 (AB q, JAB=20.4 Hz, 2H), 7.27 (d, J=7.5 Hz, 1H), 7.32 (d, J=8.0 Hz, 1H), 7.50-7.61 (m, 2H), 7.77 (d, J=7.2 Hz, 1H), 8.08 (dd, J=7.9, 1.8 Hz, 1H), 8.21 (d, J=1.7 Hz, 1H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping