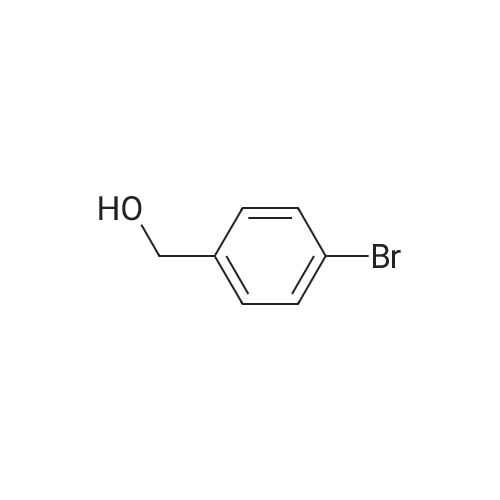
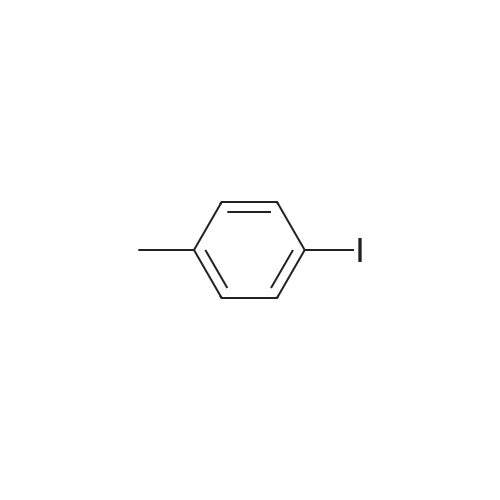
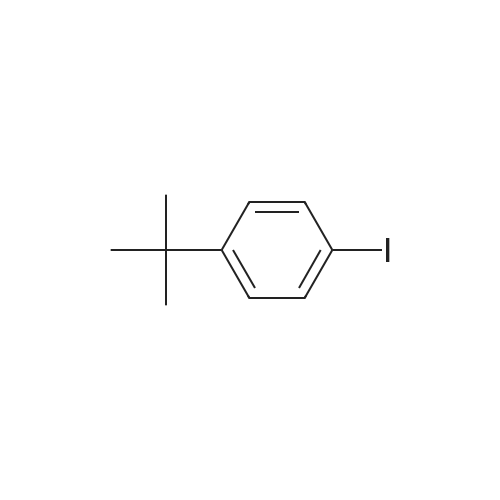
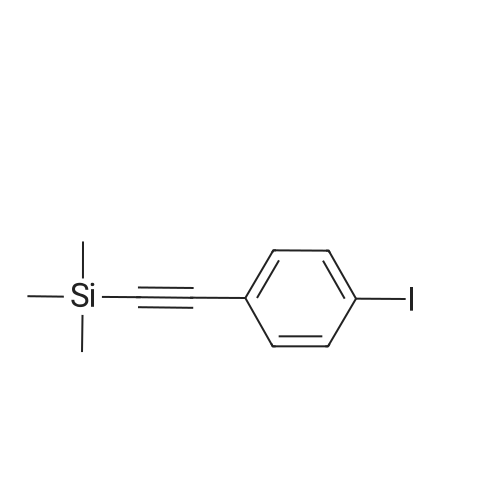
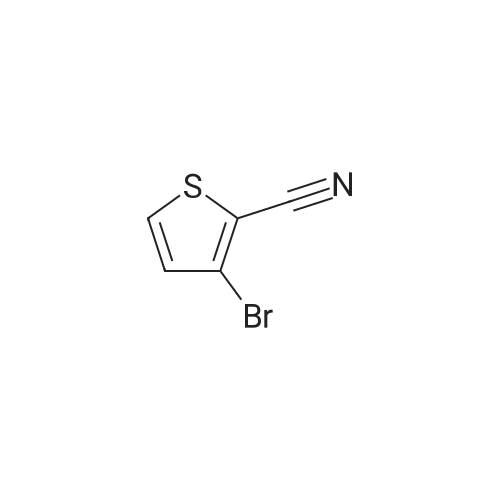
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: With n-butyllithium In diethyl ether; hexane at -78℃; for 0.5 h; Schlenk technique; Inert atmosphere Stage #2: With boric acid tributyl ester In diethyl ether; hexaneSchlenk technique; Inert atmosphere
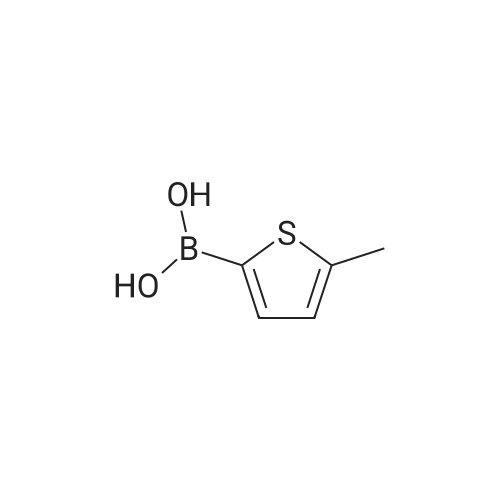
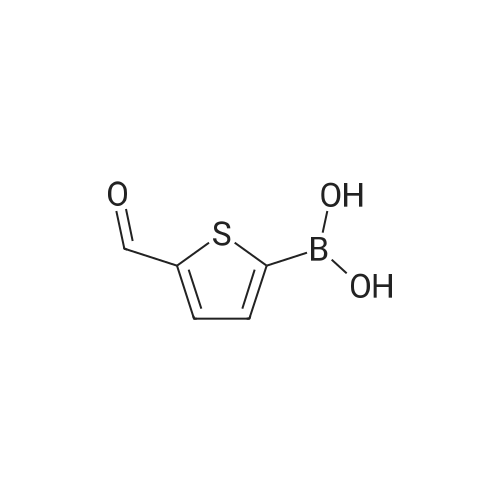
In a Schlenk flask under argon 3,5-dibromo-2-methylthiophene (12.8 g,50 mmol) was dissolved in dry diethyl ether (240 mL) and degassed.The solution was cooled to −78 °C and n-BuLi (20.4 mL, 50 mmol, 2.5M in hexane) was added dropwise. The mixture was stirred for 30 minat −78 °C.Tributyl borate (12.68 g, 50 mmol) was added and the reaction mixture was stirred overnight.1 M aq HCl (200 mL) was added, the diethyl ether phase was separated and extracted threetimes with 1 M aq NaOH solution (3 × 70 mL). To the combined NaOH phases conc. HClwas added until the mixture reached a pH of 1. In this acidic condition the product precipitatesas a beige colored solid with a yield of 76percent.
Reference:
[1] European Journal of Organic Chemistry, 2011, # 18, p. 3301 - 3312
[2] Acta Crystallographica Section C: Crystal Structure Communications, 2005, vol. 61, # 10, p. o599-o601
[3] Canadian Journal of Chemistry, 2007, vol. 85, # 1, p. 12 - 20
[4] Journal of Molecular Structure, 2007, vol. 833, # 1-3, p. 23 - 29
[5] Dyes and Pigments, 2010, vol. 84, # 1, p. 25 - 35
[6] Beilstein Journal of Organic Chemistry, 2016, vol. 12, p. 1103 - 1110
[7] Chimia, 2003, vol. 57, # 4, p. 169 - 172
[8] Chemistry - A European Journal, 2003, vol. 9, # 20, p. 4878 - 4886
[9] Tetrahedron, 2011, vol. 67, # 23, p. 4236 - 4242
[10] Dyes and Pigments, 2011, vol. 89, # 3, p. 246 - 253
[11] Journal of the American Chemical Society, 2011, vol. 133, # 42, p. 17027 - 17036
[12] Dyes and Pigments, 2015, vol. 115, p. 102 - 109
2
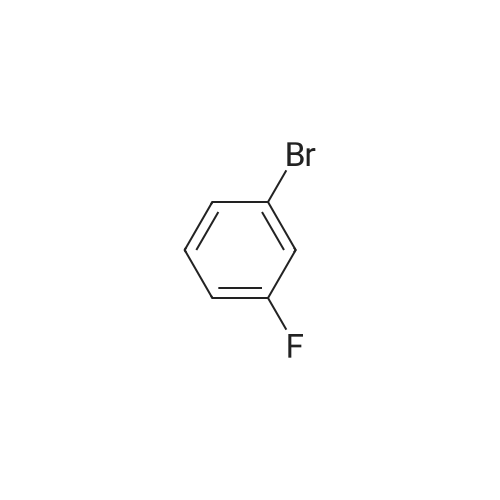
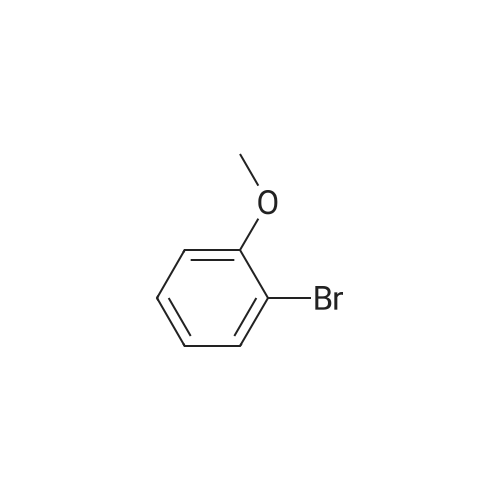
[ 29421-73-6 ]
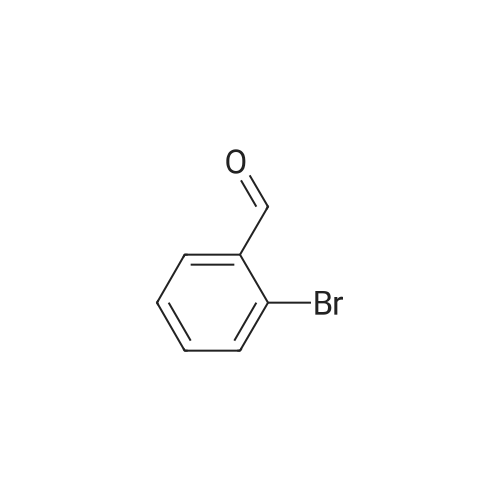
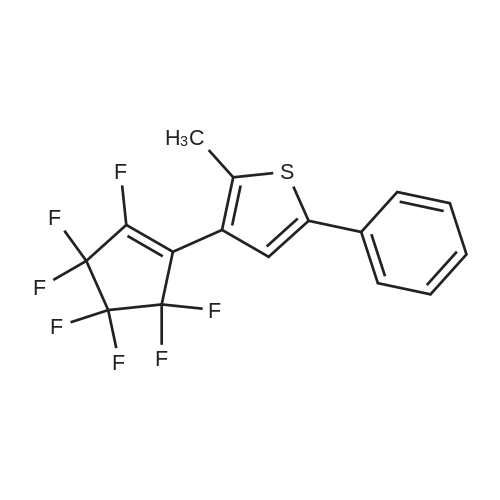
[ 688-74-4 ]
[ 154566-69-5 ]
Yield
Reaction Conditions
Operation in experiment
78.7%
Stage #1: With n-butyllithium In diethyl ether at -78.16℃; for 0.5 h; Inert atmosphere Stage #2: for 2 h; Inert atmosphere
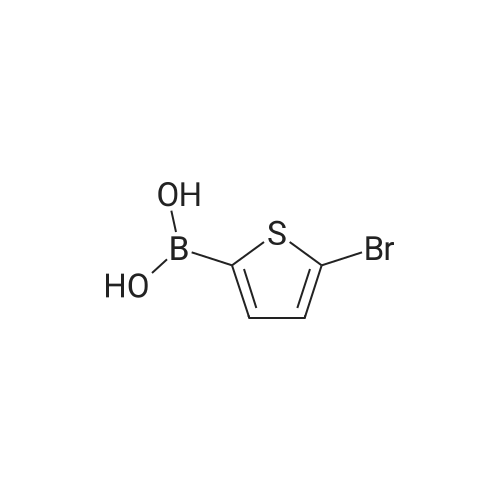
Liquid nitrogen cooling technology to control the temperature at 195K,Under the condition of nitrogen protection,Compound 6 (16.9 g, 66.4 mmol) was dissolved in purified ether and stirred.Slowly inject n-BuLi (29.2 mL, 73.0 mmol) at a concentration of 2.5 mol / L,After half an hour of low temperature reaction,Tributyl borate (20.66 mL, 76.96 mmol) was slowly added dropwise,Natural warming continue to react 2 hours later,Add 4percent dilute HCl solution 50mL to terminate the reaction, the liquid was discarded to the aqueous phase, the organic phase was extracted with 4percent dilute NaOH, the combined aqueous phase before and after the addition of 10percent dilute HCl solution acidified to neutral no longer produce a white flocculent precipitation, As an aside, the precipitate was washed with 4percent dilute HCl,Vacuum drying gave a pale yellow solid 11.5 g, 78.7percent yield;
71.5%
Stage #1: With n-butyllithium In diethyl ether at -78℃; for 0.5 h; Inert atmosphere Stage #2: for 1.5 h;
Under nitrogen and -78 & lt; 0 & gt; C,5 (17.03 g, 66.53 mmol) was dissolved in exquisite anhydrous ether,Stirring, slow injection concentration of 2.50 molL-1ofn-BuLi (26.61 mL, 66.53 mmol),Low temperature reaction after half an hour,Dropping tributyl borate,Natural temperature to continue to respond for 1.5h.The reaction was quenched by the addition of dilute HCl, the aqueous phase was separated, the organic phase was extracted with dilute NaOH,The aqueous phase is acidified with dilute HCl solution until neutr is no longer white precipitate. The precipitate was washed with dilute HCl and dried in vacuo to give 10.10 g of a pale yellow solid, yield: 71.5percent
66.7%
Stage #1: With n-butyllithium In diethyl ether at -78℃; for 0.5 h; Inert atmosphere Stage #2: at -78℃; for 1.5 h; Inert atmosphere Stage #3: With hydrogenchloride In water
And -78 deg.] C under nitrogen conditions,Compound 2 (20.6 g, 80.5 mmol)Was dissolved in anhydrous ether, stirred, slowly injected with n-BuLi (84.5 mmol)After half an hour of low temperature reaction, tributyl borate was added,After heating for 1.5 h,Adding dilute HCl to stop the reaction,The aqueous phase was separated and the organic phase was extracted with dilute NaOH, and the aqueous phase was acidified with dilute HCl solution until the neutral precipitate was no longer produced.The precipitate was washed with dilute HCl and dried in vacuo to give 11.8 g of compound 3 as a pale yellow solid in 66.7percent yield;
66%
Stage #1: With n-butyllithium In diethyl ether at -78.16℃; for 0.5 h; Inert atmosphere Stage #2: for 2 h;
Liquid nitrogen cooling technology to control the temperature at 195K,3,5-Dibromo-2-methylthiophene (19.18 g, 75.00 mmol)Added to the refined ether, with rapid stirring,Slowly inject n-BuLi (32.26 mL, 84.00 mmol) at a concentration of 2.4 mol / L,After the reaction at low temperature for 0.5h, tributyl borate (20.2mL, 5.00mmol) was slowly added dropwise. After the reaction for 2h, 50.0mL of 4percent hydrochloric acid solution was added to stop the reaction.The liquid was separated and the organic phase was extracted three more times with 4percent sodium hydroxide solution.After combining all the aqueous phases, quickly add 10percent hydrochloric acid solution acidification and vigorous stirring,Until the white flocculation precipitate completely precipitated, suction filtered,White flocculent precipitate was washed with 4percent hydrochloric acid solution, completely dried at room temperature,11.23 g of pale yellow solid was obtained in a yield of 66percent.
Reference:
[1] Canadian Journal of Chemistry, 2007, vol. 85, # 1, p. 12 - 20
[2] Journal of Molecular Structure, 2007, vol. 833, # 1-3, p. 23 - 29
[3] Patent: CN104292234, 2017, B, . Location in patent: Paragraph 0024; 0036; 0037; 0038
[4] Patent: CN104892588, 2017, B, . Location in patent: Paragraph 0030; 0035; 0036; 0037
[5] Patent: CN104098555, 2017, B, . Location in patent: Paragraph 0060; 0065; 0066; 0067
[6] Patent: CN106905312, 2017, A, . Location in patent: Paragraph 0016; 0034; 0041
[7] Journal of the Chemical Society, Chemical Communications, 1993, # 18, p. 1439 - 1442
[8] Inorganica Chimica Acta, 2006, vol. 359, # 13, p. 4281 - 4288
[9] Patent: US2014/191168, 2014, A1, . Location in patent: Paragraph 0127
3
[ 29421-73-6 ]
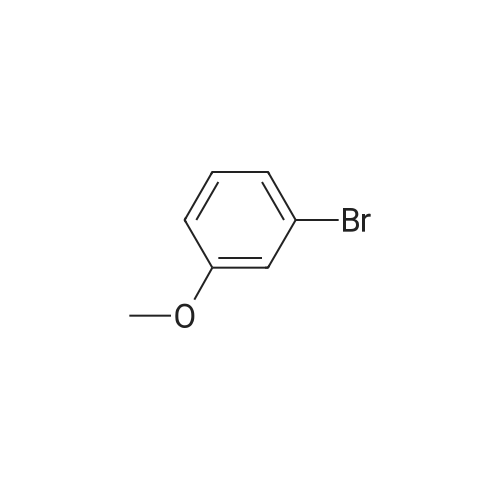
[ 5419-55-6 ]
[ 154566-69-5 ]
Yield
Reaction Conditions
Operation in experiment
82%
Stage #1: With n-butyllithium In diethyl ether; hexane at -78℃; for 0.5 h; Inert atmosphere Stage #2: With hydrogenchloride In diethyl ether; hexane; water at -78 - 20℃; for 19 h;
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 70℃; for 15h;
This compound was synthesized by the same method as that reported in our previous works.[8], 8(a), 8(b), [9] and 9(a) Compound 7a was prepared by reacting compound 6 (4.0 g, 18.1 mmol) with 4-bromoanisole (3.4 g, 18.1 mmol) in the presence of Pd(PPh3)4 and Na2CO3 (6.40 g, 60 mmol) in tetrahydrofuran (80 mL containing 10% water) for 15 h at 70 C. The product 7a was purified by column chromatography on SiO2 using hexane as an eluent and 3.8 g obtained as a pale yellow solid in 78% yield. Mp 107-108 C. Anal. Calcd for C12H11BrOS (%): calcd C, 50.90; H, 3.92. Found C, 50.72; H, 4.04; 1H NMR (400 MHz, CDCl3): delta 2.40 (s, 3H, -CH3), 3.83 (s, 3H, -OCH3), 6.90 (d, 2H, J=8.0 Hz, phenyl-H), 6.99 (s, 1H, thienyl-H), 7.43 (d, 2H, J=8.0 Hz, phenyl-H); 13C NMR (100 MHz, CDCl3, TMS): delta 14.78, 55.38, 109.63, 114.35, 124.48, 126.04, 126.39, 126.68, 132.57, 136.09, 141.10, 159.41; IR (nu, KBr, cm-1): 797, 818, 946, 1033, 1111, 1161, 1179, 1255, 1288, 1309, 1455, 1541, 1603, 2917.
78%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 69.84℃; for 15h;
Compound 5a was prepared by reacting compound 4 (4.0 g, 18.1 mmol) with 4-bromoanisole (3.4 g, 18.1 mmol) in the presence of Pd(PPh3)4 and Na2CO3 (6.40 g, 60 mmol) in tetrahydrofuran (80 mL containing 10% water) for 15h at 343 K. The product 5a was puri?ed by column chromatography on SiO2 using hexane as an eluent and 3.8 g obtained as a pale yellow solid in 78% yield. 1H NMR (400 MHz, CDCl3): delta 2.40 (s, 3H, -CH3), 3.83 (s, 3H, -OCH3 ), 6.90 (d, 2H, J = 8.0 Hz, phenyl-H), 6.99 (s, 1H, thienyl-H), 7.43 (d, 2H, J = 8.0 Hz, phenyl-H).
78%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 94.84℃; for 15h;
7a was prepared by reacting compound 6 (4.00g, 18.1mmol) with 4-bromoanisole (3.38g, 18.1mmol) in the presence of Pd(PPh3)4 and Na2CO3 (6.40g, 60mmol) in THF (80mL containing 10% water). After refluxing for 15h at 368K, the product was extracted with CH2Cl2. The organic layer was dried over MgSO4, filtrated, and evaporated. Column chromatography on SiO2 with petroleum ether as the eluent afforded 3.99g of 7a as a yellowish solid in 78% yield. M.p. 378-379K; 1H NMR (400MHz, CDCl3) delta 2.40 (s, 3H, -CH3), 3.83 (s, 3H, -OCH3), 6.90 (d, 2H, J=8.0Hz, phenyl-H), 6.99 (s, 1H, thienyl-H), 7.43 (d, 2H, J=8.0Hz, phenyl-H).
76%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; at 69.84℃; for 18h;
General procedure: Compound 6a was prepared by reacting 3-bromo-2-methyl-5-thienylboronic acid [50] (4.2 g, 19.0 mmol) with 1-bromo-2-methoxybenzene (3.91 g, 20.9 mmol) in the presence of Pd(PPh3)4 (0.73 g, 0.63 mmol) and Na2CO3 (2 mol L-1, 10 mL) in tetrahydrofuran (THF) (80 mL) for 18 h at 343 K. The reaction was allowed to cool to room temperature. After being extracted with ether, the organic layer was dried over MgSO4, filtered, and concentrated. The crude product was purified by column chromatography on silica gel using petroleum ether as eluent to afford 3.87 g of 6a visible as a buff solid with a 72% yield.
70%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; for 16h;Reflux;
Compound 9a was prepared byreacting 3-bromo-2-methyl-5-thienylboronic acid 8 (3.00 g,13.6 mmol) with 3-bromoanisole (2.79 g, 14.9 mmol) in the presence ofPd(PPh3)4 (0.15 g) and Na2CO3 (2.0 mol L-1,60.0 mL) in tetrahydrofuran (THF). After refluxing for 16 h, the product wasextracted with dichloromethane. The organic layer was dried over MgSO4,filtrated, and evaporated. The crude product was purified by columnchromatography on SiO2 using petroleum ether as the eluent and 2.73 g of 9a obtainedas a yellowish solid in 70% yield. Mp: 380-381 K. 1H NMR (400 MHz,CDCl3): delta 2.40 (s, 3H, -CH3),3.83 (s, 3H, -OCH3), 6.90 (d, 2H, J = 8.0 Hz, phenyl-H), 6.99 (s, 1H, thienyl-H), 7.43 (d, 2H, J = 8.0 Hz, phenyl-H).
70%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; for 16h;Reflux;
General procedure: compound 7a was prepared by reacting 3-bromo-2-methyl-5-thienylboronic acid 6 (3.00 g, 13.6 mmol) with 3-bromoanisole (2.79 g, 14.9 mmol) in the presence of Pd(PPh3)4 (0.15 g) and Na2CO3(2.0 mol L-1, 60.0 mL) in tetrahydrofuran (THF). After refluxing for 16 h, the product was extracted with dichloromethane. The organic layer was dried over Na2SO4, filtrated, and evaporated. The crude product was purified by column chromatography on SiO2 using petroleum ether as the eluent and 2.73 gof 7a obtained as a yellowish solidin 70% yield. Mp: 380-381 K. 1H NMR (400 MHz, CDCl3): delta 2.40 (s, 3H, -CH3), 3.83(s, 3H, -OCH3), 6.90 (d, 2H, J= 8.0 Hz, phenyl-H), 6.99 (s, 1H, thienyl-H), 7.43 (d, 2H, J = 8.0 Hz, phenyl-H).
60.9%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; for 16h;Inert atmosphere; Reflux;
P-Methoxybromobenzene (2.8 g, 14.9 mmol) was dissolved in THF solution,Under nitrogen protection,New and formed Pd (PPh3) 4 (0.5 g) was added,Was stirred at room temperature for 20 minutes was added 7 (3.0 g, 13.6 mmol)And the concentration of 2.0 mol / L Na2CO3 solution 60 mL,Continue nitrogen protection and heated to reflux for 16 hours,After the reaction was over, the mixture was cooled to room temperature, the THF solution was removed by rotary evaporation in vacuo, extracted with methylene chloride, washed with water, dried over anhydrous magnesium sulfate, and purified by silica gel column chromatography using petroleum ether as eluent.A white solid 2.7 g was obtained with a yield of 60.9%;
53.1%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; for 16h;Inert atmosphere; Reflux;
Under nitrogen protection,P-methoxybromobenzene (2.1 g, 11.3 mmol) andPd (PPh3) 4 (0.5 g)Was dissolved in 80 mL of THF,After stirring for 20 min, 3 (2.5 g, 11.3 mmol)And the concentration of 2.0 mol / L Na2CO3 solution 50 mL,Heating reflux for 16 h,Stop the reaction and cool to room temperature.The mixture was extracted with dichloromethane and the combined organic phases were dried over anhydrous magnesium sulfate.The filtrate was distilled off to remove the solvent and silica gel column chromatography (petroleum ether) to give a white solid (1.7 g, 6.0 mmol) in 53.1% yield;
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; sodium carbonate; In tetrahydrofuran; water; at 80℃;Schlenk technique; Inert atmosphere;
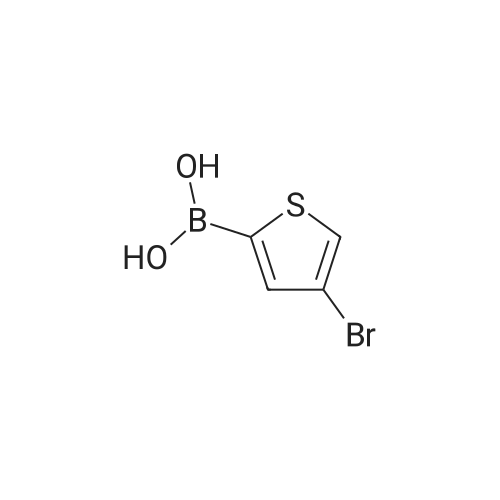
In a Schlenk flask under argon 4-bromo-5-methylthiophene-2-ylboronicacid (1.5 g, 6.8 mmol) was dissolved in THF (13 mL) and degassed.Iodbenzene (2.2 g, 8.2 mmol), Pd(dppf)Cl2 (393 mg, 0.34 mmol) andaq Na2CO3 solution (20%, 13 mL) were added and the mixture wasstirred under reflux overnight at 80 C.The reaction mixture was extracted three times with DCM, the combined organic phases werewashed with water and brine, dried over MgSO4, filtered and the solvents was removed underreduced pressure. Purification by flash column chromatography (pure cyclohexane) afforded awhite solid with a yield of 94%.
In a Schlenk flask under argon 3,5-dibromo-2-methylthiophene (12.8 g,50 mmol) was dissolved in dry diethyl ether (240 mL) and degassed.The solution was cooled to -78 C and n-BuLi (20.4 mL, 50 mmol, 2.5M in hexane) was added dropwise. The mixture was stirred for 30 minat -78 C.Tributyl borate (12.68 g, 50 mmol) was added and the reaction mixture was stirred overnight.1 M aq HCl (200 mL) was added, the diethyl ether phase was separated and extracted threetimes with 1 M aq NaOH solution (3 × 70 mL). To the combined NaOH phases conc. HClwas added until the mixture reached a pH of 1. In this acidic condition the product precipitatesas a beige colored solid with a yield of 76%.
2-bromobenzaldehyde (2.50 g, 13.57 mmol) dissolved in 100 mL THF and Pd(PPh3)4 (0.15 g) were added to 250 mL three-necked flask protected with argon, and stirred at room temperature for 20 min. <strong>[154566-69-5](4-bromo-5-methylthiophen-2-yl)boronic acid</strong> (3.00 g, 13.57 mmol) and 50 mL of 2 mol L-1 aqueous Na2CO3 solution were added to the above-mentioned three-necked flask, stirred, reflux reaction for 16 h, cooled to room temperature, vacuum rotary evaporation was carried out to remove THF solution, extracted three times with methylene chloride, organic phase was combined, washed three times with saturated brine and distilled water, dried using anhydrous MgSO4 overnight, evaporated to remove the solvent, and silica gel column chromatography was carried out with petroleum ether as eluent to give a pale yellow solid product of 3.00 g, yield: 78.95%, product structure identification.
55%
In a 250.0 mL three-necked flask, under argon atmosphere,A solution of 4-allylbromobenzene (2.23 g, 12.12 mmol) in 100.0 mL of tetrahydrofuran was added to 0.13 g of Pd (PPh3) 4 catalyst just prepared,Stir at room temperature for about 20 min and add the compound of formula 8 separately<strong>[154566-69-5]2-methyl-3-bromo-5-boronic acid thiophene</strong> (3.10 g, 13.84 mmol) and60.0mL (2.0mol L-1) aqueous solution of sodium carbonate, the reaction was continued after reflux for about 12h, the reaction was terminated, the solution was suspended, and then extracted with methylene chloride several times,The resulting organic phase was washed three times with saturated brine and distilled water, respectively,Dry with anhydrous sodium sulfate for 12h. With petroleum ether / ethyl acetate (v / v = 20: 1)As eluent, separated and purified by silica gel column, the product was pale yellow solid 8.09g,Yield: 55%.
3-bromo-2-methyl-5-(4-fluorophenyl ) thiophene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 70℃; for 15h;
General procedure: This compound was synthesized by the same method as that reported in our previous works.[8], 8(a), 8(b), [9] and 9(a) Compound 7a was prepared by reacting compound 6 (4.0 g, 18.1 mmol) with 4-bromoanisole (3.4 g, 18.1 mmol) in the presence of Pd(PPh3)4 and Na2CO3 (6.40 g, 60 mmol) in tetrahydrofuran (80 mL containing 10% water) for 15 h at 70 C. The product 7a was purified by column chromatography on SiO2 using hexane as an eluent and 3.8 g obtained as a pale yellow solid in 78% yield.
77%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 94.84℃; for 15h;
General procedure: 7a was prepared by reacting compound 6 (4.00g, 18.1mmol) with 4-bromoanisole (3.38g, 18.1mmol) in the presence of Pd(PPh3)4 and Na2CO3 (6.40g, 60mmol) in THF (80mL containing 10% water). After refluxing for 15h at 368K, the product was extracted with CH2Cl2. The organic layer was dried over MgSO4, filtrated, and evaporated. Column chromatography on SiO2 with petroleum ether as the eluent afforded 3.99g of 7a as a yellowish solid in 78% yield.
70.3%
Under a nitrogen atmosphere, fluorobromobenzene (3.51 g, 20.05 mmol) and Pd (PPh3) 4 (0.5 g) were dissolved in 80 ml of THFin,Stir for 20 minFollowed by addition of 6 (4.45 g, 20.07 mmol)And the concentration of 2.0mol·L-1Na2CO3 solution 60ml,Heated to reflux for 16 h, stop the reaction, cool to room temperature. The aqueous phase was extracted with ether and the combined organic phases were dried over anhydrous magnesium sulfate. Filter, filterThe solvent was distilled off and the silica gel column chromatography petroleum ether was separated to give a white solid (4.52 g, 14.07 mmol) in a yield of 70.03%.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; for 16h;Inert atmosphere; Reflux;
Under nitrogen protection,Bromobenzene (3.0 g, 19 mmol) andPd (PPh3) 4 (0.5 g)Was dissolved in 80 mL of THF,After stirring for 20 min, 3 (4.0 g, 17 mmol)And the concentration of 2.0 mol / L Na2CO3 solution 50 mLHeated to reflux for 16 h, stop the reaction, cool to room temperature. The aqueous phase was extracted with ether and the combined organic phases were dried over anhydrous magnesium sulfate.The filtrate was distilled off to remove the solvent and silica gel column chromatography (petroleum ether) to give 3.5 g as a pale yellow solid in 81.4% yield.
77%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 70℃; for 15h;
General procedure: This compound was synthesized by the same method as that reported in our previous works.[8], 8(a), 8(b), [9] and 9(a) Compound 7a was prepared by reacting compound 6 (4.0 g, 18.1 mmol) with 4-bromoanisole (3.4 g, 18.1 mmol) in the presence of Pd(PPh3)4 and Na2CO3 (6.40 g, 60 mmol) in tetrahydrofuran (80 mL containing 10% water) for 15 h at 70 C. The product 7a was purified by column chromatography on SiO2 using hexane as an eluent and 3.8 g obtained as a pale yellow solid in 78% yield.
77%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 69.84℃; for 15h;
General procedure: Compound 5a was prepared by reacting compound 4 (4.0 g, 18.1 mmol) with 4-bromoanisole (3.4 g, 18.1 mmol) in the presence of Pd(PPh3)4 and Na2CO3 (6.40 g, 60 mmol) in tetrahydrofuran (80 mL containing 10% water) for 15h at 343 K. The product 5a was puri?ed by column chromatography on SiO2 using hexane as an eluent and 3.8 g obtained as a pale yellow solid in 78% yield.
77%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 94.84℃; for 15h;
General procedure: 7a was prepared by reacting compound 6 (4.00g, 18.1mmol) with 4-bromoanisole (3.38g, 18.1mmol) in the presence of Pd(PPh3)4 and Na2CO3 (6.40g, 60mmol) in THF (80mL containing 10% water). After refluxing for 15h at 368K, the product was extracted with CH2Cl2. The organic layer was dried over MgSO4, filtrated, and evaporated. Column chromatography on SiO2 with petroleum ether as the eluent afforded 3.99g of 7a as a yellowish solid in 78% yield.
72%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; at 69.84℃; for 18h;
General procedure: Compound 6a was prepared by reacting 3-bromo-2-methyl-5-thienylboronic acid [50] (4.2 g, 19.0 mmol) with 1-bromo-2-methoxybenzene (3.91 g, 20.9 mmol) in the presence of Pd(PPh3)4 (0.73 g, 0.63 mmol) and Na2CO3 (2 mol L-1, 10 mL) in tetrahydrofuran (THF) (80 mL) for 18 h at 343 K. The reaction was allowed to cool to room temperature. After being extracted with ether, the organic layer was dried over MgSO4, filtered, and concentrated. The crude product was purified by column chromatography on silica gel using petroleum ether as eluent to afford 3.87 g of 6a visible as a buff solid with a 72% yield.
71%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; for 16h;Reflux;
General procedure: Compound 9a was prepared byreacting 3-bromo-2-methyl-5-thienylboronic acid 8 (3.00 g,13.6 mmol) with 3-bromoanisole (2.79 g, 14.9 mmol) in the presence ofPd(PPh3)4 (0.15 g) and Na2CO3 (2.0 mol L-1,60.0 mL) in tetrahydrofuran (THF). After refluxing for 16 h, the product wasextracted with dichloromethane. The organic layer was dried over MgSO4,filtrated, and evaporated. The crude product was purified by columnchromatography on SiO2 using petroleum ether as the eluent and 2.73 g of 9a
71%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; for 16h;Reflux;
General procedure: compound 7a was prepared by reacting 3-bromo-2-methyl-5-thienylboronic acid 6 (3.00 g, 13.6 mmol) with 3-bromoanisole (2.79 g, 14.9 mmol) in the presence of Pd(PPh3)4 (0.15 g) and Na2CO3(2.0 mol L-1, 60.0 mL) in tetrahydrofuran (THF). After refluxing for 16 h, the product was extracted with dichloromethane. The organic layer was dried over Na2SO4, filtrated, and evaporated. The crude product was purified by column chromatography on SiO2 using petroleum ether as the eluent and 2.73 gof 7a obtained as a yellowish solid.
71.2%
Under nitrogen protection,Bromobenzene (3.24 g, 20.62 mmol) and Pd (PPh3) 4 (0.5 g)Was dissolved in 80 ml of THF and stirredMix for 20minFollowed by the addition of 6 (4.57 g, 20.63 mmol)And the concentration of 2.0mol·L-1Na2CO3 solution 60ml,Heated to reflux for 16 h, stop the reaction, cool to room temperature. The aqueous phase was extracted with ether,The organic phases were combined and dried over anhydrous magnesium sulfate. The filtrate was distilled off to remove the solvent and separated with petroleum ether to give a pale yellow solid (3.72 g, 14.69 mmol) in 71.2% yield.
3-bromo-2-methyl-5-(3-fluorophenyl)thiophene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
67%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; for 16h;
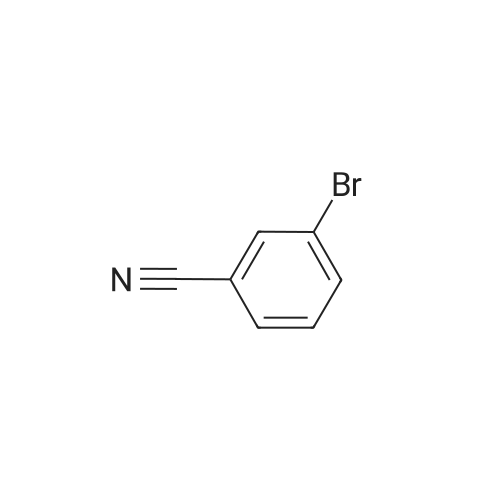
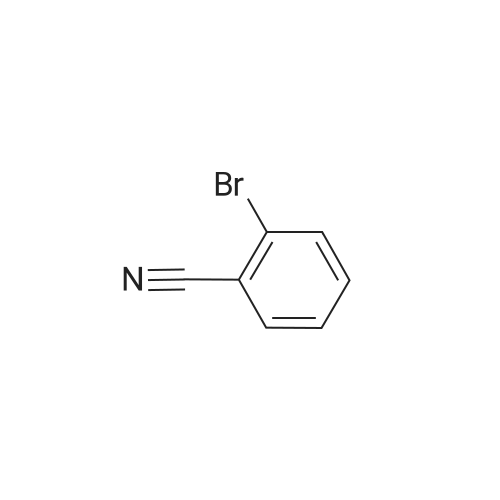
General procedure: Compound 7a was prepared by reacting 3-bromo-2-methyl-5-thienylboronic acid (3.0g, 11.3mmol) with 3-bromobenzonitrile (2.47g, 13.6mmol) in the presence of Pd(PPh3)4 (0.15g, 0.01mmol) and Na2CO3 (2.0 mol L-1, 50 mL) in tetrahydrofuran (THF) (80mL containing 10% water). After refluxing for 16h, the product was extracted with diethyl ether. The organic layer was dried over MgSO4, filtrated, and evaporated. The crude product was purified by column chromatography on SiO2 using petroleum ether as the eluent and 2.0g of 7a obtained as a yellowish solid in 53% yield.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; at 69.84℃; for 18h;
General procedure: Compound 6a was prepared by reacting 3-bromo-2-methyl-5-thienylboronic acid [50] (4.2 g, 19.0 mmol) with 1-bromo-2-methoxybenzene (3.91 g, 20.9 mmol) in the presence of Pd(PPh3)4 (0.73 g, 0.63 mmol) and Na2CO3 (2 mol L-1, 10 mL) in tetrahydrofuran (THF) (80 mL) for 18 h at 343 K. The reaction was allowed to cool to room temperature. After being extracted with ether, the organic layer was dried over MgSO4, filtered, and concentrated. The crude product was purified by column chromatography on silica gel using petroleum ether as eluent to afford 3.87 g of 6a visible as a buff solid with a 72% yield.
53%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; for 16h;
General procedure: Compound 7a was prepared by reacting 3-bromo-2-methyl-5-thienylboronic acid (3.0g, 11.3mmol) with 3-bromobenzonitrile (2.47g, 13.6mmol) in the presence of Pd(PPh3)4 (0.15g, 0.01mmol) and Na2CO3 (2.0 mol L-1, 50 mL) in tetrahydrofuran (THF) (80mL containing 10% water). After refluxing for 16h, the product was extracted with diethyl ether. The organic layer was dried over MgSO4, filtrated, and evaporated. The crude product was purified by column chromatography on SiO2 using petroleum ether as the eluent and 2.0g of 7a obtained as a yellowish solid in 53% yield.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; at 69.84℃; for 18h;
Compound 6a was prepared by reacting 3-bromo-2-methyl-5-thienylboronic acid [50] (4.2 g, 19.0 mmol) with 1-bromo-2-methoxybenzene (3.91 g, 20.9 mmol) in the presence of Pd(PPh3)4 (0.73 g, 0.63 mmol) and Na2CO3 (2 mol L-1, 10 mL) in tetrahydrofuran (THF) (80 mL) for 18 h at 343 K. The reaction was allowed to cool to room temperature. After being extracted with ether, the organic layer was dried over MgSO4, filtered, and concentrated. The crude product was purified by column chromatography on silica gel using petroleum ether as eluent to afford 3.87 g of 6a visible as a buff solid with a 72% yield. 1H NMR (400 MHz, CDCl3): delta 2.41 (s, 3H, -CH3), 3.91 (s, 3H, -CH3), 6.95-6.99 (m, 2H, phenyl-H), 7.27 (s, 1H, thienyl-H), 7.30 (s, 1H, phenyl-H), 7.54 (d, 1H, J=8.0 Hz, phenyl-H).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; for 16h;
General procedure: Compound 7a was prepared by reacting 3-bromo-2-methyl-5-thienylboronic acid (3.0g, 11.3mmol) with 3-bromobenzonitrile (2.47g, 13.6mmol) in the presence of Pd(PPh3)4 (0.15g, 0.01mmol) and Na2CO3 (2.0 mol L-1, 50 mL) in tetrahydrofuran (THF) (80mL containing 10% water). After refluxing for 16h, the product was extracted with diethyl ether. The organic layer was dried over MgSO4, filtrated, and evaporated. The crude product was purified by column chromatography on SiO2 using petroleum ether as the eluent and 2.0g of 7a obtained as a yellowish solid in 53% yield.
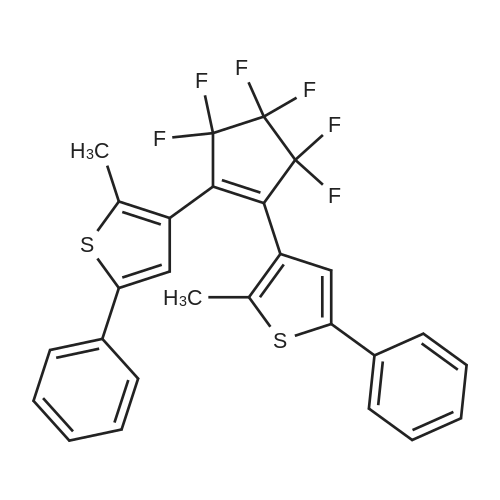
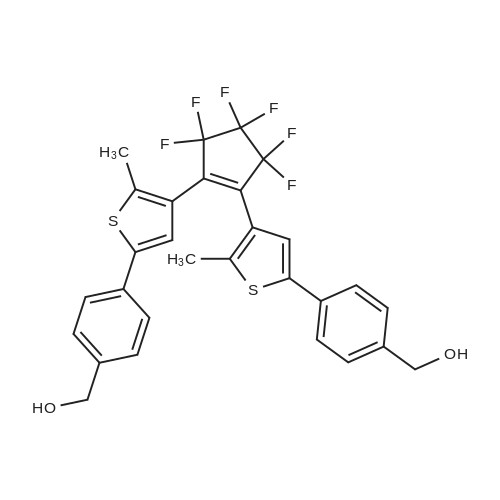
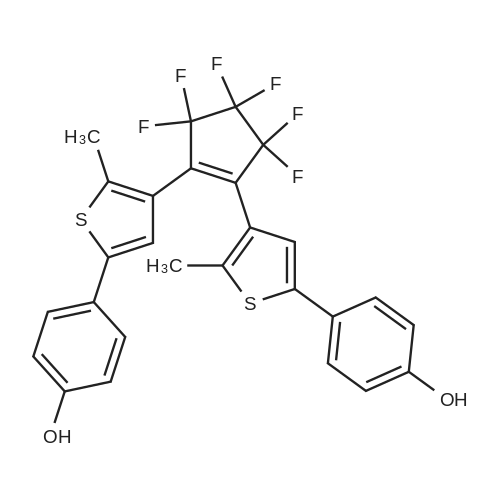
(S)-1-(4-{4-[3,3,4,4,5,5-Hexafluoro-2-(2-methyl-5-phenyl-thiophen-3-yl)-cyclopent-1-enyl]-5-methyl-thiophen-2-yl}-phenyl)-3-methyl-pentan-1-ol[ No CAS ]
1-[5'-(4''-methoxyphenyl)-2'-methylthien-3'-yl]-2-[2'-methyl-5'-(4''-triisopropylsilylethynylphenyl)thien-3'-yl]-3,3,4,4,5,5-hexafluorocyclopentene[ No CAS ]
4-[15-{4-[4-(4-{3,3,4,4,5,5-hexafluoro-2-[5-(4-methoxy-phenyl)-2-methyl-thiophen-3-yl]-cyclopent-1-enyl}-5-methyl-thiophen-2-yl)-phenylethynyl]-phenyl}-10,20-bis-(2,4,6-trimethyl-phenyl)-porphyrin-5-yl]-benzoic acid methyl ester[ No CAS ]
Liquid nitrogen cooling technology to control the temperature at 195K,Under the condition of nitrogen protection,Compound 6 (16.9 g, 66.4 mmol) was dissolved in purified ether and stirred.Slowly inject n-BuLi (29.2 mL, 73.0 mmol) at a concentration of 2.5 mol / L,After half an hour of low temperature reaction,Tributyl borate (20.66 mL, 76.96 mmol) was slowly added dropwise,Natural warming continue to react 2 hours later,Add 4% dilute HCl solution 50mL to terminate the reaction, the liquid was discarded to the aqueous phase, the organic phase was extracted with 4% dilute NaOH, the combined aqueous phase before and after the addition of 10% dilute HCl solution acidified to neutral no longer produce a white flocculent precipitation, As an aside, the precipitate was washed with 4% dilute HCl,Vacuum drying gave a pale yellow solid 11.5 g, 78.7% yield;
71.5%
Under nitrogen and -78 & lt; 0 & gt; C,5 (17.03 g, 66.53 mmol) was dissolved in exquisite anhydrous ether,Stirring, slow injection concentration of 2.50 molL-1ofn-BuLi (26.61 mL, 66.53 mmol),Low temperature reaction after half an hour,Dropping tributyl borate,Natural temperature to continue to respond for 1.5h.The reaction was quenched by the addition of dilute HCl, the aqueous phase was separated, the organic phase was extracted with dilute NaOH,The aqueous phase is acidified with dilute HCl solution until neutr is no longer white precipitate. The precipitate was washed with dilute HCl and dried in vacuo to give 10.10 g of a pale yellow solid, yield: 71.5%
68.5%
Under nitrogen protection, 2-methyl-3,5-dibromothiophene (5.1 g) was dissolved in 80.0 mL of anhydrous ether, kept at -78 C, and 10.0 mL of n-butyllithium was added at a rate of 1 drop / s. Add dropwise to the resulting mixed solution and hold at -78 C for 0.5 h.Maintain the reaction temperature, titrate the tributyl borate solution into the obtained mixed solution, and then stir the reaction at room temperature for 6 hours. After the reaction is complete, add 10.0 mL of 10% HCL solution to the system to complete the reaction; then extract the ether layer with 10% NaOH It was made basic, and the aqueous phase was collected and neutralized with 5% HCL solution to pH = 5, and filtered to obtain 2-methyl-3-bromo-5-boronic acid thiophene (3.0 g). Yield: 68.5%.
66.7%
And -78 deg.] C under nitrogen conditions,Compound 2 (20.6 g, 80.5 mmol)Was dissolved in anhydrous ether, stirred, slowly injected with n-BuLi (84.5 mmol)After half an hour of low temperature reaction, tributyl borate was added,After heating for 1.5 h,Adding dilute HCl to stop the reaction,The aqueous phase was separated and the organic phase was extracted with dilute NaOH, and the aqueous phase was acidified with dilute HCl solution until the neutral precipitate was no longer produced.The precipitate was washed with dilute HCl and dried in vacuo to give 11.8 g of compound 3 as a pale yellow solid in 66.7% yield;
66%
Liquid nitrogen cooling technology to control the temperature at 195K,3,5-Dibromo-2-methylthiophene (19.18 g, 75.00 mmol)Added to the refined ether, with rapid stirring,Slowly inject n-BuLi (32.26 mL, 84.00 mmol) at a concentration of 2.4 mol / L,After the reaction at low temperature for 0.5h, tributyl borate (20.2mL, 5.00mmol) was slowly added dropwise. After the reaction for 2h, 50.0mL of 4% hydrochloric acid solution was added to stop the reaction.The liquid was separated and the organic phase was extracted three more times with 4% sodium hydroxide solution.After combining all the aqueous phases, quickly add 10% hydrochloric acid solution acidification and vigorous stirring,Until the white flocculation precipitate completely precipitated, suction filtered,White flocculent precipitate was washed with 4% hydrochloric acid solution, completely dried at room temperature,11.23 g of pale yellow solid was obtained in a yield of 66%.
Synthesis of 3-bromo-2-methyl-5-thienylboronic acid 3 [0127] n-Butyl lithium in hexane is added to a stirred solution of compound 2 in dry ether (150 mL) at -78 C. under argon atmosphere. After stirring for 30 min, boric acid tri-butyl ester is quickly added to the reaction mixture. The mixture is extracted with 4% aq. NaOH (100 mL), the extract is collected and neutralized by with 10% HCl. The solid residue is washed, filtrated, and dried. Compound 3 is obtained as a yellowish solid. M.p. 229 C.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; sodium carbonate; In tetrahydrofuran; water; at 80℃;Schlenk technique; Inert atmosphere;
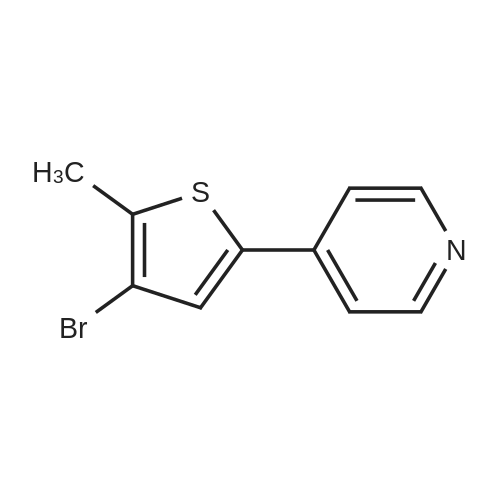
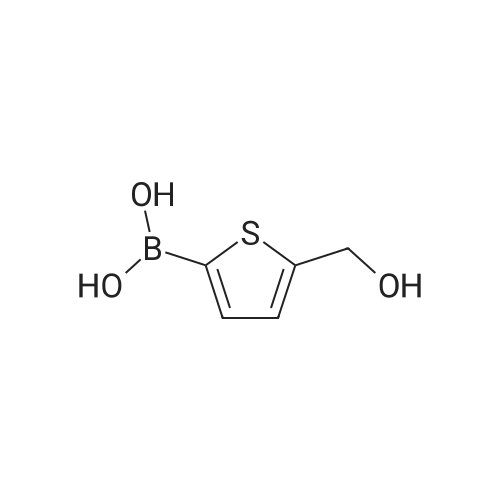
In a Schlenk flask under argon 4-bromo-5-methylthiophene-2-ylboronicacid (3.2 g, 14.14 mmol) was dissolved in THF (25 mL) and degassed.4-bromopyridine hydrochlorid (4.43 g, 22.68 mmol), Pd(dppf)Cl2(740 mg, 0.91 mmol) and aq Na2CO3 solution (20%, 25 mL) wereadded and the mixture was stirred under reflux overnight at 80 C.The reaction mixture was extracted three times with DCM, the combined organic phases werewashed with water and brine, dried over MgSO4, filtered and the solvents was removed underreduced pressure. Purification by flash column chromatography (pure cyclohexane + 1%NEt3) afforded a brownish solid with a yield of 51%.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; for 16h;
General procedure: Compound 7a was prepared by reacting 3-bromo-2-methyl-5-thienylboronic acid (3.0g, 11.3mmol) with 3-bromobenzonitrile (2.47g, 13.6mmol) in the presence of Pd(PPh3)4 (0.15g, 0.01mmol) and Na2CO3 (2.0 mol L-1, 50 mL) in tetrahydrofuran (THF) (80mL containing 10% water). After refluxing for 16h, the product was extracted with diethyl ether. The organic layer was dried over MgSO4, filtrated, and evaporated. The crude product was purified by column chromatography on SiO2 using petroleum ether as the eluent and 2.0g of 7a obtained as a yellowish solid in 53% yield. Mp 377-378K; 1H NMR (400MHz, CDCl3, ppm): delta 2.46 (s, 3H, -CH3), 7.18 (s, 1H, thiophene-H), 7.50 (t, 1H, benzene-H, J=8.0Hz), 7.57 (d, 1H, benzene-H, J=8.0Hz), 7.73 (d, 1H, benzene-H, J=8.0Hz), 7.79 (s, 1H, benzene-H).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; for 16h;Inert atmosphere; Reflux;
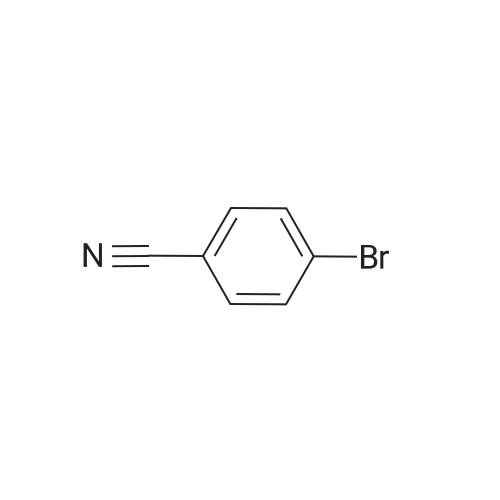
Under nitrogen protection,4-Bromo cyanobenzene (3.8 g, 13.6 mmol) andPd (PPh3) 4 (0.5 g)Was dissolved in 80 mL of THF,After stirring for 20 min, 3 (3.0 g, 13.6 mmol)And the concentration of 2.0 mol / L Na2CO3 solution 50 mL,Heating reflux for 16 h,Stop the reaction and cool to room temperature.The aqueous phase was extracted with ether and the combined organic phases were dried over anhydrous magnesium sulfate. Filtration, spin-off solvent, silica gel column chromatography (petroleum ether) separation,To give 2.9 g of a pale yellow solid, 89.8% yield;
83.50%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; for 15h;Reflux;
General procedure: Compound 3-bromo-5-(4-cyanophenyl)-2-methylthiophene was prepared by reacting 3-bromo-2-methyl-5-thienylboronic acid (2.21 g, 10.00 mmol) with 1-bromo-4-cyanobenzene (1.82 g, 10.00 mmol) in the presence of Pd(PPh3)4 (0.27 g,0.23 mmol) and Na2CO3 (6.36 g, 60.00 mmol) in tetrahydrofuran (THF) (80.0 mL containing 10% water). After refluxing for 15 h, the product was extracted with ether, dried over MgSO4, filtrated, and evaporated. The crude product was purified by column chromatographyon silica gel using hexane as the eluent to yield a white solid 2.32 g, in 83.50% yield. 1H NMR (400 MHz, CDCl3,TMS): d 2.44 (s, 3H, ACH3), 7. 22 (s, 1H, thiophene-H), 7.58 (d,J = 8.0 Hz, 2H, benzene-H), 7.65 (d, J = 8.0 Hz, 2H, benzene-H).
79%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 70℃; for 15h;
General procedure: This compound was synthesized by the same method as that reported in our previous works.[8], 8(a), 8(b), [9] and 9(a) Compound 7a was prepared by reacting compound 6 (4.0 g, 18.1 mmol) with 4-bromoanisole (3.4 g, 18.1 mmol) in the presence of Pd(PPh3)4 and Na2CO3 (6.40 g, 60 mmol) in tetrahydrofuran (80 mL containing 10% water) for 15 h at 70 C. The product 7a was purified by column chromatography on SiO2 using hexane as an eluent and 3.8 g obtained as a pale yellow solid in 78% yield.
79%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; for 16h;Reflux;
General procedure: Compound 9a was prepared byreacting 3-bromo-2-methyl-5-thienylboronic acid 8 (3.00 g,13.6 mmol) with 3-bromoanisole (2.79 g, 14.9 mmol) in the presence ofPd(PPh3)4 (0.15 g) and Na2CO3 (2.0 mol L-1,60.0 mL) in tetrahydrofuran (THF). After refluxing for 16 h, the product wasextracted with dichloromethane. The organic layer was dried over MgSO4,filtrated, and evaporated. The crude product was purified by columnchromatography on SiO2 using petroleum ether as the eluent and 2.73 g of 9a
3-bromo-2-methyl-5-(4-methylphenyl)thiophene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 70℃; for 15h;
General procedure: This compound was synthesized by the same method as that reported in our previous works.[8], 8(a), 8(b), [9] and 9(a) Compound 7a was prepared by reacting compound 6 (4.0 g, 18.1 mmol) with 4-bromoanisole (3.4 g, 18.1 mmol) in the presence of Pd(PPh3)4 and Na2CO3 (6.40 g, 60 mmol) in tetrahydrofuran (80 mL containing 10% water) for 15 h at 70 C. The product 7a was purified by column chromatography on SiO2 using hexane as an eluent and 3.8 g obtained as a pale yellow solid in 78% yield.
80%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 94.84℃; for 15h;
General procedure: 7a was prepared by reacting compound 6 (4.00g, 18.1mmol) with 4-bromoanisole (3.38g, 18.1mmol) in the presence of Pd(PPh3)4 and Na2CO3 (6.40g, 60mmol) in THF (80mL containing 10% water). After refluxing for 15h at 368K, the product was extracted with CH2Cl2. The organic layer was dried over MgSO4, filtrated, and evaporated. Column chromatography on SiO2 with petroleum ether as the eluent afforded 3.99g of 7a as a yellowish solid in 78% yield.
72.5%
Under nitrogen protection,willP-methylBromobenzene (3.06 g, 17.87 mmol) and Pd (PPh3) 4(0.5 g) was dissolved in 80 ml of THF,Stir for 20 minFollowed by the addition of 6 (3.92 g, 17.90 mmol)And the concentration of 2.0mol·L-1Na2CO3 solution 60ml,Heated to reflux for 16 h, stop the reaction, cool to room temperature.The aqueous phase was extracted with ether and the combined organic phases were dried over anhydrous magnesium sulfate. The filtrate was distilled off to remove the solvent and silica gel column chromatography (petroleum ether) to give a white solid (3.67 g, 12.96 mmol) in yield:72.5%
68.20%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; for 15h;Reflux;
General procedure: Compound 3-bromo-5-(4-cyanophenyl)-2-methylthiophenewas prepared by reacting 3-bromo-2-methyl-5-thienylboronicacid [53] (2.21 g, 10.00 mmol) with 1-bromo-4-cyanobenzene(1.82 g, 10.00 mmol) in the presence of Pd(PPh3)4 (0.27 g,0.23 mmol) and Na2CO3 (6.36 g, 60.00 mmol) in tetrahydrofuran(THF) (80.0 mL containing 10% water). After refluxing for 15 h,the product was extracted with ether, dried over MgSO4, filtrated,and evaporated. The crude product was purified by column chromatographyon silica gel using hexane as the eluent to yield awhite solid 2.32 g, in 83.50% yield.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping