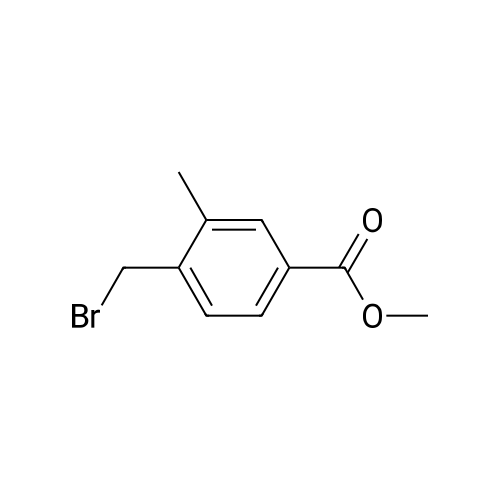
| 97% |
With dimethylsulfide borane complex; In tetrahydrofuran; at 0 - 20℃; |
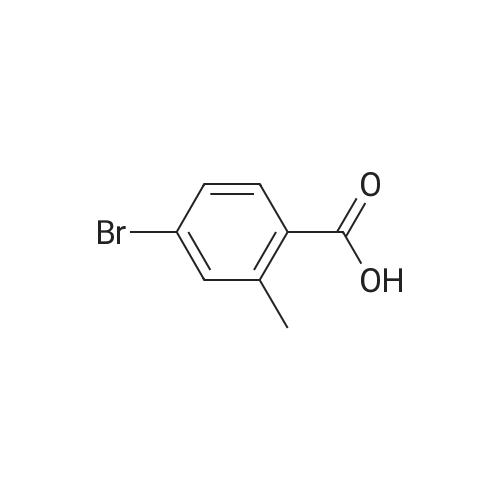
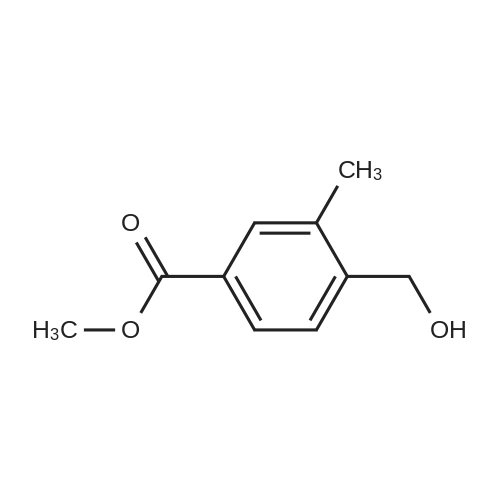
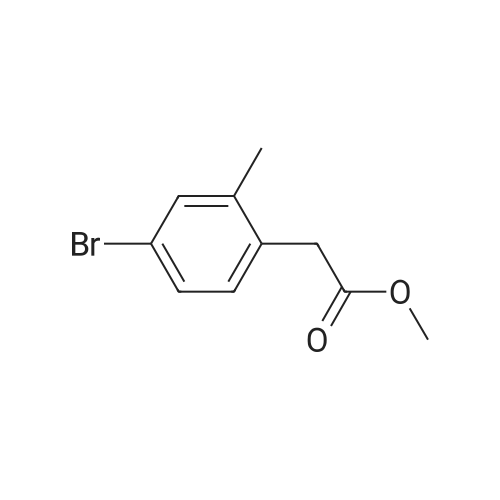
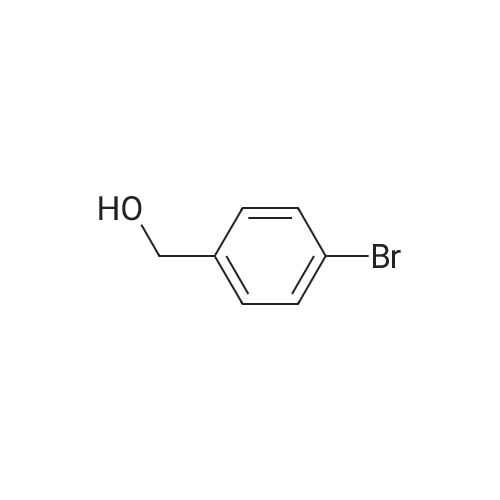
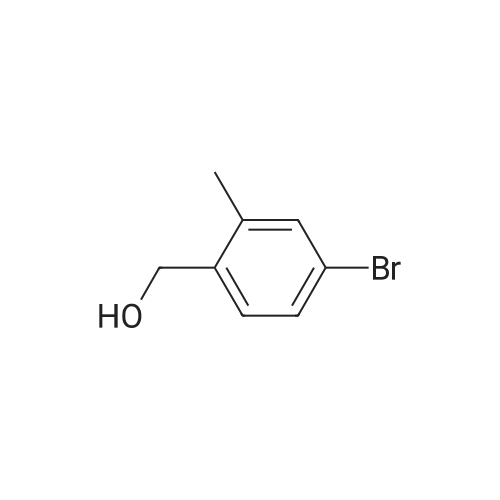
Scheme 1, step A: Borane-dimethyl sulfide complex(2M in THF; 1150 mE, 2.3 mol) is added over 1.5 hours tosolution of 4-bromo-2-methylbenzoic acid (250 g, 1.16 mol)in anhydrous tetrahydroffiran (1500 mE) at 0C. The reaction is allowed to warm slowly to ambient temperature and stirred overnight. The solution is cooled to -10 C. and water (500 mE) is added very slowly. Further water (5000 mE) is added and the mixture is extracted with ethyl acetate (2x5000 mE). The combined organic layers are washed with saturated aqueous NaC1 solution (5000 mE) and dried over Na2504. Filtration and concentration under reduced pressure provides the title compound (226 g, 97% yield). |
| 96% |
With dimethylsulfide borane complex; In tetrahydrofuran; at 0 - 20℃; for 5h;Inert atmosphere; |
BH3Me25 (2M in THF, 46.7mL, 93.40 mmol, 2.0 eq) was added to the solution of 4-bromo-2-methylbenzoic acid (10.0 g,46.70 mmol, 1.0 eq) in THF (150 mL) at 0 C under nitrogen atmosphere and the solution wasstirred at ambient temperature for 5 h. After complete consumption of starting material, the reaction mixture was diluted with EtOAc and washed with water followed by brine. The organic extract was then dried over anhydrous sodium sulfate, filtered, and solvent evaporated from the filtrated under reduced pressure to obtain a crude, which was purified by flash chromatography on silica gel, 230-400 mesh, using gradient of ethyl acetate in hexanes as eluent to obtain (4-bromo-2-methylphenyl)methanol as white solid (9.0 g, 96%). |
| 91.2% |
With borane-THF; In tetrahydrofuran; at 3 - 20℃; for 1.16667h; |
Borane-dimethyl sulfide complex (2M in THF; 116 mL, 0.232 mol) is added slowly to a solution of 4-bromo-2-methylbenzoic acid (24.3 g, 0.113 mol) in anhydrous tetrahydrofuran (THF, 146 mL) at 3 C. After stirring cold for 10 min the cooling bath is removed and the reaction is allowed to warm slowly to ambient temperature. After 1 hour, the solution is cooled to 5C, and water (100 mL) is added slowly. Ethyl acetate (100 mL) is added and the phases are separated. The organic layer is washed with saturated aqueous NaHC03 solution (200 mL) and dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by filtration through a short pad of silica eluting with 15% ethyl acetate/iso-hexane to give the title compound (20.7 g, 91.2% yield). MS (m/z): 183/185 (M+l-18). |
| 90% |
With borane-THF; In tetrahydrofuran; at 0 - 20℃; for 3h; |
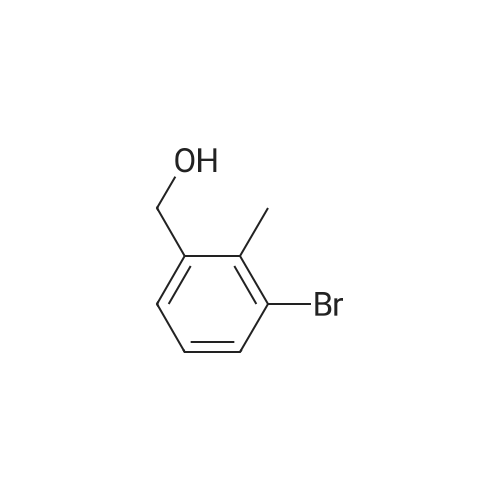
4-Bromo-2-methyl-benzoic acid (9.88 g, 45.9 mmol) was dissolved in THF (100 ml), and the solution was cooled to 0 C. To this solution, a solution of 1M BH3THF (91.89 ml, 91.89 mmol) was added at 0 C, and the solution was vigorously stirred for 3 hours at room temperature. The reaction mixture was diluted with cold water (20 mL), washed with a saturated solution of NaHCO3, then extracted with ether 3 times (300 mL). The combined organic ether was washed with brine (250 mL), dried over MgSO4, and concentrated in Vacua. The residue was purified via flash column chromatography (25% to 55% EtOAc in Hexane) to give white solid (8.24 g, 90% yield). 1H NMR (400 MHz, CDCI3) 5: 2.32 (2 H, s) 4.64 (1 H, d, J=5.81 Hz) 7.20 - 7.26 (2 H, m) 7.30 - 7.36 (1 H, m). |
| 81% |
|
Intermediate 12: (4-bromo-2-methylphe ol; To a solution of commercially available 4-bromo-2-methylbenzoic acid (250 g, 1.16 mol, in 4 batches, 62.5 g*4) in THF (300ml*4) was added a solution of borane-dimethyl sulfide (44.2g *4, 0.58 mol*4) in THF (75 ml*4) at 00C. After addition, the reaction mixture was stirred for 5 min, then the mixture was heated to 75C for 48hours. The reaction mixture was cooled to RT and then a saturated solution of K2CO3 was added to the reaction mixture. The separated organic layer was dried over sodium sulphate and concentrated in vacuo. The crude of 4 batches were purified together by flash chromatography (pentane/ AcOEt:5/1 ) to give the title compound (4- bromo-2-methylphenyl)methanol,. (190 g, 81% yield). 1H NMR: (d6-DMSO, 400MHZ) delta 7.30 (d,2H), 7.25 (d, 1 H), 5.10 (t, 1 H), 4.39 (d, 2H), 2.17 (s, 3H) . |
| 80% |
With lithium aluminium tetrahydride; In diethyl ether; for 4h; |
To a solution of 4-bromo-2-methylbenzoic acid (1.0 g, 4.65 mmol) in Et2O (15 ml.) was added lithium aluminiumhydride (370 mg, 10.23 mmol) and the mixture was stirred for 4 hours. The reaction mixture was diluted with water (100 ml.) and extracted with EtOAc (4x100 ml_). The combined organic fractions were dried (MgSO4) and concentrated in vacuo. The residue was purified by flash chromatography (silica, petrol ether/EtOAc) to give the title compound as a colorless gum (752 mg, 80%). 1H NMR (CDCI3, 400 MHz) delta 7.35-7.30 (2 H, m), 7.24 (1 H, m), 4.65 (2H, s), 2.32 (3H, s). |
| 75% |
With dimethylsulfide borane complex; In tetrahydrofuran;Inert atmosphere; |
Method 10F: using the appropriate benzylic acid.Acid ReductionTo a solution of the acid (1eq) in anhydrous tetrahydrofuran under argon atmosphere were added boron dimethylsulfite 2M solution in tetrahydrofuran drop by drop. The reaction mixture was stirred for 48 hours at room temperature. The tetrahydrofuran was evaporated and the residue was taken up in water and extracted with dichloromethane. The organic layer was washed with brine, dried over magnesium sulfate and evaporated under reduced pressure. The product was used without any further purification.10.9.1 (4-bromo-2-methylphenyl)methanol Prepared following the ester reduction method previously described (Method 10F) using 4-bromo-3-methylbenzoic acid. The product was obtained as a colorless oil. Yield: 75% Rf (petroleum ether/ethyl acetate 90/10): 0.75 NMR 1H (CDCl3): 1.95 (s, 1H); 2.33 (s, 3H); 4.63 (s, 2H); 7.22-7.36 (m, 3H). |
| 67% |
|
Step A: (4-Bromo-2-methyl-phenyl)-methanol4-Bromo-2-methylbenzoic acid (100 g, 460 mmol) was dissolved in THF (300 ml_), and cooled in an ice-bath to 0C. Borane (1 M in THF, 500 ml_, 1.1 eq.) was added dropwise over a period of 30 minutes, while keeping the temperature below 20C. After complete addition, the reaction mixture was stirred for 1 hour at room temperature, and was then carefully added to saturated aq. K2CO3 (250 mL). The obtained suspension was diluted with H20 (500 mL). The THF layer was separated, and concentrated under reduced pressure. The aqueous layer was extracted with EtOAc (3 x 300 mL). The residue from the concentrated THF layer was dissolved in the combined organic layer, which was washed with brine. The organic layer was dried (Na2S04), filtered, and concentrated in vacuo yielding the title compound (62 g, 308 mmol, 67%) with acceptable purity according to 1 H NMR as a yellow oil. 1 H NMR (CDCI3, 300 MHz) delta ppm 2.31 (s, 3H); 4.63 (s, 2H); 7.22 (d, 1 H); 7.33 (s, 2H) |
| 62% |
With lithium aluminium tetrahydride; In tetrahydrofuran; at 0℃; for 1h; |
step 1: To a solution of 4-bromo-2-methylbenzoic acid 1 (215.0 g, 1.0 mol) in THF (1 L) was added LiAlH4 (76.0 g, 1.0 mol) portions at 0 C. The reaction was stirred at this temperature for 1 h and then the reaction was quenched with 1 N HCl (2.0 L). The mixture was extracted with EtOAc (2×1000 mL) and the organic layers were washed with brine (2×1000 mL) and dried over anhydrous (Na2SO4), filtered and concentrated in vacuo. The residue was purified by SiO2 chromatography eluting with EtOAc/PE (1/5) to afford (4-bromo-2-methylphenyl)methanol as yellow solid (125 g, 62% yield). 1H NMR (400 MHz, CDCl3): delta 7.31 (m, 2H), 7.19 (d, J=8.8 Hz, 1H), 4.59 (s, 2H), 2.28 (s, 3H), 1.95 (s, 1H). |
|
With dimethylsulfide borane complex; In tetrahydrofuran; at 0 - 75℃; for 48h; |
To a solution of 4-bromo-2-methylbenzoic acid (10 g) in tetrahydrofuran (60 mL) was added borane-dimethylsulfide complex (7.07 g) under ice-cooling. The reaction mixture was stirred at room temperature for 5 minutes, and stirred at 75C for 2 days. The reaction mixture was cooled to room temperature. A saturated aqueous potassium carbonate solution was added to the reaction mixture, and the organic layer was separated. The organic layer was washed with water and brine, and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure to give the title compound (9.0 g).1H-NMR (CDCl3) delta ppm: 1.55-1.65 (1H, m), 2.36 (3H, s), 4.64 (2H, d, J=5.4Hz), 7.2-7.25 (1H, m), 7.3-7.35 (2H, m) |
|
|
EXAMPLE XII 4-Bromo-2-methylbenzyl alcohol STR22 The title compound is obtained from 10 g (46.5 mmol) of 4-bromo-2-methylbenzoic acid analogously to the instructions of Example XI. Yield: 10 g (crude, 107% of theory) Rf: 0.73 (petroleum ether/ethyl acetate=2:1) |
|
|
Step 1 (4-Bromo-2-methyl-phenyP)-methanol; A solution of BH3 (1 M in THF, 720 mL, 0.7194 mol) was slowly added to 4-bromo-2- methyl benzoic acid (51.57 g, 0.2398 mol) at O0C. The ice-bath was removed and the mixture was stirred overnight at room temperature. The reaction mixture was cooled to O0C and water was slowly added. The reaction mixture was then stirred at room temperature for 30 minutes. The resulting mixture was extracted with EtOAc, and the combined organic extracts were washed with NaHCO3 (saturated solution), water and brine; dried over MgSO4, filtered and evaporated under reduced pressure affording (4-bromo-2-methyl-phenyl)- methanol, which was used directly in step 2 without further purification. |
|
|
[0451] Experimental Details: 15ml of IM BH3/THF was added dropwise into a solution of 3g (13.95mmol) of 4- bromo-2-methyl-benzoic acid in 20ml of THF at 0C. The reaction solution was allowed to reach room temperature for lhour and quenched by the dropwise addition of 50ml of 50% aqueous THF. The mixture was treated with Na2CO3 and concentrated. The residue was extracted with Et2O. The organic layer was dried to give compound 2. |
|
|
4-bromo-2-methylbenzoic acid (5g, 22.8 mmol) is dissolved in dry THF(IOO ml) and cooled to O0C. A 1M solution of BH3THF in THF (34mL, 34 mmol) is slowly added. The resulting colorless suspension is then stirred overnight, allowing it to gradually reach room temperature. K2CO3 solid (1.1g) and H2O (100 ml) are added to the colorless solution and the mixture is stirred for an additional 30 minutes. THF is then evaporated and the residue extracted with EtOAc (30 ml). The organic phase is washed with 1 N HCI (3x50 ml), dried over Na2SO4 and evaporated. The residue gives the title compound which is used in the next step without further purification. |
|
|
III.1 ; (4-bromo-2-methylphenyl) acetonitrile; Step 1 : (4-bromo-2-methyrphenyl) methanol; Borane-dimethyl sulfide complexe (2.5 eq.) was added to a stirred solution of commercially available 4-bromo-2-methylbenzoic acid (1 eq.) in THF (0.3M). The mixture was reflux for 3 h. at without a condensor allowing the SMe2 to escape. The final reaction concentration was 0.5M. The mixture was cooled to O0C and quenched with a slow addition of HCl IN. The resulting mixture was extracted with Et2O. The organic extract was washed with water, brine, dried over MgSO4 filtered and concentrated to afford the desired material as a yellow solid. |
|
|
Reference Example 82 4-Bromo-2-methylbenzyl alcohol To a solution of 4-bromo-2-methylbenzoic acid (10 g) in tetrahydrofuran (60 mL) was added borane-dimethylsulfide complex (7.07 g) under ice-cooling. The reaction mixture was stirred at room temperature for 5 minutes, and stirred at 75C for 2 days. The reaction mixture was cooled to room temperature. A saturated aqueous potassium carbonate solution was added to the reaction mixture, and the organic layer was separated. The organic layer was washed with water and brine, and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure to give the title compound (9.0 g). 1H―NMR (CDCl3) delta ppm: 1.55-1.65 (1H, m), 2.36 (3H, s), 4.64 (2H, d, J=5.4Hz), 7.2-7.25 (1H, m), 7.3-7.35 (2H, m) |
|
|
Example 18 Experimental Details: 15 ml of 1M BH3/THF was added dropwise into a solution of 3 g (13.95 mmol) of 4-bromo-2-methyl-benzoic acid in 20 ml of THF at 0 C. The reaction solution was allowed to reach room temperature for 1 hour and quenched by the dropwise addition of 50 ml of 50% aqueous THF. The mixture was treated with Na2CO3 and concentrated. The residue was extracted with Et2O. The organic layer was dried to give compound 2. |
|
|
To a solution of 4-bromo-2-methylbenzoic acid (50.0 g, 233 mmol) in tetrahydrofuran (300 mL) at 0 C. was added BH3THF (1.0 M solution in THF, 465 mL, 465 mmol) dropwise. The reaction was then allowed to warm to room temperature and stir overnight. The reaction mixture was cooled to 0 degrees C. and quenched by dropwise addition of water. The bulk of the solvent was removed in vacuo and the residue was partitioned between ethyl acetate and water. The organics were washed with brine, dried over sodium sulfate, and concentrated in vacuo. Chromatography (20 to 35% ethyl acetate/hexane) provided the product, (4-bromo-2-methylphenyl)methanol, as a white solid. 1H NMR (400 MHz, CDCl3) delta 2.31 (s, 3H), 4.64 (s, 2H), 7.18-7.27 (m, 1H), 7.28-7.37 (m, 2H) |
|
With borane-THF; In tetrahydrofuran; at 20℃; for 4h;Inert atmosphere; |
The (4-bromo-2-methylphenyl)methanol used in the above process was prepared by dropwise addition of BH3.THF (27.76 mL, 27.76 mmoL) to a stirred solution of 4-bromo- 2-methylbenzoic acid (2.99 g, 13.88 mmol) in THF (20 mL) under N2 at 0 C. After complete addition of BH3.THF the reaction was stirred at RT, for 4 h and then cold water (10 mL) was added. The reaction was washed with satd. NaHC03 (60 mL) and the aqueous phase washed with EtOAc (3 x 100 mL). The combined organic layers were dried (MgSC>4) and the solvent was removed under reduced pressure to give the desired material (2.83 g). 1H NMR (d6-DMSO) delta ppm 7.51-7.32 (3H, m), 5.25 (1 H, t), 4.51 (2H,d), 2.31 (3H, s). |
| 32.9 g |
With borane-THF; In tetrahydrofuran; at 20℃; for 18h; |
Scheme 1, step A: Add borane-tetrahydrofuran complex (0.2 mol, 200 mL, 1.0 M solution) to a solution of 4-bromo-2-methylbenzoic acid (39 g, 0.18 mol) in tetrahydrofuran (200 mL). After 18 hours at room temperature, remove the solvent under the reduced pressure to give a solid. Purify by flash chromatography to yield the title compound as a white solid (32.9 g, 0.16 mol). 1H NMR (CDCl3): delta 1.55 (s, 1H), 2.28 (s, 3H), 4.61 (s, 2H), 7.18-7.29 (m, 3H). |
|
With potassium carbonate; In tetrahydrofuran; water; |
(1) To a stirred solution of 4-bromo-2-methylbenzoic acid (10.75 g) in tetrahydrofuran (50 ml) was added boranetetrahydrofuran complex (1 M in tetrahydrofuran, 150 ml) by syringe under nitrogen atmosphere at 5 C. and the mixture was heated under reflux for 18 hours. after cooling, water (50 ml) and potassium carbonate (20 g) were added to the solution at 5 C. The organic layer was separated and the aqueous layer was extracted with ethyl acetate. The combined extract was washed with water, dried over magnesium sulfate, and concentrated under reduced pressure to give 4-bromo-2-methylbenzyl alcohol (9.55 g). This compound was used to the next reaction without further purification. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping