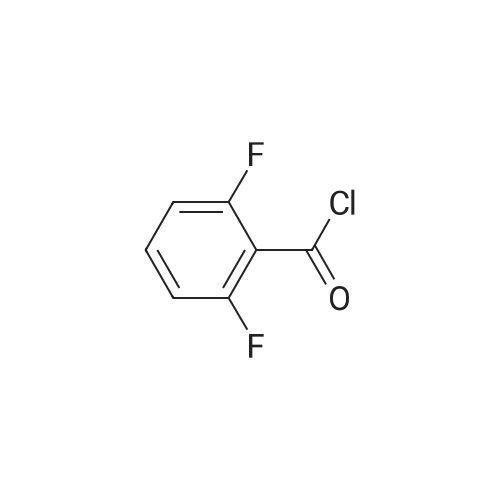
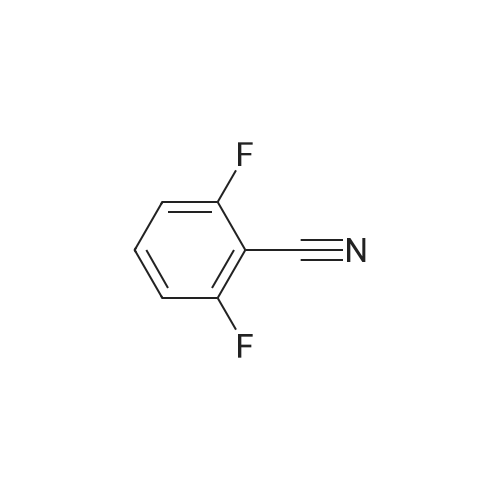
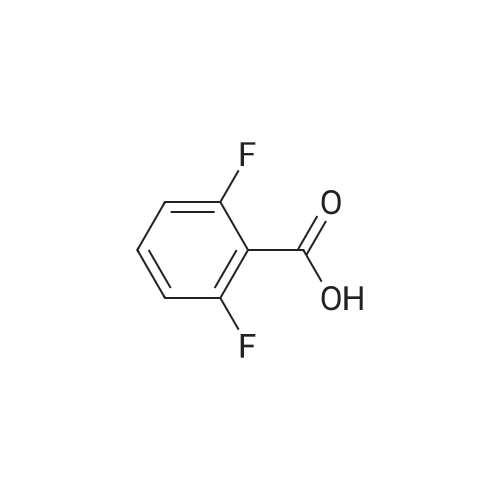
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of Medicinal Chemistry, 1968, vol. 11, # 4, p. 814 - 819
[2] Russian Journal of Organic Chemistry, 1998, vol. 34, # 2, p. 202 - 204
[3] Molecules, 2010, vol. 15, # 6, p. 4267 - 4282
[4] Journal of Agricultural and Food Chemistry, 2010, vol. 58, # 5, p. 3037 - 3042
[5] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 10, p. 2544 - 2546
[6] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 14, p. 3263 - 3270
[7] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 16, p. 4127 - 4132
2
[ 75-52-5 ]
[ 1897-52-5 ]
[ 18063-03-1 ]
Yield
Reaction Conditions
Operation in experiment
80%
With copper(l) iodide; caesium carbonate; 1,8-diazabicyclo[5.4.0]undec-7-ene In water at 20 - 100℃; for 1 h;
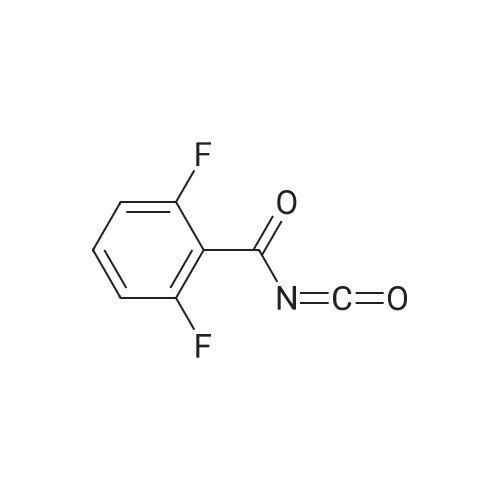
To a nitromethane (0.1 mL) solution of 2,6-difluorobenzonitrile (1j) (30 mg, 0.216 mmol) were addedH2O (1.0 mL), DBU (66 mg, 0.431 mmol), copper (I) iodide (8.2 mg, 0.0431 mmol), cesium (I)carbonate (35 mg, 0.108 mmol) at room temperature. The reaction mixture was heated at 100 °C for1 h and then poured into water (50 mL). The organic layer was separated and the aqueous layer wasextracted with AcOEt. The combined organic layer was dried over MgSO4. The solvent wasremoved under reduced pressure. The residue was purified by preparative TLC on silica gel elutingwith AcOEt-n-hexane (1:1) to give 2,6-difluorobenzamide (2j)S8 (27 mg, 80percent) as pale yellow powdersand (Z)-1-(2,6-difluorophenyl)-2-nitroethen-1-amine (3j)S2 (1 mg, 2percent) as pale yellow powders.mp 138-140 °C,
(1c) 2,6-Difluorobenzamide To 240 g of concentrated sulfuric acid and 24 ml of water are added dropwise, at room temperature, 100 g (0.72 mol) of 2,6-difluorobenzonitrile. The reaction mixture is stirred at 80°-85° C. for 12 hours, and then poured into 1.2 kg of ice-water; the mixture is stirred for 30 minutes and subsequently filtered. The crystals are washed neutral with water, and are afterwards dried at 85° C. in vacuo; yield: 91.0 g (80.6percent), melting point: 140°-141.5° C.
Reference:
[1] Tetrahedron, 1989, vol. 45, # 5, p. 1347 - 1354
[2] Polish Journal of Chemistry, 1991, vol. 65, # 5-6, p. 1049 - 1053
[3] Tetrahedron Letters, 1995, vol. 36, # 47, p. 8657 - 8660
[4] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2003, vol. 42, # 11, p. 2814 - 2819
[5] Patent: US4560770, 1985, A,
[6] Journal of Medicinal Chemistry, 2009, vol. 52, # 17, p. 5295 - 5298
[7] Tetrahedron Letters, 2010, vol. 51, # 13, p. 1639 - 1641
4
[ 385-00-2 ]
[ 18063-03-1 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2006, vol. 16, # 1, p. 162 - 166
[2] Russian Journal of Organic Chemistry, 1998, vol. 34, # 2, p. 202 - 204
[3] Journal of Medicinal Chemistry, 1968, vol. 11, # 4, p. 814 - 819
[4] Journal of Agricultural and Food Chemistry, 2010, vol. 58, # 5, p. 3037 - 3042
[5] Letters in Drug Design and Discovery, 2016, vol. 13, # 4, p. 329 - 334
[6] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 10, p. 2544 - 2546
[7] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 14, p. 3263 - 3270
[8] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 16, p. 4127 - 4132
[9] European Journal of Medicinal Chemistry, 2018, vol. 149, p. 170 - 181
5
[ 1897-52-5 ]
[ 18063-03-1 ]
[ 385-00-2 ]
Reference:
[1] Tetrahedron Letters, 1995, vol. 36, # 52, p. 9561 - 9564
[2] Journal of the Chemical Society - Perkin Transactions 1, 1997, # 8, p. 1099 - 1104
[3] Journal of the Chemical Society - Perkin Transactions 1, 1997, # 8, p. 1099 - 1104
6
[ 19064-16-5 ]
[ 18063-03-1 ]
Reference:
[1] Russian Journal of Organic Chemistry, 1998, vol. 34, # 2, p. 202 - 204
7
[ 104-12-1 ]
[ 79-34-5 ]
[ 18063-03-1 ]
[ 35367-38-5 ]
Reference:
[1] Patent: US4117009, 1978, A,
8
[ 372-18-9 ]
[ 18063-03-1 ]
Reference:
[1] Russian Journal of Organic Chemistry, 1998, vol. 34, # 2, p. 202 - 204
[2] Journal of Medicinal Chemistry, 1968, vol. 11, # 4, p. 814 - 819
9
[ 216064-18-5 ]
[ 18063-03-1 ]
Reference:
[1] Russian Journal of Organic Chemistry, 1998, vol. 34, # 2, p. 202 - 204
10
[ 135133-56-1 ]
[ 18063-03-1 ]
Reference:
[1] Russian Journal of Organic Chemistry, 1998, vol. 34, # 2, p. 202 - 204
With Triethoxysilane; [cis-Fe(H)(SPh)(PMe3)4]; In tetrahydrofuran; at 60℃; for 24h;Inert atmosphere;
General procedure: To a 25ml Schlenk tube containing a solution of 1 in 2ml of THF was added amide (1.0 mmol) and (EtO)3SiH (0.50 g, 3.0 mmol). The reaction mixture was stirred at 60 C until there was no amide left (monitored by TLC and GC-MS). The product was purified according to literature procedures by Beller [27].
81%
With Triethoxysilane; [(2,5-F2C6H2-CH=N-C10H6)Co(III)(H)(PMe3)2]; In tetrahydrofuran; at 60℃; for 24h;Schlenk technique;
General procedure: To a 25 mL Schlenk tube containing a solution of 2 in 2 mL of THF was added amide (1.0 mmol) and (EtO)3SiH (0.50 g, 3.0 mmol). The reaction mixture was stirred at 60 C until there was no amide left (monitored by TLC and GC-MS). The product was purified according to literature procedures by Beller
General procedure: A solution of thionyl chloride (25 mmol) was addeddropwisely to 1 (20 mmol) in toluene(15 mL). The mixture was heated to reflux for 3 h. Excess thionyl chloride wasremoved in vacuo. The remained mixture was added dropwise to ammonia waterbelow 10 C, then stirred at 10 C for 1 h. The intermediate 3 was obtained as white solid by filtration.
With ammonium hydroxide; In dichloromethane; at 0 - 20℃; for 3.5h;
General procedure: The reaction of substituted benzoic acid M4 (10 mmol) with oxalyl chloride (2.52 g, 20 mmol) gavesubstituted benzoyl chloride. A solution of the substituted benzoyl chloridewas added dropwise to the solution of ammonium hydroxide (5 mL) indichloromethane (10 mL) at 0 oC. Then the reaction mixture wasstirred for 3.5 h at room temperature. Dichloromethane (10 mL) was added to themixture and the mixture was washed with water, a solution of sodium hydroxide(1 M), a solution of diluted hydrochloric acid (1 M) and brine, dried oversodiumsulfate and ltered. The solvent was evaporated under reduced pressure toget crude products. The crude products were recrystallized with dichloromethaneto give the pure compounds M5, whichwere used directly for preparation of isocyanates M6. The key intermediates isocyanates M6wereprepared by the usual method. Substituted benzamides M5 (5mmol), and to this 10mmol of oxalyl chloride was addeddropwisefor 10 min at ice-bath. After addition, the resulting clearsolution was heatedat about 75 oC for 6-8 h, and then the excessive oxalyl chloride wasremoved under reduced pressure to givea clear solution of substituted benzoylisocyanate M6, which was usedfor thenext step reaction without further purication.
With ammonium hydroxide; In tetrahydrofuran; at 0 - 20℃;
(i) A solution of 2,6-difluorobenzoic acid (0.32 mol) in thionyl chloride (100 mL) was heated to reflux for 2 h. The resulting solution was concentrated and acid chloride intermediate was used in the next step without additional purification. To a solution of the acid chloride in anhydrous THF (100 mL) was added ammonium hydroxide (79 mL) at 0 C. After stirring at room temperature for 0.5 h, the reaction mixture was concentrated under reduced pressure. Then the reaction mixture was poured into cooled water (50 mL), extracted with ethyl acetate (100 mL x 3), and washed with brine (150 mL x 2). The organic layer was dried and concentrated to give the intermediate 2 in 90% yield which used in next step without purification. (ii) To a solution of 2,6-difluorobenzamide (0.28 mol) in concentrated sulfuric acid (90 mL) was added fuming nitric acid (12 mL) by dropwise under 0 C. The mixture was stirred for 2 h at room temperature. The pH was adjusted to 6 with 30% sodium hydroxide solution, then filtered and the filtrate was extracted with ethyl acetate (100 mL x 3),and washed with brine (150 mL x 2). The organic layer was dried and concentrated in vacuo to give the intermediate 3 as yellow solid in 91.40%yield. (iii) To a solution of 2,6-difluoro-3-nitrobenzamide (0.25 mol) in ethanol (300 mL) was added ammonium hydroxide (25 mL). The reaction mixture was stirred at room temperature overnight and the precipitate was collected by filtration, washed with isopropanol and dried in vacuum to give 4 31.5 g as yellow solid, yield 77.6%. (iv) A suspension of 2-amino-6-fluoro-3-nitrobenzamide (0.05 mol) in ethanol (100 mL) was reduced by hydrogen in the presence of palladium on carbon (10%, 1.00 g). After stirring at room temperature for 12 h, the reaction mixture was filtered. Solvent was removed under reduced pressure and the residue was subjected to silica gel column chromatography using dichloromethane/methanol (3:1) as eluent to give 55.00 g as light yellow solid, yield 58.9%. (v) To a solution of 3-pipecolinic acid (0.06 mol) and 2,3-diamino-6-fluorobenzamide (0.06 mol) in DMF (100 mL) was treated with PyBOP (0.06 mol) and N,N-diisopropylethylamine (0.18 mol).The reaction mixture was stirred at room temperature overnight. The solvent was removed using high vacuum. The residue was subjected to flash column chromatography using methylene chloride/methanol (30:1) to give the intermediate 6 as a white solid. The intermediate 6 was dissolved in glacial acetic acid (30 mL) and refluxed for 4 h until the reaction was complete (monitoring by TLC). The solvent was removed and the solid residue was purified by column chromatography using methylene chloride/methanol(80:1) as eluent to give pure 7a-7e in 50-72% yield. (vi) A solution of 7a-7e (25 mmol) in methanol (100 mL) was reduced with hydrogen in the presence of palladium on carbon (10%, 1.00 g). After stirring at room temperature for 12 h, the reaction mixture was filtered, and the filtrate was concentrated to give pure target compounds 8a-8e in 52-80% yield.
Intermediate 1: 2,6-difluorobenzoyl isocyanate STR3 A mixture of 0.52 g of <strong>[18063-03-1]2,6-difluorobenzamide</strong> and 0.33 ml of oxalyl chloride was stirred under reflux in 15 ml 1,2-dichloroethane overnight. Solvent was removed under vacuum and 10 ml 1,2-dichloroethane was added. Solvent was removed under vacuum to leave the title intermediate, which could be used directly or dissolved in 1,2-dichloroethane and stored for future use.
In 1,2-dichioroethane; 1,2-dichloro-ethane;
2,6-difluorobenzoyl isocyanate STR10 A mixture of 0.52 g of <strong>[18063-03-1]2,6-difluorobenzamide</strong> and 0.33 ml of oxalyl chloride was stirred under reflux in 15 ml, 1,2-dichloroethane overnight. Solvent was removed under vacuum and 10 mL 1,2-dichloroethane was added. Solvent was removed under vacuum to leave the title intermediate, which could be used directly or dissolved in 1,2-dichioroethane and stored for future use.
In 1,1-dichloroethane;
REFERENCE EXAMPLE 3 2,6-Difluorobenzoyl isocyanate In 150 ml of dichloroethane was suspended 13.5 g of <strong>[18063-03-1]2,6-difluorobenzamide</strong>, and 14.0 g of oxalyl chloride was little by little added to the suspension, followed by heating under reflux at about 110 C. for 15 hours. The reaction mixture was concentrated under reduced pressure, and the resultant oily substance was distilled under reduced pressure to give 12 g of the subject compound as an oily material of boiling point of 123 C./60 mmHg.
In toluene;
EXAMPLE 4 2,6-Difluorobenzoyl isocyanate Twenty-eight grams (0.22 mol) of oxalyl chloride were added dropwise to a solution of 31.4 gm (0.20 mol) of <strong>[18063-03-1]2,6-difluorobenzamide</strong> in 250 ml of toluene. The solution thus obtained was boiled under reflux for five hours, during which time hydrogen chloride and carbon monoxide were liberated. The solvent was then distilled off, in vacuo at the end. The product which remained was distilled in an oil pump vacuum. Yield: 28.5 gm (0.156 mol; 77% of theory), B.p.: 54-56 C./0.7 mbar.
In 1,1-dichloroethane;
EXAMPLE 2 Preparation of N-(2,6-difluorobenzoyl)-N'-[3-ethoxy-carbonyl-4-(3-chloro-5-trifluoromethyl-2-pyridyloxy)phenyl]urea. A solution of 1.7 g. of <strong>[18063-03-1]2,6-difluorobenzamide</strong> in 50 cc of dichloroethane was heated to 50 C. and 4.2 g. of oxalyl chloride was added dropwise with stirring and the reaction was continued for 1 hour with stirring to obtain 2,6-difluorobenzoyl isocyanate. Then, dichloroethane and excess of oxalyl chloride were distilled off under a reduced pressure.
In toluene;
Example 4 2,6-Difluorobenzoyl isocyanate Twenty-eight grams (0.22 mol) of oxalyl chloride were added dropwise to a solution of 31.4 gm (0.20 mol) of <strong>[18063-03-1]2,6-difluorobenzamide</strong> in 250 ml of toluene. The solution thus obtained was boiled under reflux for five hours, during which time hydrogen chloride and carbon monoxide were liberated. The solvent was then distilled off, in vacuo at the end. The product which remained was distilled in an oil pump vacuum. Yield: 28.5 gm (0.156 mol; 77% of theory), B.p.: 54-56 C./0.7 mbar.
In 1,2-dichloro-ethane;
EXAMPLE 1 Preparation of 2,6-Difluorobenzoylisocyanate A mixture containing 30.26 g (0.193 moles) of <strong>[18063-03-1]2,6-difluorobenzamide</strong> in 250 ml of 1,2-dicloroethane was placed under an atmosphere of nitrogen and cooled to 5 C. (internal temp.). To this was then added dropwise a solution of 24.0 ml (34.8 g, 0.27 moles) of oxalyl chloride in 50 ml of 1,2-dichloroethane. During this addition the internal temperature was maintained below 10 C. The resulting reaction mixture was then stirred for 1/2 hour in an ice bath. The ice bath was removed and the reaction mixture allowed to warm to room temperature. After stirring at room temperature to 11/2 hours the reaction mixture was slowly heated to reflux. After refluxing for 5 hours the reaction mixture was allowed to cool to room temperature. Removal of the solvent in vacuo afforded 31.77 g of an orange oil. NMR (CDCl3): delta6.6-7.7 (m); IR (CHCl3): 3370,2990,2220,1770,1747,1685,1605,1450 cm-1.
37.4 g (89%)
In dichloromethane;
(1d) 2,6-Difluorobenzoylisocyanate 36.1 g (0.23 mol) of <strong>[18063-03-1]2,6-difluorobenzamide</strong> are suspended in 360 ml of methylene chloride, and 53.7 g (0.42 mol) of oxalyl chloride are then added dropwise in the course of 20 minutes. After the release of hydrochloric acid has finished, the mixture is well stirred overnight under reflux. The solvent is subsequently distilled off, and the residue is distilled through a 10 cm Vigreux column at about 80 C./0.3 mm; yield 37.4 g (89%) of a slightly yellow liquid.
In 1,2-dichloro-ethane;
EXAMPLE 1 This Example illustrates the preparation of 2,6-difluorobenzoyl isocyanate. Oxalyl chloride (5.5 g) was added slowly to a suspension of <strong>[18063-03-1]2,6-difluorobenzamide</strong> (6.0 g) in 1,2-dichloroethane maintained at a temperature within the range -10 to -5 C. When the addition was complete the mixture was allowed to warm to the ambient temperature, and then gradually heated to the reflux temperature and maintained thereat for a period of 16 hours. The solvent was then removed by evaporation under reduced pressure and the residual oil purified by distillation to yield 2,6-difluorobenzoyl isocyanate, b.p. 60-61/0.7 mm.
In 1,2-dichloro-ethane; for 6h;Reflux; Inert atmosphere;
General procedure: Oxalylchloride (30 mmol) was added dropwise to a solution of 3 (10 mmol) in 1,2-dichloroethane(25 mL) and the mixture was heated to reflux for 6h under nitrogen atmosphere.Excess oxalylchloride and 1,2-dichloroethane was evaporated under reducedpressure to afford the crude intermediate4.
In 1,2-dichloro-ethane; at 75℃;Cooling with ice;
General procedure: The reaction of substituted benzoic acid M4 (10 mmol) with oxalyl chloride (2.52 g, 20 mmol) gavesubstituted benzoyl chloride. A solution of the substituted benzoyl chloridewas added dropwise to the solution of ammonium hydroxide (5 mL) indichloromethane (10 mL) at 0 oC. Then the reaction mixture wasstirred for 3.5 h at room temperature. Dichloromethane (10 mL) was added to themixture and the mixture was washed with water, a solution of sodium hydroxide(1 M), a solution of diluted hydrochloric acid (1 M) and brine, dried oversodiumsulfate and ltered. The solvent was evaporated under reduced pressure toget crude products. The crude products were recrystallized with dichloromethaneto give the pure compounds M5, whichwere used directly for preparation of isocyanates M6. The key intermediates isocyanates M6wereprepared by the usual method. Substituted benzamides M5 (5mmol), and to this 10mmol of oxalyl chloride was addeddropwisefor 10 min at ice-bath. After addition, the resulting clearsolution was heatedat about 75 oC for 6-8 h, and then the excessive oxalyl chloride wasremoved under reduced pressure to givea clear solution of substituted benzoylisocyanate M6, which was usedfor thenext step reaction without further purication.
In 1,2-dichloro-ethane; at 20℃; for 8.5h;Reflux;
3.2g <strong>[18063-03-1]2,6-difluorobenzamide</strong> was added to a 100mL flask. To this was added 50mL 1,2-dichloroethane and 4g oxalyl chloride. Then, install reflux condenser tube, dryers and exhaust gas absorption device. It was stirred at room temperature for 30min. The reaction was then heated at reflux for 8h until no hydrogen chloride gas evolution. The condensed reflux device into a distillation apparatus, The excess oxalyl chloride and the solvent was distilled off dichloroethane to give a yellow liquid. It was used directly in the next reaction without purification.
In dichloromethane;Inert atmosphere; Schlenk technique; Reflux;
General procedure: The appropriate amide 32a-p, benzhydrazide (40a), or p-toluenesulfonylhydrazide (43a) (1.0 mmol, 1.0 equiv) was placed in a SchlenkKjeldahl reaction flask and the flask was evacuated/argon re-filledthree times. Subsequently, anhyd CH2Cl2 (25 mL) was added and themixture was stirred at r.t. for 10 min before dropwise addition of oxalylchloride (3.0 mmol, 3.0 equiv) followed. The reaction mixturewas then stirred at reflux for 2.5-3.0 h before cooling to r.t. and thesolvent was evaporated in vacuo. Subsequently, the appropriate nucleophile32, 34, 36, 38, 40, 42, 43, or 45 (1.1-1.25 mmol, 1.1-1.25equiv) was rapidly added and the flask was evacuated/argon re-filledbefore anhyd toluene (12 mL) was added. The reaction mixture wasthen stirred at reflux for 2.5-3 h before cooling to r.t. and concentrationto about 1/3 of the initial volume on rotavapor. Hexane was addedto the residue and the obtained precipitate (often sonicated) wascollected by filtration under reduced pressure to yield the crudeproduct. When necessary, the isolated material was purified either bycrystallization from MeOH or flash chromatography on silica gel withhexane-EtOAc as the eluent.
In 1,2-dichloro-ethane; at 0 - 70℃; for 5h;
In a 250 ml four-necked flask,Add 18.3 g of <strong>[18063-03-1]2,6-difluorobenzamide</strong> as raw material,Soluble in 150ml dichloroethane solvent,A solution of 28.2 g of oxalyl chloride in 30 ml of dichloroethane was added dropwise with stirring at less than 0C.After adding oxalyl chloride,The reaction was performed at room temperature for 2 h, then slowly heated to 70 C for 3 h.The light brown liquid after vacuum desolvation,That is, the desired raw material is 2,6-difluorobenzoyl isocyanate.
In 1,2-dichloro-ethane; at 0℃; for 5h;Inert atmosphere; Reflux;
General procedure: Compounds 1-30 were synthesized according to [20,21]. Substituted benzamide (5.0 mmol) and1,2-dichloroethane (10 mL) were added to a 100 mL three-necked flask under N2. The reaction mixturewas cooled to 0 C and oxalyl chloride (1.27 g, 10.0 mmol) was added dropwise under stirring, thenthe mixture was stirred at room temperature for 1 h, left standing at 60 C for 3 h and refluxed for1 h. The solvent and excess oxalyl chloride were evaporated under reduced pressure to give a yellowtransparent liquid. Anhydrous dichloromethane (5 mL) was added to the residue, then a substituted2-aminopyrimidine (2.5 mmol) was added to the system and reacted at room temperature for 12 h.The solvent was removed under reduced pressure and the residue was purified by flash columnchromatography on silica gel to give compounds 1-30.
(1c) 2,6-Difluorobenzamide To 240 g of concentrated sulfuric acid and 24 ml of water are added dropwise, at room temperature, 100 g (0.72 mol) of 2,6-difluorobenzonitrile. The reaction mixture is stirred at 80-85 C. for 12 hours, and then poured into 1.2 kg of ice-water; the mixture is stirred for 30 minutes and subsequently filtered. The crystals are washed neutral with water, and are afterwards dried at 85 C. in vacuo; yield: 91.0 g (80.6%), melting point: 140-141.5 C.
General procedure: A solution of starting material 1 (20 mmol), thionyl chloride(SOCl2) (40 mmol) and dichloromethane (30 mL) was stirred underreflux for 3 h, then the reaction mixture was concentrated underreduced pressure, the residue was added to the aqueous ammonia(20 mL) at room temperature or below. Upon reaction completion(as monitored by TLC), the reaction was terminated with water,followed by extraction with ethyl acetate (3 10 mL), dried overanhydrous sodium sulphite and then filtered to obtain a crudeproduct and recrystallized to get compounds 2.
2,6-Difluoro-N-{3-[3-{2-[(3-fluorophenyl)amino]-4-pyrimidinyl}-6-(trifluoromethyl)pyrazolo[1,5-a]pyridin-2-yl]phenyl}benzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
30%
With caesium carbonate;tris-(dibenzylideneacetone)dipalladium(0); 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 170℃; for 0.25h;Microwave irradiation;
Step H: 2,6-Difluoro-N-{3-[3-{2-[(3-fluorophenyl)amino]-4-pyrimidinyl}-6-(trifluoromethyl)pyrazolo[1,5-a]pyridin-2-yl]phenyl}benzamide (title compound); To a solution of 4-[2-(3-bromophenyl)-6-(trifluoromethyl)pyrazolo[1,5-a]pyridin-3-yl]-N-(3-fluorophenyl)-2-pyrimidinamine (125 mg, 0.24 mmol) in dioxane (10 mL) was added Pd2 dba3 (7 mg, 0.007 mmol), Cs2CO3 (94 mg, 0.34 mmol), Xantphos (7 mg, 0.011 mmol), and <strong>[18063-03-1]2,6-difluorobenzamide</strong> (44 mg, 0.28 mmol). The reaction mixture was then subjected to microwave irradiation (Personal Chemistry Optimizer, 170 C., 15 min). The reaction was then cooled and the mixture partitioned in EtOAc (100 mL) and water (25 mL). The organic layer was separated, dried (over MgSO4) and concentrated. The crude material was purified by silica gel column chromatography (20-50% EA/Hex) to provide the title compound (43 mg, 30%) as an beige solid. 1H NMR (400 MHz, d6-DMSO): delta 10.95 (s, 1H), 9.88 (s, 1H), 9.54 (s, 1H), 8.54 (d, J=9.3 Hz, 1H), 8.38 (d, J=5.3 Hz, 1H), 8.07 (s, 1H), 7.80 (d, J=8.8 Hz, 1H), 7.71 (m, 2H), 7.59 (m, 1H), 7.49 (t, J=8.0 Hz, 1H), 7.43 (d, J=9.3 Hz, 1H), 7.35 (d, J=7.7 Hz, 1H), 7.25 (m, 3H), 6.71 (dt, J=2.3, 8.4 Hz, 1H), 6.65 (d, J=5.1 Hz, 1H) ppm. ESIMS (M+H)+=605.
2,6-difluoro-N-[3-(3-{5-fluoro-2-[(3-fluorophenyl)amino]-4-pyrimidinyl}pyrazolo[1,5-a]pyridin-2-yl)phenyl]benzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
61%
With caesium carbonate;tris-(dibenzylideneacetone)dipalladium(0); 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 160℃; for 0.25h;Microwave irradiation;
Step E: 2,6-difluoro-N-[3-(3-{5-fluoro-2-[(3-fluorophenyl)amino]-4-pyrimidinyl}pyrazolo[1,5-a]pyridin-2-yl)phenyl]benzamide (title compound); A solution 4-[2-(3-bromophenyl)pyrazolo[1,5-a]pyridin-3-yl]-5-fluoro-N-(3-fluorophenyl)-2-pyrimidinamine (40 mg, 84 mmol), Pd2(dba)3 (15.3 mg, 0.016 mmol), xantphos (14.5 mg, 0.025 mmol), Cs2CO3 (95 mg, 0.29 mmol), and <strong>[18063-03-1]2,6-difluorobenzamide</strong> (39 mg, 0.25 mmol) in anhydrous 1,4-dioxane, that had been previously degassed with nitrogen, was heated at 160 C. in the microwave for 15 min. At this time, the crude reaction was adsorbed to silica gel and purified by LC (DCM to 5% MeOH/DCM) to afford the product (28.3 mg, 61%) as a white solid. ESIMS (M+H)+=555. 1H NMR (400 MHz, DMSO-d6) delta 6.63-6.67 (m, 1H), 7.09-7.24 (m, 4H), 7.29-7.49 (m, 3H), 7.54-7.61 (m, 1H), 7.68-7.72 (m, 1H), 7.81 (s, 1H), 7.90-7.94 (m, 1H), 8.04 (m, 1H), 8.09 (m, 1H), 8.53-8.54 (m, 1H), 8.87-8.89 (m, 1H), 9.91 (s, 1H), 10.84 (s, 1H); ESIMS (M+H)+=555.
With sodium hydride; In N,N-dimethyl-formamide; at 20℃;
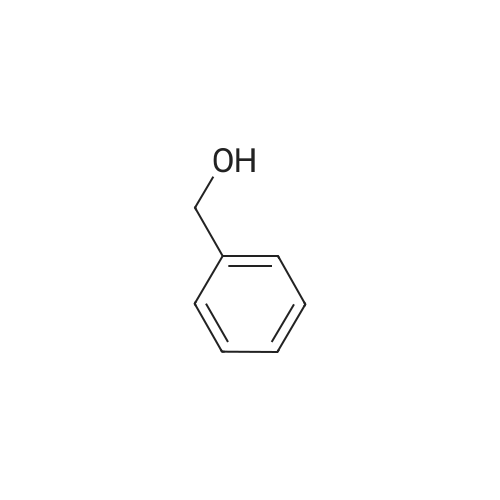
Benzyl alcohol (24 ml, 0.229 mol) is added portionwise to a suspension of sodium hydride (9.16 g, 0.229 mol) in DMF (300 ml) and stirred at room temperature for 1 h. 2, 6-Difluorobenzamide (30 g, 0.190 mol) is added in one portion and stirring is continued overnight at room temperature. The reaction mixture is poured into 1N HCl (1. 5 1) and extracted with dichloromethane. The extract is washed with water and saline, dried (MgS04), and evaporated to give product as a white solid suitable for use in the next step (39.6 g, 85%); MS for C14Hl2FN02 ilzlz 246 (M+H) +.
85%
Step 1: Preparation of 2-(benzyloxy)-6-fluorobenzamide benzyl alcohol (24 ml, 0.229 mol) is added portionwise to a suspension of sodium hydride (9.16 g, 0.229 mol) in DMF (300 ml) and stirred at room temperature for 1 h. <strong>[18063-03-1]2,6-Difluorobenzamide</strong> (30 g, 0.190 mol) is added in one portion and stirring is continued overnight at room temperature.The reaction mixture is poured into 1N HCl (1.5 l) and extracted with dichloromethane.The extract is washed with water and saline, dried (MgSO4), and evaporated to give product as a white solid suitable for use in the next step (39.6 g, 85%); MS for C14H12FNO2 m/z 246 (M+H)+.
With potassium phosphate; copper(l) iodide; ethylenediamine; In 1,4-dioxane; at 100℃; for 24h;
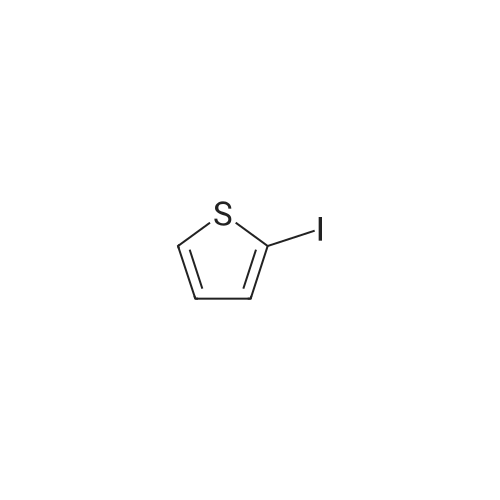
Compound 117: 2,6-Difluoro-N~[5-(3-trifluoromethy[-phenyi)-thiophen-2-yl]- benzamide; A stirred mixture of 2-iodothiophene (d, 12 mmol), <strong>[18063-03-1]2,6-difluorobenzamide</strong> (e, 10 mmol), copper(l) iodide (1.0 mmol), ethylenediamine (1.0 mmol), potassium phosphate (20 mmol) in dioxane (20 mL) was heated at 1000C for 24 h. The mixture was diluted with EtOAc (100 mL), filtered through celite, washed with water and purified by column chromatography to give f as white solid (1.0 g).A mixture of above amide (c, 2.1 mmol), iodine (1.0 mmol), UHP (1.0 mmol) in acetic anhydride (3 mL), acetic acid (1.5 mL) was stirred at room temperature for 16 h. The mixture was diluted with EtOAc, washed with water and Na2SO3 solution and dried. Removal of solvents and purification of the resulting oil gave intermediate g as grayish solid (0.42 g).A mixture of the above intermediate g (60 mg), 3-trifluoromethylbenzeneboronic acid (100 mg), Pd(PPh3J4 (0.05 mmol), K2CO3 (1 mmol) in dioxane (2 mL) was heated in EPO <DP n="98"/>a sealed tube at 120 0C for 30 min. The mixture was taken up with EtOAc (100 mL), washed with water (2x100 mL) and dried (Na2SO4). The oil obtained on concentration was purified by flash chromatography to give Compound 117 as a white solid (15 mg). 1H-NMR (CDCl3) delta 8.5 (br, 1 H), 7.7 (m, 2H), 7.5 (m, 3H), 7.2 (m, 1 H), 7.0 (t, 2H, J =8), 6.8 (m, 1 H) ppm; ESMS calcd for C18H10F5NOS: 383.0; found: 384.0 (M + H+).
EXAMPLE 1 Preparation of N-2,6-difluorobenzoyl-O-methylcarbamate <strong>[18063-03-1]2,6-Difluorobenzamide</strong> (79.5 g, 0.5 mol), dimethylcarbonate (67 g, 0.76 mol) and acetone (250 ml) were stirred together at 0 C. Sodium hydride (16.6 g, 0.69 mol) was added. Reaction was monitored by collection and measurement of hydrogen. After 2 hours, during which 13 liters of hydrogen were collected, the reaction mixture was allowed to warm to ambient temperature (20 C.) and when evolution of hydrogen ceased, formic acid (98%, 35 g) was added. Further hydrogen evolved, to an overall total of 15 liters. The resulting mixture was filtered, to remove precipitated sodium formate, and the resulting acetone solution was evaporated to yield N-2,6-difluorobenzoyl-O-methylcarbamate as a white solid (109 g, 97%,.97.2% pure by high performance liquid chromatography (HPLC), the remainder being difluorobenzamide). NMR, in CDCl3, delta (ppm): 3.75,s,3H; 6.95,m,2H; 7.40,m,1H; 8.7,s,1H.
85.5%
With formic acid; In ethyl acetate; isopropyl alcohol; toluene;
EXAMPLE 2 Preparation of N-2,6-difluorobenzoyl-O-methylcarbamate <strong>[18063-03-1]2,6-Difluorobenzamide</strong> (79.5 g, 0.5 mol), dimethylcarbonate (67 g, 0.76 mol) and ethyl acetate (250 ml) were stirred together at 0 C. Sodium hydride (16.6 g, 0.69 mol) was added, followed by isopropanol (30 g) to initiate evolution of hydrogen. Reaction was monitored by collection and measurement of hydrogen, as in Example 1. When evolution of hydrogen ceased at 0 C., the reaction mixture was allowed to warm to ambient temperature (20 C.) and formic acid (35 g) was added. After evolution of hydrogen ceased, and after filtration to remove precipitated sodium formate, solvent was evaporated off to yield crude product as a white solid (106 g). The crude product was added to 170 ml toluene and warmed until a clear solution was obtained. On cooling to ambient temperature, a white solid precipitated out and was filtered to give pure N-2,6-difluorobenzoyl-O-methylcarbamate (92 g, 85.5%), m.p. 123 to 123.5 C.
N-2,6-difluorobenzoyl-O-methylcarbamate, sodium salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
99%
With hydrogen; In 5,5-dimethyl-1,3-cyclohexadiene; isopropyl alcohol;
EXAMPLE 6 Preparation of N-2,6-difluorobenzoyl-O-methylcarbamate, sodium salt <strong>[18063-03-1]2,6-Difluorobenzamide</strong> (79 g, 0.5 mol), dimethylcarbonate (56 g, 0.6 mol) and xylene (400 ml) were stirred together at ambient temperature (20 C.), and sodium hydride (13 g, 0.54 mol) was added. Isopropanol (45 g) was added to initiate reaction and the reaction mixture was cooled to 0 C. and stirred at 0 C. for 20 hours, until hydrogen evolution ceased (12 liters of hydrogen were evolved). The reaction mixture was allowed to warm to ambient temperature (20 C.) and was filtered. The resulting white solid was washed with xylene (50 ml*3) and then with cyclohexane (50 ml*3) to give N-2,6-difluorobenzoyl-O-methylcarbamate, sodium salt (120 g, 99%). NMR, in D6 acetone, delta (ppm): 3.44,s,3H; 6.80,m,2H; 7.20,m,1H.
EXAMPLE 4 Preparation of N-2,6-difluorobenzoyl-O-methylcarbamate <strong>[18063-03-1]2,6-Difluorobenzamide</strong> (79.2 g, 0.5 mol), dimethyl carbonate (65 g, 0.72 mol) and 2-butanol (250 ml) were stirred together at 0 C., and sodium hydride (16.8 g, 0.7 mol) was added. After 12 liters of hydrogen had evolved, reaction ceased, the reaction mixture was allowed to warm to ambient temperature (20 C.) and formic acid (32.5 g) was added. The mixture, which was in the form of a thick slurry, was heated to 80 C. and filtered (at 80 C.) to remove precipitated sodium formate. Evaporation of solvent yielded crude product as a white solid (111 g, 91.7% pure by HPLC). Recrystalisation from toluene (170 ml) yielded pure N-2,6-difluorobenzoyl-O-methylcarbamate (87 g, 81%), m.p. 123 to 123.5 C.
With potassium hydroxide; acetic acid; In toluene;
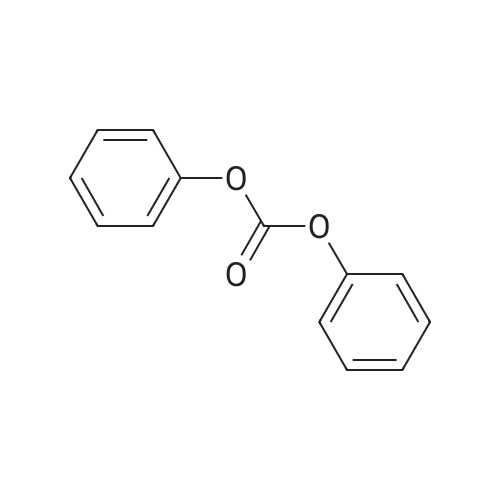
EXAMPLE 9 Preparation of N-2,6-difluorobenzoyl-O-phenylcarbamate A mixture of <strong>[18063-03-1]2,6-difluorobenzamide</strong> (48 g, 0.3 mol), potassium hydroxide pellets (19.68 g, 85.5%), oleic acid (1.38 g) and toluene was subjected to azeotropic distillation for 11/2 hours. Diphenylcarbonate (66 g, 0.31 mol) was added, and after a further 15 minutes at distillation temperature the mixture was allowed to cool to ambient temperature. The precipitated solid was filtered off from the reaction mixture and was treated with aqueous acetic acid, as in Example 7, to give N-2,6-difluorobenzoyl-O-phenylcarbamate (64 g, 77%) mp 149 C.
EXAMPLE 7 Preparation of N-2,6-difluorobenzoyl-O-phenylcarbamate <strong>[18063-03-1]2,6-Difluorobenzamide</strong> (48 g, 0.3 mol), diphenyl carbonate (96 g, 0.45 mol) and toluene (400 g) were stirred together at 10 C., and sodium hydride (8 g, 0.33 mol; as 16 g 50% w/w dispersion of sodium hydride in mineral oil) was added. The temperature of the reaction mixture rose spontaneously to 44 C. and the mixture grew full of solid matter. After the temperature of the reaction mixture began to fall, the precipitated solid was filtered off to give N-2,6-difluorobenzoyl-O-phenylcarbamate, sodium salt spectrum in mull (cm-1) 3450 broad small, 3400 broad small, 2740 sharp small, 2680 sharp small, 1690 broad large, 1630 sharp small, 1550 broad large, 1240 sharp small, 1170 broad large, 1115 broad medium, 1005 sharp small, 970 sharp small, 905 sharp medium, 810 sharp small, 765 shoulder, 725 sharp medium, 695 sharp medium). This salt was stirred in 10% w/w aqueous acetic acid (400 ml) at ambient temperature giving N-2,6-difluorobenzoyl-O-phenyl-carbamate (80 g, 95%) m.p. 149 C.
With potassium hydroxide; sodium hydroxide;palladium-carbon; In methanol; water; benzyl alcohol;
EXAMPLE 8 261.2 g (1.66 mol) of 2,6-difluorobenzamide are suspended in 232.3 g (2.15 mol) of benzyl alcohol, the suspension is heated to 55° C., and 233.3 g (2.08 mol) of 50percent potassium hydroxide solution are added dropwise within the space of 2 h. After this time, the temperature is increased to 60° C., and stirring is continued for a further 3 h. Subsequently, a further 600 g of water, 280 g (7 mol) of sodium hydroxide and 185 g (1.71 mol) of benzyl alcohol are added at 10° C., and chlorine is passed in (15-18 l/h) at 40° C. for 3 h. The phases are separated, 500 ml of methanol and 7 g of Pd/C (5percent Pd, 50percent moist) are added to the organic phase and hydrogenation is carried out as described in Example 1b. After working up in analogy with Example 1b, 123.5 g (0.97 mol, 59percent) of 2-amino-3-fluorophenol are obtained as pale-gray shiny flakes.
2-fluoro-4-(1,1,2,2-tetrafluoroethoxy)phenyl isocyanate[ No CAS ]
[ 18063-03-1 ]
N-2,6-difluorobenzoyl-N'-[2-fluoro-4-(1,1,2,2-tetrafluoroethoxy)phenyl]urea[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
60%
In 5,5-dimethyl-1,3-cyclohexadiene;
PRODUCTION EXAMPLE 2 0.16 Gram of <strong>[18063-03-1]2,6-difluorobenzamide</strong>, 0.25 g of 2-fluoro-4-(1,1,2,2-tetrafluoroethoxy)phenyl isocyanate and 20 ml of xylene were added to a reactor and stirred under reflux for 24 hours. The reaction solution was cooled and concentrated to obtain a crude product. This crude product was subjected to chromatography on silica gel to obtain 0.24 g of N-2,6-difluorobenzoyl-N'-[2-fluoro-4-(1,1,2,2-tetrafluoroethoxy)phenyl]urea as white crystals. Yield: 60%. m.p.: 172-173 C.
4-bromo-2-chloro-5-trifluoromethylphenyl isocyanate[ No CAS ]
[ 18063-03-1 ]
N-(4-bromo-2-chloro-5-trifluoromethylphenyl)-N'-(2,6-difluorobenzoyl)urea[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
In toluene;
SYNTHESIS EXAMPLE 3 Production of N-(4-bromo-2-chloro-5-trifluoromethylphenyl)-N'-(2,6-difluorobenzoyl)urea: 4.8 g (0.016 mol) of 4-bromo-2-chloro-5-trifluoromethylphenyl isocyanate and 50 ml of toluene were added to 2.36 g (0.015 mol) of <strong>[18063-03-1]2,6-difluorobenzamide</strong> and the mixture was heated under refluxing for 20 h. After cooling the precipitate was formed and filtered. After the recrystallization from ethyl acetate, 5.8 g (85% yield) of colorless, crystals were obtained. m.p.: 184 to 187 C.
With potassium carbonate; In water; at 20℃; for 13.5h;
Production Example 4 A mixture of 3.00 g of 2, 6-difluorobenzamide, 3.6 ml of a 36% aqueous formalin solution and 0.10 g of potassium carbonate was stirred at room temperature for 13 hours. The reaction mixture was added to 10 ml of water, the resulting mixture was stirred for 30 minutes, and filtered, and the resulting solid was washed with 40 ml of water.The resulting solid was dried over diphosphorus pentaoxide under reduced pressure to obtain 2.05 g of 2, 5-difluoro-N- hydroxymethylbenzamide .1H-NMR (DMSO-d6 )delta[ppm] : 4.63-4.66 (2H, m) , 5.87-5.91 (IH, <n="240"/>m) , 7.13-7.17 (2H, m) , 7.48-7.52 (IH, m) , 9.25 (IH, brm)
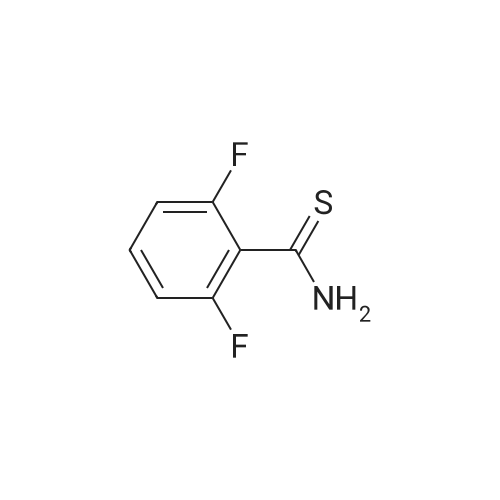
With Lawessons reagent; In toluene; at 90℃; for 14h;
A solution of <strong>[18063-03-1]2,6 difluorobenzamide</strong> (1 eq) and Lawesson's reagent (0.5 eq.) in toluene (0.2 M) was heated at 90 C. for 14 hours. Upon cooling the volatiles were removed in vacuo and purified by SiO2 chromatography (25% EtOAc/hexanes) yielding 2,6-difluorobenzothioamide as a light yellow solid (99%). LCMS (m/z): 174.1 (MH+); LC Rt=2.19 min.
99%
With Lawessons reagent; In toluene; at 90℃; for 14h;
Synthesis of 2 -difluorobenzothioamide[00138] A solution of 2, 6 difluorobenzamide (1 eq) and Lawesson's reagent (0.5 eq.) in toluene (0.2 M) was heated at 90C for 14 hours. Upon cooling the volatiles were removed in vacuo and purified by Si02 chromatography (25%EtOAc/hexanes) yielding 2,6-difluorobenzothioamide as a light yellow solid (99%>). LCMS (m/z): 174.1 (MH+); LC R, = 2.19 min.
99%
With Lawessons reagent; In toluene; at 90℃; for 14h;
A solution of 2, 6 difluorobenzamide (1 eq) and Lawesson's reagent (0.5 eq.) in toluene (0.2 M) was heated at 9O0C for 14 hours. Upon cooling the volatiles were removed in vacuo and purified by SiO2 chromatography (25% EtOAc/hexanes) yielding2,6-difluorobenzothioamide as a light yellow solid (99%). LCMS (m/z): 174.1 (MH+);LC Rt = 2.19 min.
Example 111N-[6-chloro-4-(1H-pyrrolo[2,3-b]pyridin-3-yl)pyridin-2-yl]-<strong>[18063-03-1]2,6-difluorobenzamide</strong>; A mixture of Example 7a (0.100 g, 0.247 mmol), <strong>[18063-03-1]2,6-difluorobenzamide</strong> (0.066 g, 0.421 mmol), cesium carbonate (0.105 g, 0.322 mmol), palladium (II) acetate (2.78 mg, 0.012 mmol), and Xantphos (10.73 mg, 0.019 mmol) in dioxane (3 mL) and DMF (0.1 mL) was heated at 150 C. for 30 minutes in a Biotage microwave reactor. The mixture was concentrated. The solid was suspended in dioxane (3 mL), treated with NaOH (20%, 0.10 mL), and heated at 50 C. for 3 hours. The reaction mixture was concentrated. The residue was stirred in water for 15 minutes, filtered, and purified by reverse-phase HPLC as described in Example 56 to give the title compound (8.5 mg) as trifluoroacetic acid salts. 1H NMR (500 MHz, DMSO-d6) ppm 7.24 (t, J=8.24 Hz, 2H), 7.30 (dd, J=8.09, 4.73 Hz, 1H), 7.52-7.65 (m, 2H), 7.70 (s, 1H), 8.34-8.41 (m, 3H), 8.65 (s, 1H), 11.66 (s, 1H), 12.41 (s, 1H). MS (ESI+) m/z 384.9 (M+H)+.
[bis(methyl)tris(trimethylphosphane)nickel(II)][ No CAS ]
[ 201230-82-2 ]
[ 18063-03-1 ]
[ 666701-53-7 ]
Ni(CO)2(PMe)3[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50.3%
Method (a): A solution of 1 (0.46 g, 2.96 mmol) in 30 mL of THF was combined with a solution of NiMe2(PMe3)3 (0.94 g, 2.96 mmol) in THF (20 mL) at -78 C. The reaction mixture was warmed to ambient temperature and stirred for 40 h. During this period the pale yellow mixture turned shiny red in color. Then the shiny red mixture was stirred under 1 bar of CO for 48 h and the solution slowly turned claret. The solvent was removed under reduced pressure, the orange residue was extracted with pentane (30 mL) and diethyl ether (40 mL), respectively. Compound 2 as white powder was obtained (washed with diethyl ether (2 x 15 mL)). Yield: 0.57 g (50.3%).
General procedure: 2-(trifluoromethyl)-benzamide 1a. To a nitrogene purged screw-cap vial containing 1a (250 mg, 1.3 mmol, 1.0 equiv) and 2.0 ml of anhydrous toluene were added PMHS (320 mul, 5.4 mmol, 4.2 equiv) and Ti(OiPr)4 (400 mul, 1.3 mmol, 1.0 equiv) at rt. The mixture was stirred and heated at 100 C until analysis by tlc showed complete consumption of the starting material (ca. overnight). The mixture was then diluted with Et2O (30.0 ml) and acidified using 2 M Et2O-HCl solution (2.0 ml, 3.0 equiv). The ammonium salt 4a was precipitated, filtered, and washed first with Et2O and then with pentane. The primary amine 4a was isolated as the hydrochloride salt in 89% yield.
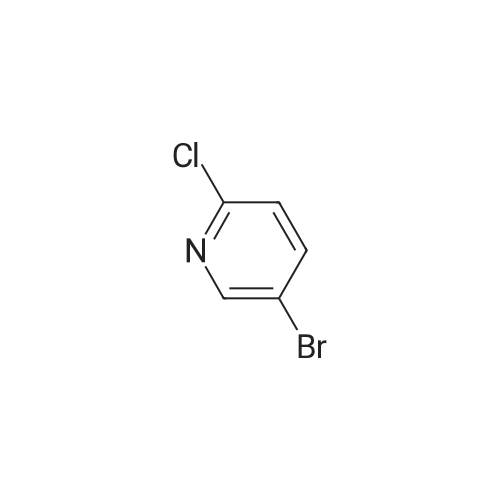
With potassium phosphate;copper(l) iodide; N,N-dimethylethylenediamine; In 1,4-dioxane; for 15h;Reflux;
Intermediate 51N-(5-Bromopyridin-2-yl)-<strong>[18063-03-1]2,6-difluorobenzamide</strong>To a mixture of 2-chloro-5-bromopyridine (370 mg, 1.9 mmol, 1.2 eq) and 2,6- difluorobenzamide (250 mg, 1.5 mmol, 1.0 eq) in dioxane (10 mL), copper iodide (151 mg, 0.75 mmol, 0.5 eq), potassium phosphate (670 mg, 3.15 mmol, 2.1 eq) and N,N- dimethylethylene diamine (0.1 mL, 1.05 mmol, 0.7 eq) were added sequentially. The resulting mixture was stirred at reflux for 15 h. The reaction was cooled to room temperature, filtered to remove the solid components and the filtrate was concentrated under vacuum. The residue was dissolved in ethyl acetate (50 mL), washed with water (20 mL), brine (20 mL), dried ( a2S04) and filtered. The filtrate was concentrated under vacuum and the crude product was purified by flash column chromatography (silica gel, ethyl acetate and hexane) to afford 300 mg of the solid product as a white solid. 1HNMR (400 MHz, DMSO-<¾ delta 1 1.22 (s, 1H, D20 exchangeable), 8.70 (d, J = 2.5 Hz, 1H), 8.18 (dd, J = 8.0, 2.5 Hz, 1H), 7.67-7.59 (m, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.29 (t, J= 8.0 Hz, 2H); ESI-MS (m/z) 313, 315 [(MH)+ Br79' 81].
With sulfuric acid; In methanol; at 20℃; for 3h;Cooling with ice;
Steps: in the 250 ml flask is sequentially added in four 14.3g (0.1mol) 2,6-difluoro-benzamide, chloroacetaldehyde compression b methanol 49.8g (0.4mol), violent stirring in the ice bath, the 1h adding concentrated sulfuric acid to dripping in 6 ml, stirring the mixture at room temperature for about 2h, TLC for tracking to 2,6-difluoro-benzamide the reaction is complete. In reaction solution is poured into the separatory funnel, adding water 150 ml, of methylene chloride 150 ml, extraction, extraction solution with 80 ml water washing secondary. The spin vaporization of the dichloromethane and excessive chloroacetaldehyde compression b methanol, hexane is added 80 ml, stirring cooling sleepovers. Separation by crystallization, filtration, drying to obtain 18.3g white compound V, yield of 73.5%.
In a single-necked flask, add <strong>[18063-03-1]2,6-difluorobenzamide</strong> and 2-chloroacetaldehyde dimethyl acetal, and stir under ice-water bath conditions for 5 minutes, and then slowly add concentrated sulfuric acid thereto. After the dropwise addition, react at room temperature overnight. After the reaction is completed, the reaction solution is poured into ice water, stirred thoroughly, and extracted with ethyl acetate 2-3 times. The organic phases are combined, and the organic phases are washed, concentrated, and purified by column to obtain intermediate 1. , 6-difluorobenzamide,The ratio of the amount of 2-chloroacetaldehyde dimethyl acetal and concentrated sulfuric acid is 1: 1.5: 15;
4-chloromethyl-2-(2,6-difluorophenyl)oxazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76%
for 4h;Reflux;
To 100 ml round bottomflask was added <strong>[18063-03-1]2,6-difluorobenzamide</strong> (10.00 g, 0.064 mol) and1,3-dichloroacetone 16.20 G, 0.128 mol), warmed to reflux and the reaction wasmaintained at reflux for 4 hours. After stopping the reaction, since Thencooled to room temperature, poured into 500 ml water and extracted with 3 × 100ml of ethyl acetate, the organic phase was 3 × 100 ml of saturated Afterwashing with aqueous sodium chloride solution, dried over anhydrous magnesiumsulfate, and concentrated by column chromatography (eluent: ethyl acetate:petroleum ether = 1: 3) to give 11.50 g of 4-chloro-2- (2,6-difluorophenyl)oxazole as a yellow solid, yield 76%.
at 130℃; for 1h;
General procedure: Benzamides 2 (6 mmol) was dissolved in dry THF (20 mL) andthereto was added P2S5 (6 mmol) and heated at 65 C for 3 h, afterwhich the reaction mixture was cooled, poured in NaHCO3 (10%,100 mL), stirred for half an hour, then the resulting solid wasfiltered, dried and recrystallized from ethyl acetate to get compounds3.
With a mechanical stirrer, thermometer, constant pressure dropping funnel,To the four-necked flask of the reflux condenser, 2.97 g of bis(trichloromethyl)carbonate and 20 ml of chlorobenzene were added and mixed at -5 to 5 C and stirred.9.43 g of <strong>[18063-03-1]2,6-difluorobenzamide</strong> was dissolved in a solution of 60 ml of chlorobenzene through a constant pressure dropping funnel, and slowly added dropwise to the above solution, and the dropping process was at a low temperature of -5 to 5 C. After the addition is completed, the mixture is stirred for 30 minutes, and slowly heated to a high temperature stage of 126 to 128 C, and a solution of 2.97 g of bis(trichloromethyl) carbonate dissolved in 20 ml of chlorobenzene is added dropwise at the reaction temperature for 2 to 3 hours. After the addition, the reaction is continued at this temperature for 2 hours, and the reaction is completed.After recovering chlorobenzene, it was distilled under reduced pressure to obtain 9.06 g of 2,6-difluorobenzoyl isocyanate in a yield of 82.45%. The purity of the product was determined by GC to be 98.21%.
<i>N</i>-cyclohexyl-2,6-difluoro-benzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
79%
With copper(I) thiophene-2-carboxylate; (4,4'-di-tert-butyl-2,2'-dipyridyl)-bis-(5-methyl-2-(4-fluorophenyl)pyridine(-1H))-iridium(III) hexafluorophosphate; N,N,N?,N?-tetramethyl-N?-tert-butylguanidine; bathophenanthroline; iodomesitylene diacetate; In 1,4-dioxane; at 20℃; for 1h;Inert atmosphere; Irradiation;
General procedure: To a 20 ml or 40 ml viale quipped with a stir bar was added photocatalyst, nitrogen nucleophile, iodomesitylene dicarboxylate, copper salt, and ligand. Dioxane was added followed by addition of the base. The solution was sonicated for 1-3 min until it became homogeneous. Next, the solution was degassed by sparging with nitrogen for 5-10 min before sealing with Parafilm. The reaction was stirred and irradiated using two 34-W blue LED lamps (3 cm away, with cooling fan to keep the reaction at room temperature) for 1 h. The reaction mixture was removed from the light, cooled to ambient temperature, diluted with water (15 ml) and ethyl acetate (25 ml), and the aqueous layer was extracted with ethyl acetate (3 × 25 ml). The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by flash chromatography on silica gel to afford the desired decarboxylative C-N coupling product. For aniline substrates, a solution of these nitrogen nucleophiles in dioxane was used; additionally, if the iodomesitylene dicarboxylate is a liquid, its solution in dioxane was used.
(Z)-1-(2,6-difluorophenyl)-2-nitroethen-1-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%; 2%
With copper(l) iodide; caesium carbonate; 1,8-diazabicyclo[5.4.0]undec-7-ene; In water; at 20 - 100℃; for 1h;
To a nitromethane (0.1 mL) solution of 2,6-difluorobenzonitrile (1j) (30 mg, 0.216 mmol) were addedH2O (1.0 mL), DBU (66 mg, 0.431 mmol), copper (I) iodide (8.2 mg, 0.0431 mmol), cesium (I)carbonate (35 mg, 0.108 mmol) at room temperature. The reaction mixture was heated at 100 C for1 h and then poured into water (50 mL). The organic layer was separated and the aqueous layer wasextracted with AcOEt. The combined organic layer was dried over MgSO4. The solvent wasremoved under reduced pressure. The residue was purified by preparative TLC on silica gel elutingwith AcOEt-n-hexane (1:1) to give 2,6-difluorobenzamide (2j)S8 (27 mg, 80%) as pale yellow powdersand (Z)-1-(2,6-difluorophenyl)-2-nitroethen-1-amine (3j)S2 (1 mg, 2%) as pale yellow powders.mp 138-140 C,
N-((4,6-bis(2,2,2-trifluoroethoxy)pyrimidin-2-yl)carbamoyl)-2,6-difluorobenzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
<strong>[18063-03-1]2,6-difluorobenzamide</strong> (0.78 g, 5.0 mmol) and 10 mL of 1,2-dichloroethane were placed in a 100 mL three-necked flask under inert gas protection. The reaction system was cooled to 0 C, and oxalyl chloride (1.27 g, 10.0 mmol) was added dropwise with stirring, stirred at room temperature for 1 h, reacted at 60 C for 3 h, and refluxed for 1 h. The solvent and excess oxalyl chloride were evaporated under reduced pressure to give a yellow transparent liquid, and 5 mL of anhydrous dichloromethane was added, and then I-a (0.73 g, 2.5 mmol) was added to the system.The reaction was carried out for 12 h at room temperature, and was monitored by TLC until the reaction was completed.Dissolve the solvent under reduced pressure,The silica gel column gave a white solid with a yield of 83%.
With sodium hypochlorite; sodium hydroxide; In water; at -5℃; for 2h;Large scale; Green chemistry;
First, 14.4 L of the prepared 30% aqueous sodium hydroxide solution and 19.0 L of sodium hypochlorite aqueous solution were added to the reaction vessel, and the mixture was cooled to -5 C.Add <strong>[18063-03-1]2,6-difluorobenzamide</strong> to a total of 4.0 kg (25 mol) with stirring. After the addition,Stirring reaction was continued for 2 h. Then, add 2.1 kg of sodium bisulfate at room temperature.After stirring for 10 min, steam distillation was carried out, and the mixture was separated and distilled under reduced pressure.The light red product 2,6-difluoroaniline was obtained and weighed 3.0 kg (23 mol).The purity was 98.3% and the yield was 92.0%.
89.6%
With sodium hypochlorite; sodium hydroxide; at -5℃; for 1.5h;Large scale;
First, 28.8 L of prepared 30% sodium hydroxide solution and 38.0 L of sodium hypochlorite solution were added to the reaction kettle, it was cooled to -5 C, a total of 8.0 kg (50 mol) of <strong>[18063-03-1]2,6-difluorobenzamide</strong> was added in portions with stirring, it was ensured that the last addition of <strong>[18063-03-1]2,6-difluorobenzamide</strong> was completely dissolved before each addition, and after the addition, continued to stir for 1.5 h. 4.1 kg of sodium bisulfate was added at room temperature, stirred for 15 min, the solution was orange, and steam distillation was carried out. The obtained distillate was separated, and the lower organic layer was separated and distilled under reduced pressure to give an Tangerine product 2,6-difluoroaniline. The weighing was 5.9 kg (46 mol), the purity was 96.5%, and the yield was 89.6%.
2-fluoro-6-(3-methylindol-1-yl)benzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76%
With potassium hydroxide; In dimethyl sulfoxide; at 100℃; for 24h;Inert atmosphere; Sealed tube;
General procedure: To a 50 mL screw-capped thick-walled Pyrex tube equipped with a magnetic stirrer, 3-methylindole(1a, 131.0 mg, 1.0 mmol), 1,2-dichlorobenzene (2a, 365.0 mg, 2.5 mmol), KOH (168.2 mg, 3.0 mmol) and DMSO (5.0 mL) were added sequentially under a nitrogen atmosphere. The tube was then sealedand stirred at 100 C for 24 h. After removal of the solvent under reduced pressure, purification was performed by flash column chromatography on silica gel with petroleum ether/ethyl acetate (gradient mixture ratio from 100:0 to 90:10) as eluent to afford N-(2-chlorophenyl)-3-methylindole (3aa, 171.8 mg,0.71 mmol, 71% yield).
Stage #1: oxalyl dichloride; 2,6-difluorobenzamide In 1,2-dichloro-ethane at 18 - 25℃; Large scale;
Stage #2: 2,5-dichloro-4-(1,1,2,3,3,3-hexafluoropropyloxy)aniline at 10 - 45℃; Large scale;
9-11 Example 9
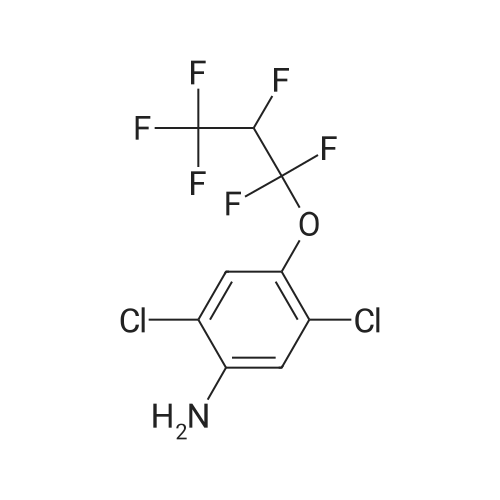
Synthesis of intermediate SMN-Z4 and synthesis of lufenuron250kg of dichloroethane is pumped into the 1000L reactor, 150kg of difluorobenzamide is added, the brine is cooled to 18-25, 167kg of oxalyl chloride is added dropwise, the dropping rate is controlled, the temperature is controlled at 18-25°C, and the dropping time is controlled at After 2-3 hours, the initial exotherm is obvious. It needs to be dripped slowly. After the dripping is completed, the cooling brine is pressed off. After stirring for half an hour, the temperature starts to rise slowly. Atmospheric distillation started. After the atmospheric distillation reached 120°C, the heating steam was turned off, the pressure was changed, and the vacuum was pulled for 1 hour and then the temperature was lowered. When the temperature is lowered to about 50°C in the reactor, 250kg of dichloroethane is added to the reactor to obtain an intermediate SMN-Z4 dichloroethane mixture.Transfer the dichloroethane mixture after the reaction of the intermediate SMN-Z4 in the previous step to a clean 1000L dry reactor, control the temperature at 38-45, add 265kg of the intermediate SMN-Z3 dropwise, stir and keep the reaction for 1-1.5h, and cool down Incubate and crystallize for 1 hour below 10°C, centrifuge, and wash with dichloroethane. Centrifuging and drying the material to obtain 385 kg of white solid, the yield is 93% (calculated based on the intermediate SMN-Z3), and the purity is 99%.
Stage #1: 2,5-dichloro-4-(1,1,2,3,3,3-hexafluoropropyloxy)aniline; oxalyl dichloride With potassium carbonate In dichloromethane at 20℃; for 3h; Cooling with ice;
Stage #2: 2,6-difluorobenzamide In dichloromethane at 50℃; for 0.5h; Inert atmosphere;
2.2 2) N-((2,5-Dichloro-4-(1,1,2,3,3,3-hexafluoropropoxy)phenyl)carbamoyl)-2,6-difluorobenzene Formamide (Formula I)
2,5-dichloro-4-(1,1,2,3,3,3-hexafluoropropoxy)aniline (14g, 42.7mmol) was dissolved in dichloromethane (150ml), add potassium carbonate (17.7g, 128.1mmol), stir, add dimethyl carbonate dropwise under ice bath (1.28g, 14.2mmol), reacted at room temperature for 3h. The resulting reaction solution was added dropwise to 2,6-difluorobenzene under argon protection In a solution of formamide (7.4 g, 46.9 mmol) in dichloromethane (100 ml), the temperature was raised to 50°C for 30 min. Concentrate the reaction solution The filtrate was concentrated and recrystallized to obtain 21.2 g of white solid, the yield was 97.2%, and the purity was 99.6%
Stage #1: 2,5-dichloro-4-(1,1,2,3,3,3-hexafluoropropyloxy)aniline; bis(trichloromethyl) carbonate With potassium carbonate In dichloromethane at 20℃; for 3h; Cooling with ice;
Stage #2: 2,6-difluorobenzamide In dichloromethane at 50℃; for 0.5h; Inert atmosphere;
4.2 2) N-((2,5-Dichloro-4-(1,1,2,3,3,3-hexafluoropropoxy)phenyl)carbamoyl)-2,6-difluorobenzene Formamide (Formula I)
2,5-dichloro-4-(1,1,2,3,3,3-hexafluoropropoxy)aniline (14g, 42.7mmol) was dissolved in dichloromethane (150ml), add potassium carbonate (17.7g, 128.1mmol), stir, add dimethyl carbonate dropwise under ice bath (1.28g, 14.2mmol), reacted at room temperature for 3h. The resulting reaction solution was added dropwise to 2,6-difluorobenzene under argon protection In a solution of formamide (7.4 g, 46.9 mmol) in dichloromethane (100 ml), the temperature was raised to 50°C for 30 min. Concentrate the reaction solution The filtrate was concentrated and recrystallized to obtain 21.2 g of white solid, the yield was 97.2%, and the purity was 99.6%
Stage #1: 2,5-dichloro-4-(1,1,2,3,3,3-hexafluoropropyloxy)aniline; carbonic acid dimethyl ester With potassium carbonate In dichloromethane at 20℃; for 3h; Cooling with ice;
Stage #2: 2,6-difluorobenzamide In dichloromethane at 50℃; for 0.5h; Inert atmosphere;
1.2-3.2 2) N-((2,5-Dichloro-4-(1,1,2,3,3,3-hexafluoropropoxy)phenyl)carbamoyl)-2,6-difluorobenzene Formamide (Formula I)
2,5-dichloro-4-(1,1,2,3,3,3-hexafluoropropoxy)aniline (14g, 42.7mmol) was dissolved in dichloromethane (150ml), add potassium carbonate (17.7g, 128.1mmol), stir, add dimethyl carbonate dropwise under ice bath (1.28g, 14.2mmol), reacted at room temperature for 3h. The resulting reaction solution was added dropwise to 2,6-difluorobenzene under argon protection In a solution of formamide (7.4 g, 46.9 mmol) in dichloromethane (100 ml), the temperature was raised to 50°C for 30 min. Concentrate the reaction solution The filtrate was concentrated and recrystallized to obtain 21.2 g of white solid, the yield was 97.2%, and the purity was 99.6%






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping