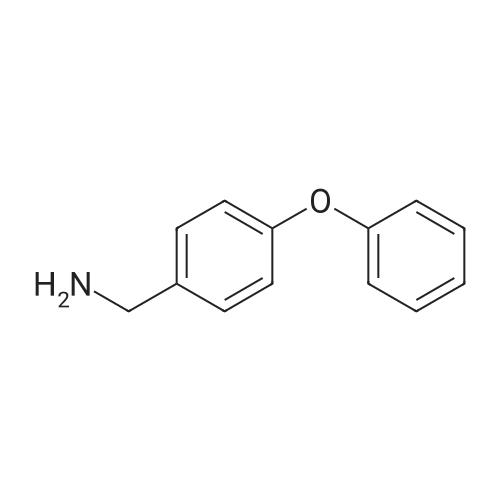
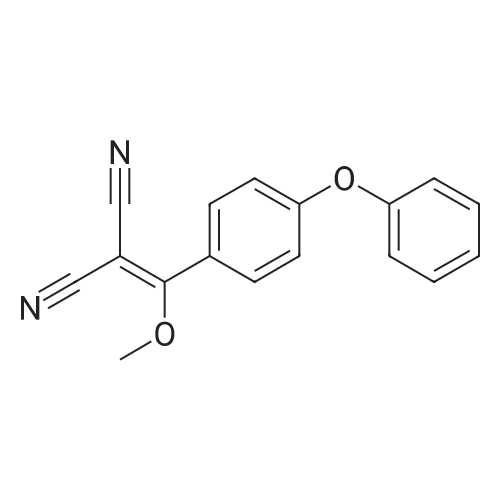
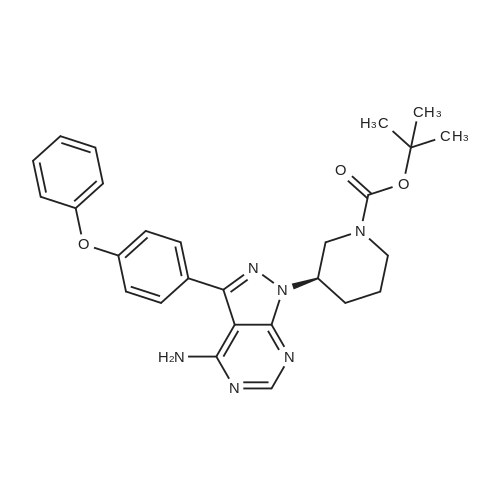
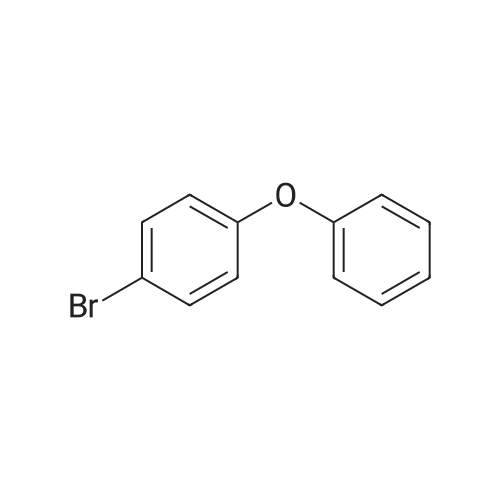
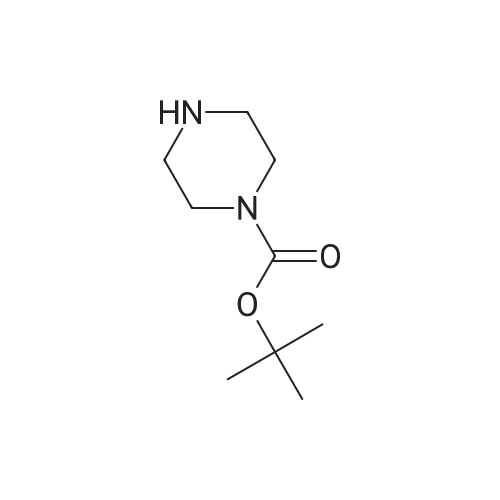
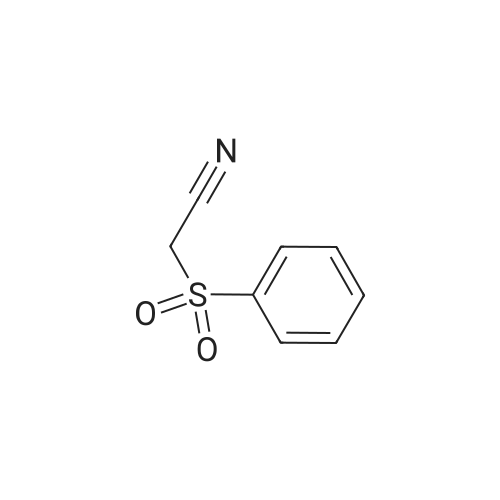
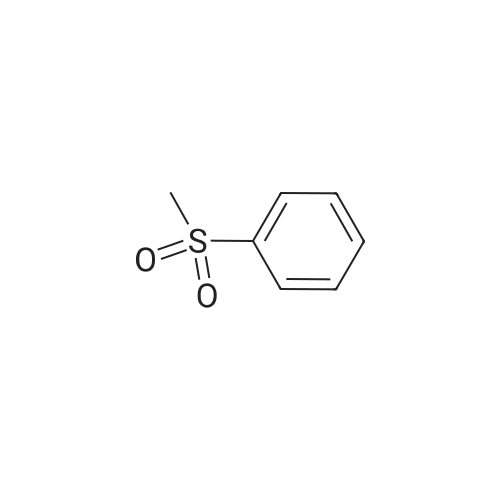
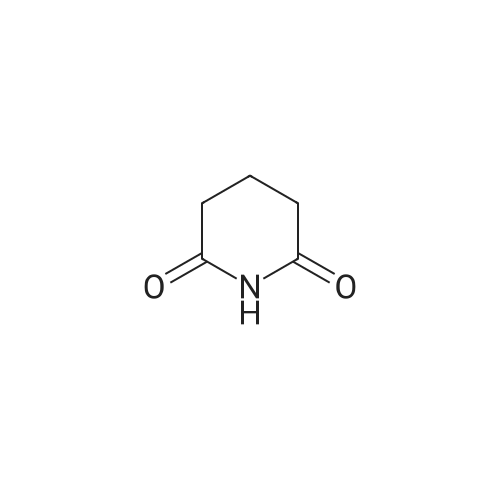
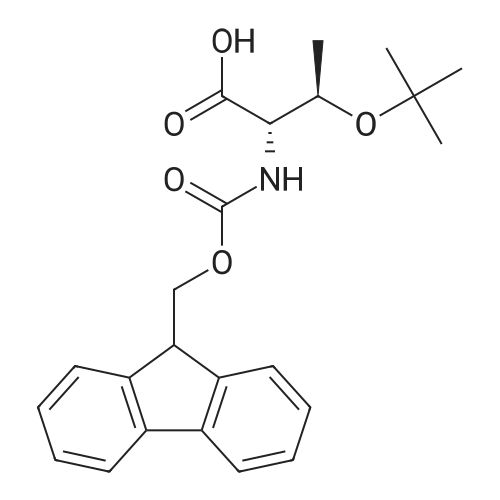
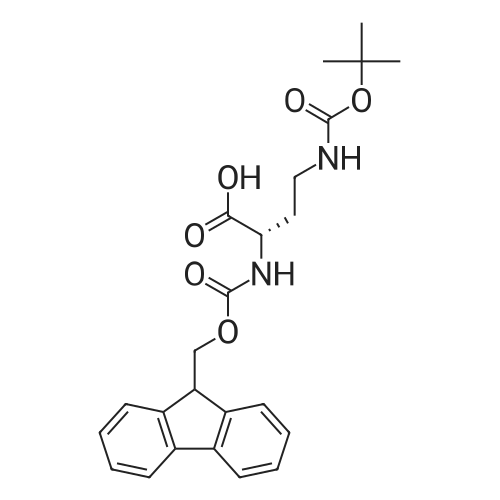
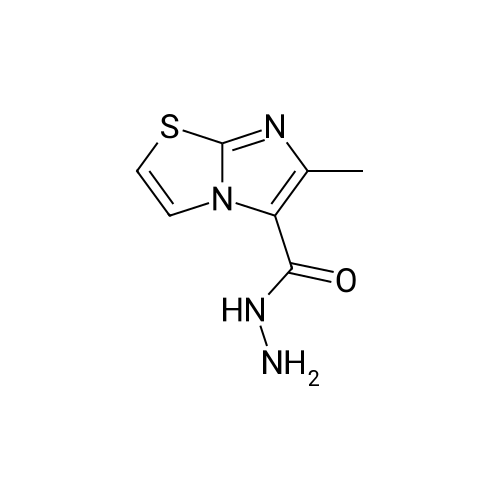
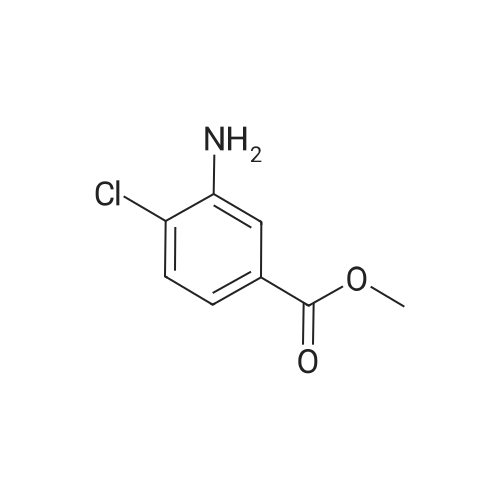
| 93% |
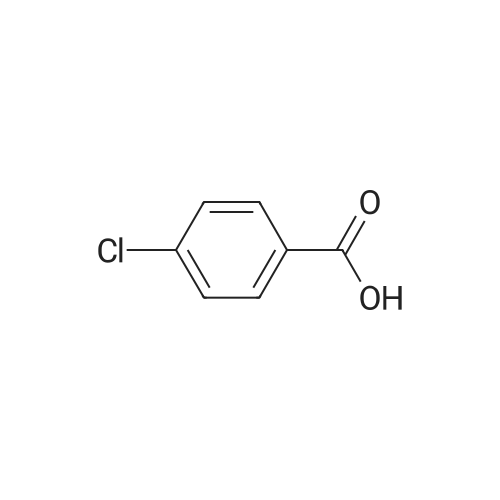
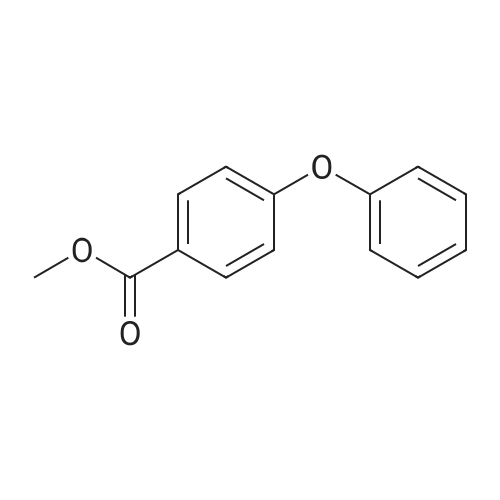
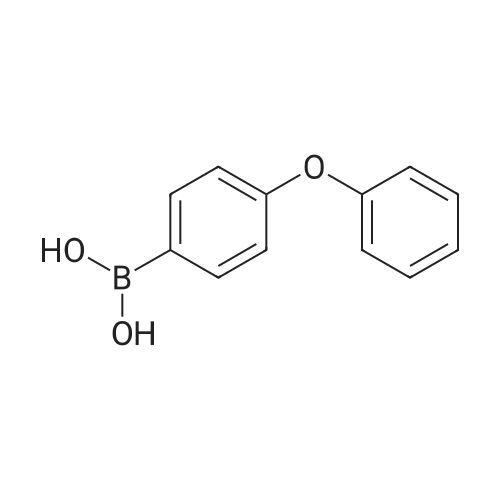
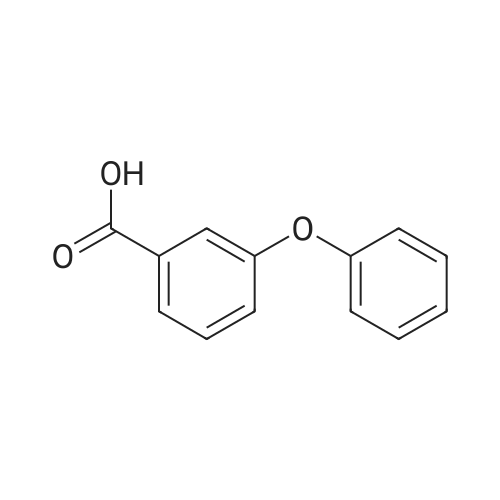
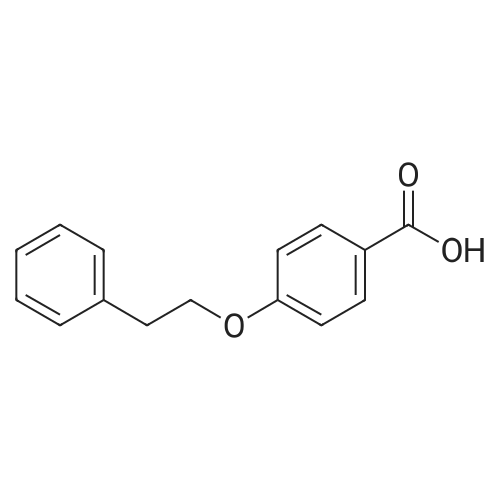
Stage #1: 4-Phenoxybenzoic acid With thionyl chloride at 80℃; for 3h;
Stage #2: malononitrile With N-ethyl-N,N-diisopropylamine In tetrahydrofuran; toluene at 0 - 5℃; for 2h; |
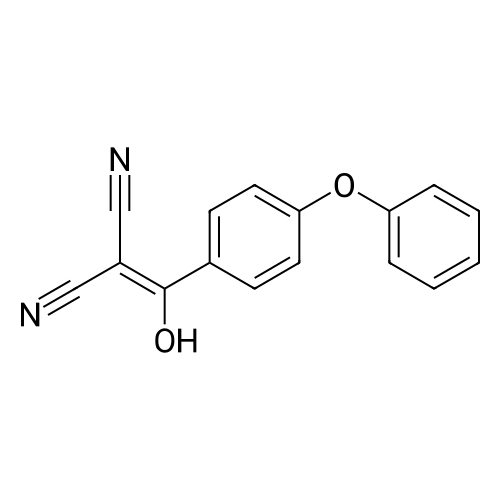
1.1 Step 1: 2-(Hydroxy(4-phenoxyphenyl)methylene)malononitrile
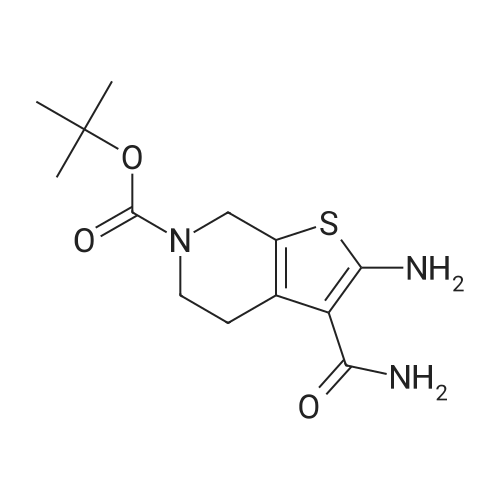
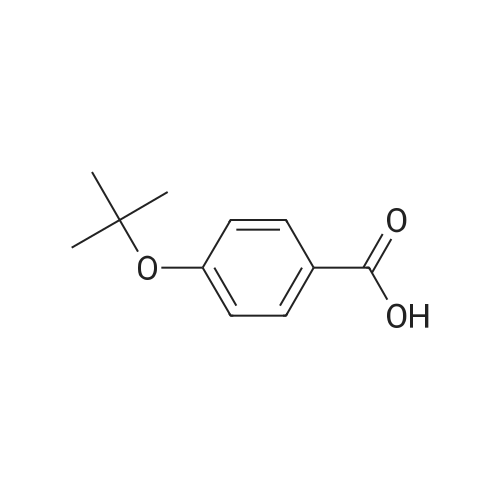
Add SOCl2 (1.2 L) to 4-phenoxybenzoic acid (300 g, 1.4 mol).Heat to 80°C and stir for 3 hours.The mixture was concentrated under reduced pressure to give the intermediate (315 g),It was used directly in the next reaction without further purification.Contains malononitrile (89.5 g, 1355 mmol) at 0-5°CTo a solution of DIEA (350 g, 2710 mmol) in THF (800 mL) was added dropwise a solution of the intermediate (315 g) in toluene (800 mL) for about 2 hours.The mixture was allowed to return to room temperature and stirred for 16 hours.The reaction was quenched with water (2.0 L) and extracted with EA (2.0 L x 3). The organic phases were combined, washed successively with dilute hydrochloric acid (3N, 1 L), saturated brine (2.0 L×3), dried over Na 2 SO 4 and concentrated. A yellow solid (330 g, 93%) was obtained. |
|
Stage #1: 4-Phenoxybenzoic acid With thionyl chloride for 1h; Reflux;
Stage #2: malononitrile With N-ethyl-N,N-diisopropylamine In tetrahydrofuran; toluene at -10 - 20℃; |
1
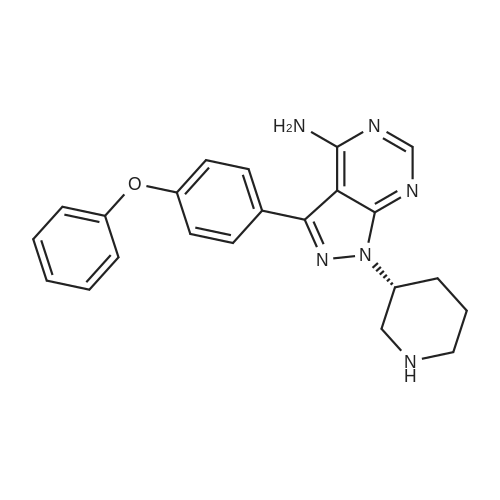
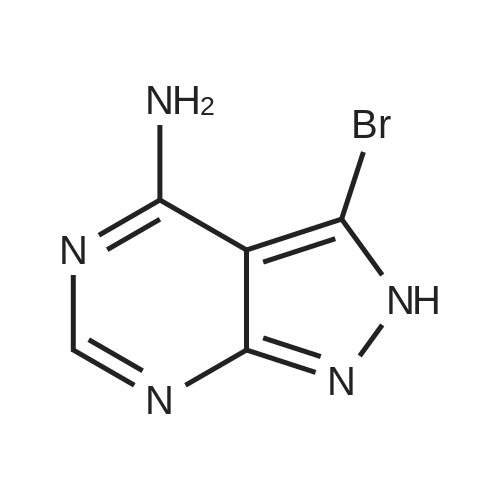
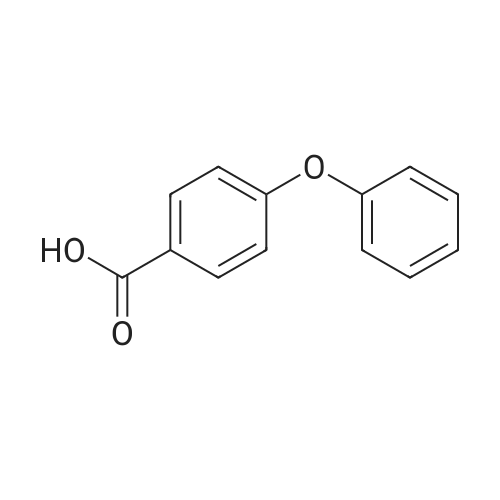
Example 1: Preparation of 4-Amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidine[00363] 4-Amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidine is prepared as disclosed in International Patent Publication No. WO 01/019829. Briefly, 4-phenoxybenzoic acid (48 g) is added to thionyl chloride (100 mL) and heated under gentle reflux for lh. Thionyl chloride is removed by distillation, the residual oil dissolved in toluene and volatile material removed at 80 °C/20 mbar. The resulting acid chloride is dissolved in toluene (200 mL) and tetrahydrofuran (35 mL). Malononitrile (14.8 g) is added and the solution and stirred at -10 °C while adding diisopropylethylethylamine (57.9 g) in toluene (150mL), while maintaining the temperature below 0 °C. After 1 h at 0 °C, the mixture is stirred at 20°C overnight. Amine hydrochloride is removed by filtration and the filtrate evaporated in vacuo. The residue is taken up in ethyl acetate (EA) and washed with 1.25 M sulphuric acid, then with brine and dried over sodium sulfate. Evaporation of the solvents gives a semisolid residue which is treated with a little EA to give 4.1 g of l,l-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a white solid (m.p. 160- 162°C). The filtrate on evaporation gives 56.58 (96%) of l,l-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a grey-brown solid, which is sufficiently pure for further use. |
|
Stage #1: 4-Phenoxybenzoic acid With thionyl chloride at 80℃; for 3h; Inert atmosphere;
Stage #2: malononitrile With N-ethyl-N,N-diisopropylamine In tetrahydrofuran; toluene at 0 - 20℃; for 18h; |
1 Preparation of 2-(hydroxy(4-phenoxyphenyl)methylene)malononitrile
A solution of 4-phenoxybenzoic acid (300 g, 1.4 mol) in SOCl2 (1.2 L) is stirred at 80° C under N2 for 3 hours. The mixture is concentrated in vacuum to give the intermediate (315 g) which is used for next step without further purification. [00455] To a solution of propanedinitrile (89.5 g, 1355 mmol) and DIEA (350 g, 2710 mmol) in THF (800 mL) is dropwise a solution of the intermediate (315 g) in toluene (800 mL) at 0-5° C. over 2 hours. The resultant mixture is allowed to warm to RT and stirred for 16 hours. The reaction is quenched with water (2.0 L) and extracted with of EA (2.0 L × 3). The combined organic layers are washed with 1000 mL of 3 N HCl aqueous solution, brine (2.0 L × 3), dried over Na2SO4 and concentrated to give the crude product (330 g, 93%). |
| 242 g |
With dmap; pivaloyl chloride; N-ethyl-N,N-diisopropylamine In tetrahydrofuran at 0 - 70℃; for 8h; Inert atmosphere; |
1 Example-i: Preparation of 2-(hydroxy(4-phenoxyphenyl)methylene)malononitrile(Formula-6)
Pivoloyl chloride (135 gms) was added to a mixture of 4-phenoxybenzoic acid compound of formula-S (200 gms), tetrahydrofuran (100 ml), malononitrile (74 gms),dimtehylaminopyridine (11.5 gms) and diisopropylethylamine (277.5 gms) at 0-5°C under nitrogen atmosphere. Heated the reaction mixture to 65-70°C and stirred for 8 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Ethyl acetate and water were added to the reaction mixture. Both the organic and aqueous layers were separated and the aqueouslayer was extracted with ethyl acetate. Combined the organic layers and then cooled to 10-15°C. The organic layer was washed with aqueous potassium carbonate solution and followed by dilute hydrochloric acid solution. Further, the organic layer was washed with aqueous sodium chloride solution. Distilled off the solvent from the organic layer completely under reduced pressure to get the title compound. Yield: 242 gms. |
|
Stage #1: 4-Phenoxybenzoic acid With thionyl chloride at 80℃; for 3h; Inert atmosphere;
Stage #2: malononitrile With N-ethyl-N,N-diisopropylamine In tetrahydrofuran at 0 - 20℃; for 18h; Inert atmosphere; |
Preparation of 2-(hydroxy(4-phenoxyphenyl)methylene)malononitrile
A solution of 4-phenoxybenzoic acid (300 g, 1.4 mol) in SOd2 (1.2 L) is stirred at 80°C under N2 for 3 hours. The mixture is concentrated in vacuum to give the intermediate (315 g)which is used for next step without further purification. To a solution of propanedinitrile (89.5 g, 1355 mmol) and N,N-diisopropylethylamine (DIEA) (350 g, 2710 mmol) in THF (800 mL) is added dropwise a solution of the intermediate (315 g) in toluene (800 mL) at 0-5 °C over 2 hours. The resultant mixture is allowed to warm to RT and stirred for 16 hours. The reaction is quenched with water (2.0 L) and extracted with of EA (2.0 L x 3). The combined organic layers are washed with 1000 mL of 3 N HC1 aqueous solution, brine (2.0 L x 3), dried over Na2SO4 and concentrated to give the crude product (330 g, 93%). |
|
Stage #1: 4-Phenoxybenzoic acid With thionyl chloride at 80℃; for 3h; Inert atmosphere;
Stage #2: malononitrile With N-ethyl-N,N-diisopropylamine In tetrahydrofuran; toluene at 0 - 20℃; for 18h; |
1 Step 1. Preparation of 2-(hydroxy(4-phenoxyphenyl)methylene)malononitrile
Step 1. Preparation of 2-(hydroxy(4-phenoxyphenyl)methylene)malononitrile A solution of 4-phenoxybenzoic acid (300 g, 1.4 mol) in SOCl2 (1.2 L) is stirred at 80° C. under N2 for 3 hours. The mixture is concentrated in vacuum to give the intermediate (315 g) which is used for next step without further purification. To a solution of propanedinitrile (89.5 g, 1355 mmol) and N,N-diisopropylethylamine (DIEA) (350 g, 2710 mmol) in THF (800 mL) is added dropwise a solution of the intermediate (315 g) in toluene (800 mL) at 0-5° C. over 2 hours. The resultant mixture is allowed to warm to RT and stirred for 16 hours. The reaction is quenched with water (2.0 L) and extracted with of EA (2.0 L*3). The combined organic layers are washed with 1000 mL of 3 N HCl aqueous solution, brine (2.0 L*3), dried over Na2SO4 and concentrated to give the crude product (330 g, 93%). |
| 79.9 kg |
With benzotriazol-1-ol; N-(3-dimethylaminopropyl)-N-ethylcarbodiimide; triethylamine In ethyl acetate at 10 - 20℃; Inert atmosphere; Large scale; |
1.1 Step 1: Synthesis of BG-2
Under nitrogen atmosphere, to a solution of EA (5 v) , HOBT (1.2 eq. ) , EDCI (1.2 eq. ) , 4-phenoxybenzoic acid (BG-1, 80 Kg, 1.0 eq. ) and malononitrile (1.2 eq. ) was added TEA (2.4 eq. ) at 10. The mixture was then stirred at RT until the reaction was completed. The mixture was then centrifuged and the cake was washed with EA. The filtrate was washed with aqueous NaHCO3 twice and NH4Cl. The organic phase was washed with 1.5 N H2SO4 twice and stirred. Concentrated, precipitated from methanol and purified water. The solid was collected by centrifugation and dried under vacuum. This gave 79.9 Kg of BG-2. 1H NMR (DMSO-d6) δ 7.62 (d, J 8.6 Hz, 2H) , 7.46-7.38 (m, 2H) , 7.18 (t, J 7.4 Hz, 1H) , 7.06 (d, J 8.0 Hz, 2H) , 6.94 (d, J 8.6 Hz, 2H) . |
| 79.9 kg |
With benzotriazol-1-ol; N-(3-dimethylaminopropyl)-N-ethylcarbodiimide; triethylamine In ethyl acetate at 20℃; Inert atmosphere; Large scale; |
1.1 Step 1: Synthesize BG-2
EA (5 v), HOBT (1.2 equivalents), EDCI (1.2 equivalents) at 10 ° C under a nitrogen atmosphereA solution of 4-phenoxybenzoic acid (BG-1, 80 Kg, 1.0 eq.) and malononitrile (1.2 eq.) was added TEA (2.4 eq.). The mixture was then stirred at room temperature until the reaction was complete. The mixture was then centrifuged and the filter cake was washed with EA. The filtrate was washed twice with aqueous NaHCO3 and washed with NH4Cl. The organic phase was washed twice with 1.5 N H2SO4 and stirred. It was concentrated from methanol and purified water and precipitated. The solid was collected by centrifugation and dried under vacuum. Thus, 79.9 Kg of BG-2 |
| 79.9 kg |
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; triethylamine In ethyl acetate at 10 - 20℃; Inert atmosphere; Large scale; |
|
|
Stage #1: 4-Phenoxybenzoic acid With thionyl chloride at 80℃; for 3h; Inert atmosphere;
Stage #2: malononitrile With N-ethyl-N,N-diisopropylamine In tetrahydrofuran; toluene at 0 - 20℃; for 18h; |
1 Step 1. Preparation of 2-(hydroxy(4-phenoxyphenyl)methylene)malononitrile
A solution of 4-phenoxybenzoic acid (300 g, 1.4 mol) in SOCl2 (1.2 L) is stirred at 80° C. under N2 for 3 hours. The mixture is concentrated in vacuum to give the intermediate (315 g) which is used for next step without further purification. To a solution of propanedinitrile (89.5 g, 1355 mmol) and DIEA (350 g, 2710 mmol) in THF (800 mL) is dropwise a solution of the intermediate (315 g) in toluene (800 mL) at 0-5° C. over 2 hours. The resultant mixture is allowed to warm to RT and stirred for 16 hours. The reaction is quenched with water (2.0 L) and extracted with of EA (2.0 L*3). The combined organic layers are washed with 1000 mL of 3 N HCl aqueous solution, brine (2.0 L*3), dried over Na2SO4 and concentrated to give the crude product (330 g, 93%). |
| 79.9 kg |
With benzotriazol-1-ol; N-(3-dimethylaminopropyl)-N-ethylcarbodiimide; triethylamine In ethyl acetate at 10 - 20℃; Inert atmosphere; Large scale; |
1.1 Step 1: Synthesis of BG-2
[0106] Under nitrogen atmosphere, to a solution of EA (5 v), HOBT (1.2 eq.), EDCI (1.2 eq.), 4-phenoxybenzoic acid (BG-1, 80 Kg, 1.0 eq.) and malononitrile (1.2 eq.) was added TEA (2.4 eq.) at 10°C. The mixture was then stirred at RT until the reaction was completed. The mixture was then centrifuged and the cake was washed with EA. The filtrate was washed with aqueous NaHCO3 twice and NH4Cl. The organic phase was washed with 1.5 N H2SO4 twice and stirred. Concentrated, precipitated from methanol and purified water. The solid was collected by centrifugation and dried under vacuum. This gave 79.9 Kg of BG-2. 1H NMR (DMSO-d6) δ 7.62 (d, J= 8.6 Hz, 2H), 7.46-7.38 (m, 2H), 7.18 (t, J= 7.4 Hz, 1H), 7.06 (d, J= 8.0 Hz, 2H), 6.94 (d, J= 8.6 Hz, 2H). |
|
Stage #1: 4-Phenoxybenzoic acid With thionyl chloride at 80℃; for 3h; Inert atmosphere;
Stage #2: malononitrile With N-ethyl-N,N-diisopropylamine In tetrahydrofuran; toluene at 0 - 20℃; for 18h; |
1 Step 1.
Preparation of 2-(hydroxy(4-phenoxyphenyl)methylene)malononitrile
A solution of 4-phenoxybenzoic acid (300 g, 1.4 mol) in SOCl2 (1.2 L) is stirred at 80° C. under N2 for 3 hours. The mixture is concentrated in vacuum to give the intermediate (315 g) which is used for next step without further purification. To a solution of propanedinitrile (89.5 g, 1355 mmol) and N,N-diisopropylethylamine (DIEA) (350 g, 2710 mmol) in THF (800 mL) is added dropwise a solution of the intermediate (315 g) in toluene (800 mL) at 0-5° C. over 2 hours. The resultant mixture is allowed to warm to RT and stirred for 16 hours. The reaction is quenched with water (2.0 L) and extracted with of EA (2.0 L*3). The combined organic layers are washed with 1000 mL of 3 N HCl aqueous solution, brine (2.0 L*3), dried over Na2SO4 and concentrated to give the crude product (330 g, 93%). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping