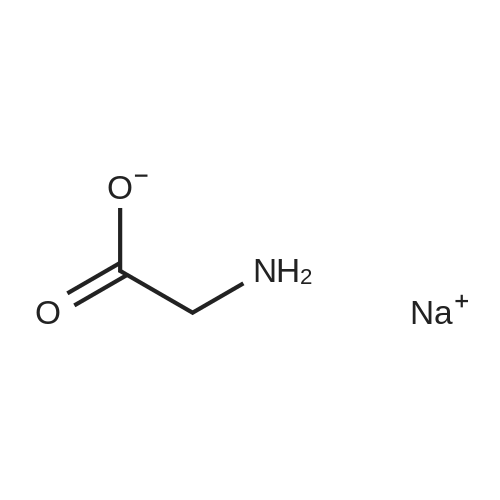
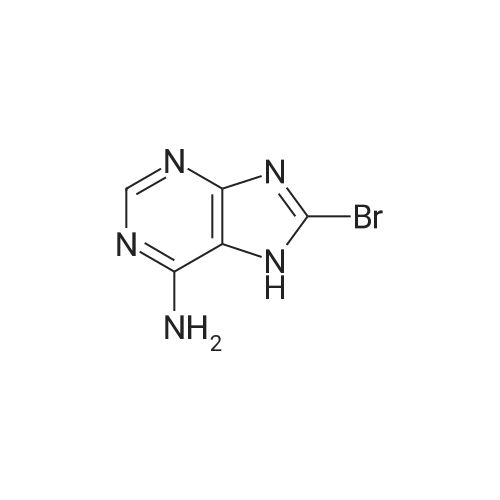
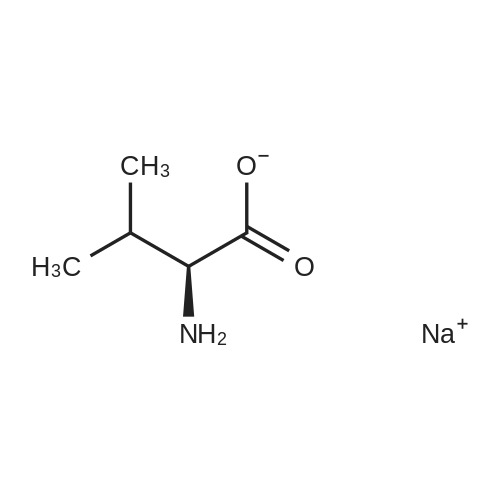
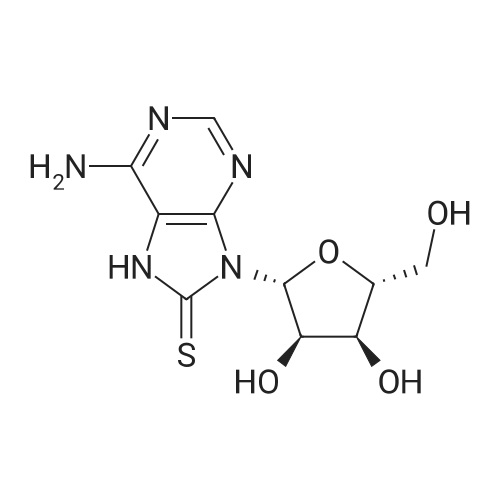
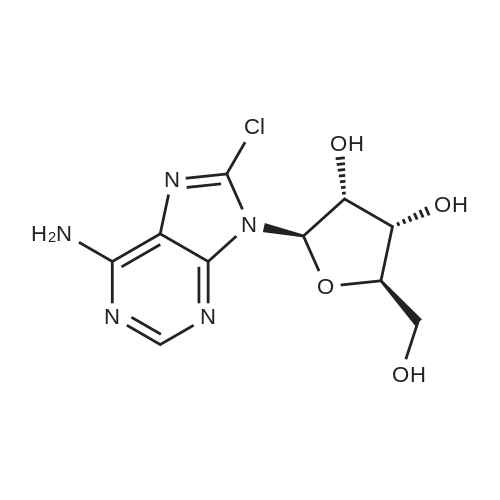
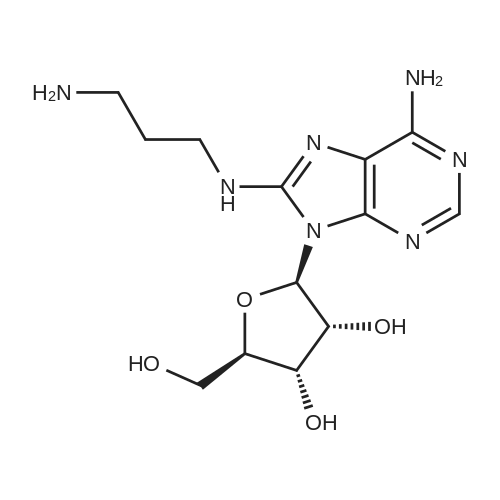
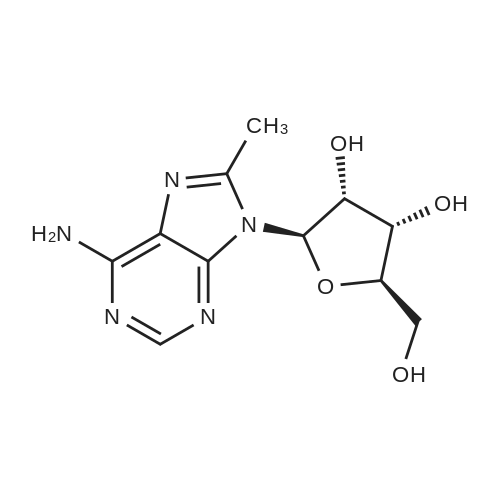
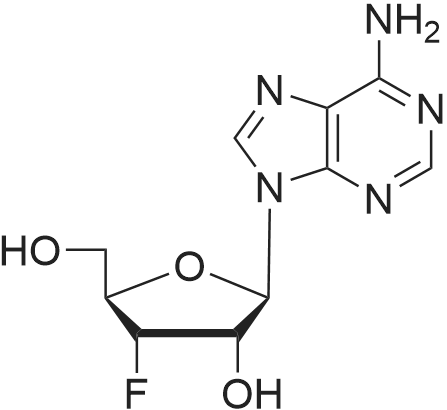
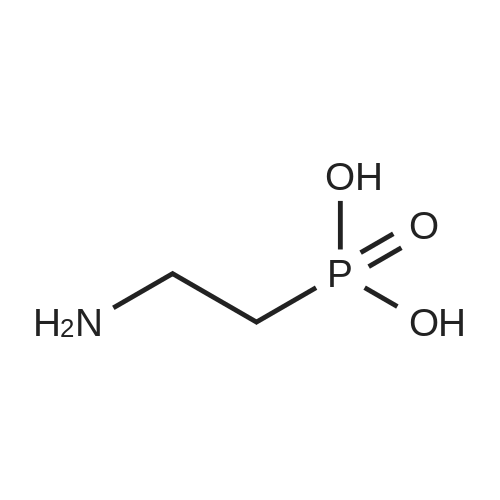
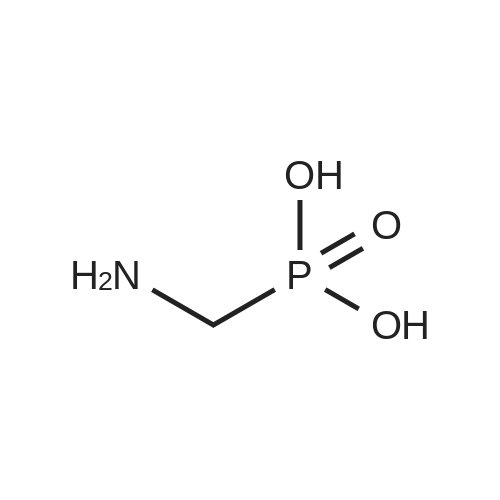
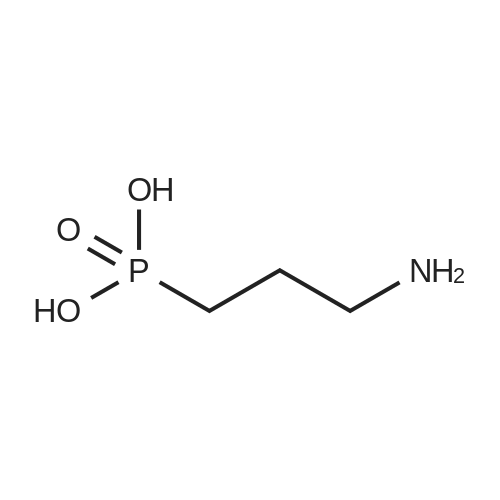
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
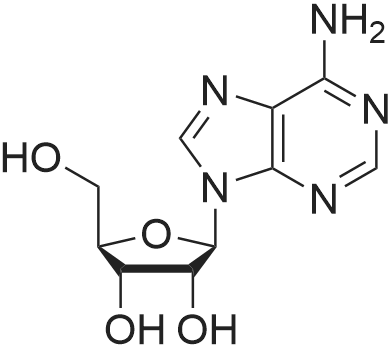
8-Mercaptoadenosine (10). NaSH (0.8 g, 10 eq) was added to a solution of 8-bromoadenosine (0.5 g, 1.44 mmol) in DMF (7 mL). The mixture was warmed to 100° C. and a few drops of water were added to improve solubility. The mixture was stirred at 100° C. overnight. The solvent was evaporated under high vacuum and the residue was coevaporated repeatedly with MeOH, until the residue turned into a solid. The residue was dissolved in water and neutalized with NaOH. After freeze drying, the product was purified on a silica gel column (CHCl3:MeOH 10:1). The product was obtained as a yellowish powder (100percent yield, mp 169-170° C.). 1H-NMR (CD3OD, 200 MHz) 8.09 (s, 1H, H-2), 6.65 (d, J=7 Hz, 1H, H-1'), 5.01 (dd, J=7, 5.5 Hz, 1H, H-2'), 4.39 (dd, J=5.5, 2.5 Hz, 1H, H-3'), 4.13 (q, J=2.5 Hz, 1H, H-4'), 3.87 (dd, J=12.5, 2.5 Hz, 1H, H-5'), 3.71 (dd, J=1.25, 3 Hz, 1H, H-5'); 13C-NMR (CD3OD, 300 MHz) δ 167.88 (C-6), 151.92 (C-2), 148.12 (C-4), 147.88 (C-8), 107.00 (C-5), 88.62 (C-1'), 85.59 (C-4'), 70.70 (C-2'), 70.62 (C-3'), 62.13 (C-5'); MS (CI/NH3): m/z 317 M+NH4+.
Reference:
[1] Journal of Medicinal Chemistry, 2000, vol. 43, # 11, p. 2239 - 2247
[2] Patent: US6617439, 2003, B1,
[3] Journal of Medicinal Chemistry, 2014, vol. 57, # 11, p. 4677 - 4691
[4] Journal of Medicinal Chemistry, 1999, vol. 42, # 26, p. 5325 - 5337
[5] Journal of Medicinal Chemistry, 2015, vol. 58, # 15, p. 6248 - 6263
5
[ 58-61-7 ]
[ 2946-39-6 ]
Yield
Reaction Conditions
Operation in experiment
82.5%
With bromine; sodium acetate In water
Example 1: Modified siRNA Molecules8-methoxyadenosine phosphoramidite was synthesized and incorporated into the anti- sense strand of caspase 2 siRNA. Figure 7A shows a schematic of the double-stranded positive control siRNA (SEQ ID NO: 20 (top) and SEQ ID NO: 21 (bottom)) and the double-stranded negative control siRNA (SEQ ID NO: 22 (top) and SEQ ID NO: 23 (bottom). Figure 7B shows a schematic for a singly modified siRNA, wherein the modification can be, but is not limited to, 8-proparglyoxyadenosine, 8-phenethyloxyadenosine, and 8-cyclohyexylethyloxyadenosine. In Figure 7B, 4AS corresponds to SEQ ID NO: 24, 6AS corresponds to SEQ ID NO: 25, 10AS corresponds to SEQ ID NO: 26, and 15AS corresponds to SEQ ID NO: 27.The bromination of adenosine is known in the art; generally, a three to four- fold excess of bromine generates a yield of about 75percent to about 82.5percent of 8-bromoadenosine. To protect the 2'-OH group during the synthesis of ribo-phosphoramidites, the 5' -OH ofN^-benzoyladenosine was first protected; and then t-butyldimethylsilyl chloride (TBDMS-C1) was used to protect the 2' -OH. Although the addition of Ag+ ion is recommended to minimize unavoidable reaction at the 3 '-OH, the unwanted reaction generally occurs and the overall yield is significantly reduced. Therefore, to accomplish a good overall yield, a novel protecting group di-ibutylsilyl ditriflate (DTBSDT), which protects the 5' -OH and the 3' -OH simultaneously and leaves the 2'-OH ready to be protected by TBDMS-C1 was used. The two reactions were basically a one- pot reaction, which eliminated the need for tedious separation. The reaction was almost quantitative with about 96percent to about 98percent product yield observed. (See Figures 3 and 4).Synthesis of the methoxy derivative using sodium methoxide reagent in methanol yielded the 8-methoxyadenosine derivative as well as the 8-oxoadenosine derivative. The separation of these two derivates was not trivial and the yield of the desired compound was below 50percent. Thus, the methoxy anion was generated in situ by reacting n-BuLi with excess anhydrous methanol. This reaction was much more efficient and yielded about 85percent to about 93percent of the desired product. The deprotection of 5' -OH and the 3'-OH was accomplished using a special fluoride reagent, HF -pyridine, at sub-zero temperature. These DMT reactions generated low yields (about 40percent to about 50percent) of the desired product. The phosphoramidite synthesis step yielded about 95percent to about 98percent of desired product. The 8- methoxy adenosine phosphoramidite was then incorporated into the antisense strand at position 9 or 14 (opposite to positions 11 and 6 of the sense strand, respectively), or both positions 9 and 14. The propargyl moiety at position 8 of adenosine can simplify the syntheses of other position 8 substituted adenosine analogs. The alkyne moiety of 8- propargyladenosine in siRNA can be "clicked" with suitable water-soluble azides leading to the formation of desirable minor groove modification.
81%
With bromine In water at 20℃; for 47 h; aq. acetate buffer
Saturated bromine-water (18.5 mL) was added slowly to a suspension of adenosine (0.5 g, 1.9 mmol) in NaOAc buffer (11.4 mL, 0.5 M, pH 4). The mixture was stirred at r.t. for 47 h. The solution was decolorized by the addition of 5 N NaHSO3, and the pH of the solution was then adjusted to 7 with 2 N NaOH. The resulting solids were collected by filtration, successively washed with water and acetone, and dried in vacuo to yield 19 (81percent). 1H NMR (270 MHz, DMSO-d6) δ: 3.46-3.74 (2H, m, H-5'), 3.98 (1 H, dd, J = 6.3, 4.0 Hz, H-4'), 4.16-4.22 (1 H, m, H-3'), 5.08 (1 H, dd, H-2'), 5.24 (1 H, d, J = 4.6 Hz, 3'-OH), 5.47 (1H, d, J = 6.3 Hz, H-1'), 5.52 (1 H, dd, J = 8.6, 4.0 Hz, 5'-OH), 5.83 (1 H, d, J = 6.5 Hz, 2'-OH), 7.56 (2H, br s, NH2), 8.11 (1H, s, H-2). ESI MS: m/z 345.1 Da [M+H]+,C10H12BrN5O4 Mol. Wt. 346.14.
73%
With sodium azide; bromoisocyanuric acid monosodium salt In water; N,N-dimethyl-formamide at 20℃; for 0.5 h;
General procedure: 5'-O-Monomethoxytrityl-N2-phenoxyacetylguanosine (33, 0.138 g, 0.2 mmol) was dissolved inaqueous DMF solution (H2O:DMF 1:4, 5 mL) under stirring. SMBI (1.1 equiv., 0.051 g, 0.22 mmol)was added at r.t. and the mixture stirred. Progress of the reaction was followed by TLC. An additionalamount of the reagent (0.15 equiv., 0.007 g) was added into the reaction mixture after 1.5 h. Oncompletion of the reaction by 2 h, the reaction mixture was filtered, evaporated to dryness underreduced pressure and coevaporated with water (2 × 2 mL). The crude reaction mixture was purified bycolumn chromatography (4percent–5percent MeOH in DCM, v/v) to afford nucleoside 34 (0.148 g, 96percent) in pureform as a white solid.8-Bromo-5'-O-monomethoxytrityl-N2-phenoxyacetylguanosine (34).
48%
With 1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione In N,N-dimethyl-formamide at 25℃; for 5 h;
Typical procedure for the bromination of unprotected nucleosides: DBH (323 mg, 1.13 mmol) was added to a stirred solution of 1d (500 mg, 2.05 mmol) in DMF (5 mL). The resulting pale-yellow solution was stirred at room temperature for 20 minutes or until TLC showed absence of starting material and formation of less polar product. Volatiles were evaporated and the residue was coevaporated with MeCN. The resulting pale solid was crystallized from hot acetone to give 2d (500 mg, 75percent) as colorless crystals with data as reported.14
53.1 kg
Stage #1: With sodium acetate; acetic acid In water at 75℃; for 0.5 h; Industrial scale Stage #2: With bromine In water at 30℃; for 10 h; Industrial scale
First, in a 2t reactor, 50 kg (185 mol) of adenosine and 600 kg of water, acetic acid-sodium acetate buffer solution (0.4 M, pH 4.5) were sequentially added, and then the temperature was raised to 75 ° C, and the temperature was kept for 30 minutes until the reaction system was dissolved;Then, the temperature was lowered to 30 ° C, 59 kg of Br 2 was further added, and then reacted at 30 ° C for 10 h, and the reaction was carried out by a high-performance liquid phase method, and the reaction was terminated when the content of the raw material in the reaction system was less than 1percent.Finally, after the reaction is completed, the reaction system is cooled to room temperature.Unreacted Br2 was removed by adding 19.5 kg of NaHSO3, then the pH was adjusted to 7 with a 5percent NaOH solution, filtered, and the filter cake was washed with water.Drying in vacuo gave a yellow product, 8-bromoadenosine, 53.1 kg.
Reference:
[1] Journal of Organic Chemistry, 2014, vol. 79, # 21, p. 9992 - 9997
[2] European Journal of Organic Chemistry, 2009, # 10, p. 1515 - 1521
[3] Patent: WO2011/119674, 2011, A1, . Location in patent: Page/Page column 50-51
[4] Journal of the American Chemical Society, 2012, vol. 134, # 42, p. 17643 - 17652
[5] Synthesis, 2004, # 17, p. 2799 - 2804
[6] European Journal of Medicinal Chemistry, 2012, vol. 54, p. 202 - 209
[7] Journal of Medicinal Chemistry, 2011, vol. 54, # 10, p. 3492 - 3499
[8] Tetrahedron, 2007, vol. 63, # 18, p. 3782 - 3789
[9] Tetrahedron, 1993, vol. 49, # 19, p. 4035 - 4050
[10] Journal of Organic Chemistry USSR (English Translation), 1985, vol. 21, p. 1639 - 1644[11] Zhurnal Organicheskoi Khimii, 1985, vol. 21, # 8, p. 1795 - 1800
[12] Biological Chemistry, 2011, vol. 392, # 4, p. 357 - 369
[13] Molecules, 2013, vol. 18, # 10, p. 12740 - 12750
[14] Journal of the American Chemical Society, 2013, vol. 135, # 9, p. 3423 - 3438
[15] Journal of Natural Products, 2005, vol. 68, # 11, p. 1689 - 1691
[16] Tetrahedron Letters, 2012, vol. 53, # 26, p. 3333 - 3336
[17] Angewandte Chemie - International Edition, 2017, vol. 56, # 13, p. 3536 - 3540[18] Angew. Chem., 2017, p. 3590 - 3594,5
[19] Indian Journal of Chemistry - Section A Inorganic, Physical, Theoretical and Analytical Chemistry, 2013, vol. 52, # 8-9, p. 1004 - 1013
[20] Bioorganic and Medicinal Chemistry, 2006, vol. 14, # 8, p. 2653 - 2659
[21] Chemistry - A European Journal, 2007, vol. 13, # 12, p. 3441 - 3449
[22] Structural Chemistry, 2010, vol. 21, # 1, p. 245 - 254
[23] Journal of Medicinal Chemistry, 2011, vol. 54, # 7, p. 2114 - 2126
[24] Beilstein Journal of Organic Chemistry, 2017, vol. 13, p. 495 - 501
[25] Patent: CN108129535, 2018, A, . Location in patent: Paragraph 0017-0028
[26] Organometallics, 2018, vol. 37, # 22, p. 4181 - 4185
6
[ 4294-16-0 ]
[ 1427459-39-9 ]
[ 2946-39-6 ]
Yield
Reaction Conditions
Operation in experiment
22%
With bromine; sodium acetate; acetic acid In water at 45℃; for 2.5 h;
6-Benzylamino-9-(β-D-ribofuranosyl)purine (469.4 mg; 1.313 mmol) was suspended in 15 ml 1 M AcONa and 15 ml 1 M AcOH. Bromine water (12.7 ml) was added to suspension and mixture was heated for 2.5 h at 45° C. Excess bromine was eliminated by addition of solid NaHSO3 and then the mixture was neutralized by 10percent NaOH and evaporated. Residue was shaken out with water and chloroform. Organic layer was separated, dried in MgSO4 and after filtration of desiccant evaporated to dryness. The residue was purified by column chromatography in CHCl3-MeOH-NH4OH (95:5:0.5). Yield 126.5 mg 6-benzylamino-8-bromo-9-(β-D-ribofuranosyl)purine (22percent), 42.2 mg starting material (9percent), 205 mg 8-bromoadenosine (45percent) and mixture of benzaldehyde and bromobenzaldehyde. Crystallization from CHCl3-hexan; mp: 98-100° C. MS ESI+: 436.2 [M+H+]. For C17H13BrN5O4 calculated 435.0542, found 436.0680 [M+H+]. 1H NMR (400 MHz; CDCl3) δ 3.75 (dd, J=2.7 Hz, 12.7 Hz, H5′), 3.90 (dd, J=2.4 Hz, 12.7 Hz, H5′), 4.20 (d, J=1.8 Hz, H4′), 4.39 (dd, J=1.8 Hz, 5.3 Hz, H3′), 4.81 (bs, —CH2—), 5.08 (dd, J=5.3 Hz, 7.2 Hz, H2′), 6.07 (d, J=7.2 Hz, H1′), 7.24 (m, H4-Ph), 7.31 (m, H3-Ph), 7.38 (m, H2-Ph), 8.19 (s, H2). MS ESI+ (8-bromoadenosine): 346.3 [M+H+]. GC: Rt (benzaldehyde)=321 s. MS EI (benzyldehyde): 105 (100percent), 77 (25percent). Rt (bromobenzyldehyde)=722 s. MS EI: 185 (100percent), 155 (50percent), 77 (45percent).
[0075] Starting with commercially available <strong>[2946-39-6]8-bromoadenosine</strong> (1), the hydroxyl groups of the ribose moiety were transiently protected by treatment with hexamethyldisilazane and catalytic ammonium sulfate. The crude intermediate underwent palladium catalyzed coupling reaction with trimethylaluminum to install the methyl group at the 8-position. The product, without purification, was deprotected directly to afford 8-methyladenosine derivative (2). Treatment of 2 with thionyl chloride in pyridine afforded the chloride derivative 3, which was subjected to reaction with excess methylamine under microwave irradiation to yields the amine 4. Allylic <n="23"/>displacement with c/5'-l-(?-butoxycarbonylamino)-4-chloro-2-butene under catalytic halide exchange conditions proceeded to form amine 5, which was then deprotected with methanolic hydrogen chloride and purified by silica gel chromatography to afford the final product.
To a solution of 19 (0.15 g, 0.43 mmol) in dry DMF (3 mL) is added Sodium ethanethiolate (MeSNa) (0.06 g, 0.86 mmol) and the solution is stirred for 2 h at r.t.. The mixture is neutralized with 1N HCl and the solvent evaporated under reduced pressure. The residue is purified by flash chromatography (CH2Cl2/MeOH, 8.5:1.5) to give 20. Yield 75%; mp 230 C. 1H NMR (270 MHz, DMSO-d6) delta: 2.72 (3H, s), 3.55 (2H, m), 3.96 (1H, m), 4.10 (1H, m), 4.95-5.14 (2H, m), 5.35-5.69 (3H, m), 7.23 (2H, br s), 8.05 (1H, s). ESI MS: m/z 314.4 Da [M+H]+, C11H15N5O4S Mol. Wt.: 313.33.
[0097] This example provides characterization data and synthetic guidance for the synthesized compound number 6 shown here:[0098] Characterization data: 1H NMR (CD3OD, 400 MHz) delta 8.21 (s, IH), 6.01 (d, /= 6.0 Hz, IH), 5.99-5.89 (m, 2H), 5.11 (t, / = 5.6 Hz, IH), 4.53-4.49 (m, IH), 4.46- 4.44 (m, IH), 4.01 (d, / = 6.8 Hz, 2H), 3.86 (dd, /= 10.8, 12.8 Hz, IH), 3.73 (t, / = 6.8 Hz, 2H), 3.57 (dd, /= 2.4, 12.8 Hz, IH), 3.00 (q, / = 7.2 Hz, 2H), 2.90 (s, 3H), 1.43 (t, / = 7.2 Hz, 3H); MS mlz 378.4 (M + H)+. <n="32"/>[0099] In preparation of compound 6, the intermediate compound, 8-ethyl adenosine, can be synthesized by the following procedure.[0100] A solution of <strong>[2946-39-6]8-Bromoadenosine</strong> (0.5g, 1.5 mmol) and hexamethyldisilazane (5mL) in 1,4-dioxane (1OmL) was heated overnight at 80 0C. The reaction was cooled to room temperature, concentrated in vacuo, and azeotroped with toluene. The residue was taken up in NMP (3 mL) and tetraethyltin (0.59 mL, 3.0 mmol) and Pd(PPh3)4 (0.175g, 0.15 mmol) were added. The reaction was allowed to stir at 130 0C for 16h before cooling to room temperature. The reaction mixture was partitioned between H2O and EtOAc. The aqueous layer was extracted with EtOAc (2 x 20 mL). The combined organic layers were washed with brine,dried over MgSO4, and concentrated in vacuo. The residue was taken up in MeOHiH2O (4:1) (25mL) and ammonium chloride was added (400 mg). The reaction was heated at 65 0C for 3h. When judged complete by TLC, the reaction was concentrated in vacuo and was subjected to silica gel column chromatography running 0-25% MeOH:DCM with 0.1% NH4OH to afford 8-ethyl adenosine.
Example 1: Modified siRNA Molecules8-methoxyadenosine phosphoramidite was synthesized and incorporated into the anti- sense strand of caspase 2 siRNA. Figure 7A shows a schematic of the double-stranded positive control siRNA (SEQ ID NO: 20 (top) and SEQ ID NO: 21 (bottom)) and the double-stranded negative control siRNA (SEQ ID NO: 22 (top) and SEQ ID NO: 23 (bottom). Figure 7B shows a schematic for a singly modified siRNA, wherein the modification can be, but is not limited to, 8-proparglyoxyadenosine, 8-phenethyloxyadenosine, and 8-cyclohyexylethyloxyadenosine. In Figure 7B, 4AS corresponds to SEQ ID NO: 24, 6AS corresponds to SEQ ID NO: 25, 10AS corresponds to SEQ ID NO: 26, and 15AS corresponds to SEQ ID NO: 27.The bromination of adenosine is known in the art; generally, a three to four- fold excess of bromine generates a yield of about 75% to about 82.5% of 8-bromoadenosine. To protect the 2'-OH group during the synthesis of ribo-phosphoramidites, the 5' -OH ofN^-benzoyladenosine was first protected; and then t-butyldimethylsilyl chloride (TBDMS-C1) was used to protect the 2' -OH. Although the addition of Ag+ ion is recommended to minimize unavoidable reaction at the 3 '-OH, the unwanted reaction generally occurs and the overall yield is significantly reduced. Therefore, to accomplish a good overall yield, a novel protecting group di-ibutylsilyl ditriflate (DTBSDT), which protects the 5' -OH and the 3' -OH simultaneously and leaves the 2'-OH ready to be protected by TBDMS-C1 was used. The two reactions were basically a one- pot reaction, which eliminated the need for tedious separation. The reaction was almost quantitative with about 96% to about 98% product yield observed. (See Figures 3 and 4).Synthesis of the methoxy derivative using sodium methoxide reagent in methanol yielded the 8-methoxyadenosine derivative as well as the 8-oxoadenosine derivative. The separation of these two derivates was not trivial and the yield of the desired compound was below 50%. Thus, the methoxy anion was generated in situ by reacting n-BuLi with excess anhydrous methanol. This reaction was much more efficient and yielded about 85% to about 93% of the desired product. The deprotection of 5' -OH and the 3'-OH was accomplished using a special fluoride reagent, HF -pyridine, at sub-zero temperature. These DMT reactions generated low yields (about 40% to about 50%) of the desired product. The phosphoramidite synthesis step yielded about 95% to about 98% of desired product. The 8- methoxy adenosine phosphoramidite was then incorporated into the antisense strand at position 9 or 14 (opposite to positions 11 and 6 of the sense strand, respectively), or both positions 9 and 14. The propargyl moiety at position 8 of adenosine can simplify the syntheses of other position 8 substituted adenosine analogs. The alkyne moiety of 8- propargyladenosine in siRNA can be "clicked" with suitable water-soluble azides leading to the formation of desirable minor groove modification.
81%
With bromine; In water; at 20℃; for 47h;pH 4.0;aq. acetate buffer;
Saturated bromine-water (18.5 mL) was added slowly to a suspension of adenosine (0.5 g, 1.9 mmol) in NaOAc buffer (11.4 mL, 0.5 M, pH 4). The mixture was stirred at r.t. for 47 h. The solution was decolorized by the addition of 5 N NaHSO3, and the pH of the solution was then adjusted to 7 with 2 N NaOH. The resulting solids were collected by filtration, successively washed with water and acetone, and dried in vacuo to yield 19 (81%). 1H NMR (270 MHz, DMSO-d6) delta: 3.46-3.74 (2H, m, H-5'), 3.98 (1 H, dd, J = 6.3, 4.0 Hz, H-4'), 4.16-4.22 (1 H, m, H-3'), 5.08 (1 H, dd, H-2'), 5.24 (1 H, d, J = 4.6 Hz, 3'-OH), 5.47 (1H, d, J = 6.3 Hz, H-1'), 5.52 (1 H, dd, J = 8.6, 4.0 Hz, 5'-OH), 5.83 (1 H, d, J = 6.5 Hz, 2'-OH), 7.56 (2H, br s, NH2), 8.11 (1H, s, H-2). ESI MS: m/z 345.1 Da [M+H]+,C10H12BrN5O4 Mol. Wt. 346.14.
73%
With sodium azide; bromoisocyanuric acid monosodium salt; In water; N,N-dimethyl-formamide; at 20℃; for 0.5h;
General procedure: 5'-O-Monomethoxytrityl-N2-phenoxyacetylguanosine (33, 0.138 g, 0.2 mmol) was dissolved inaqueous DMF solution (H2O:DMF 1:4, 5 mL) under stirring. SMBI (1.1 equiv., 0.051 g, 0.22 mmol)was added at r.t. and the mixture stirred. Progress of the reaction was followed by TLC. An additionalamount of the reagent (0.15 equiv., 0.007 g) was added into the reaction mixture after 1.5 h. Oncompletion of the reaction by 2 h, the reaction mixture was filtered, evaporated to dryness underreduced pressure and coevaporated with water (2 × 2 mL). The crude reaction mixture was purified bycolumn chromatography (4%-5% MeOH in DCM, v/v) to afford nucleoside 34 (0.148 g, 96%) in pureform as a white solid.8-Bromo-5'-O-monomethoxytrityl-N2-phenoxyacetylguanosine (34).
48%
With 1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione; In N,N-dimethyl-formamide; at 25℃; for 5h;
Typical procedure for the bromination of unprotected nucleosides: DBH (323 mg, 1.13 mmol) was added to a stirred solution of 1d (500 mg, 2.05 mmol) in DMF (5 mL). The resulting pale-yellow solution was stirred at room temperature for 20 minutes or until TLC showed absence of starting material and formation of less polar product. Volatiles were evaporated and the residue was coevaporated with MeCN. The resulting pale solid was crystallized from hot acetone to give 2d (500 mg, 75%) as colorless crystals with data as reported.14
53.1 kg
First, in a 2t reactor, 50 kg (185 mol) of adenosine and 600 kg of water, acetic acid-sodium acetate buffer solution (0.4 M, pH 4.5) were sequentially added, and then the temperature was raised to 75 C, and the temperature was kept for 30 minutes until the reaction system was dissolved;Then, the temperature was lowered to 30 C, 59 kg of Br 2 was further added, and then reacted at 30 C for 10 h, and the reaction was carried out by a high-performance liquid phase method, and the reaction was terminated when the content of the raw material in the reaction system was less than 1%.Finally, after the reaction is completed, the reaction system is cooled to room temperature.Unreacted Br2 was removed by adding 19.5 kg of NaHSO3, then the pH was adjusted to 7 with a 5% NaOH solution, filtered, and the filter cake was washed with water.Drying in vacuo gave a yellow product, 8-bromoadenosine, 53.1 kg.
29 g
With bromine; sodium acetate; acetic acid; at 10 - 20℃; for 48h;
step 1:Adenosine (50g, 187mmol)Suspended in acetic acid / sodium acetate (pH = 4.0, 0.5M, 1L)In the buffer,Slowly add liquid bromine (60g, 374mmol),Keep the system temperature below 10 ,After the addition, the reaction system was stirred at room temperature for 48 hours.Add to the reaction solutionSaturated aqueous sodium bisulfite solution,After removing excess bromine,Adjust the pH to neutral with aqueous sodium hydroxide (1M),Under the ice water bath cooling,After stirring for 2 hours, a solid precipitated,Filter, collect solids,After drying in vacuo, intermediate 1-1 (29 g) was obtained.
With 1H-imidazole; In N,N-dimethyl-formamide; at 20℃; for 24h;
Example 15 2',3',5'-Tris-(O-tert-butyldimethylsilyl)-8- bromoadenosine (13); TBDMS-Cl (2.30 g, 15.0 mmol) was added to a solution of <strong>[2946-39-6]8-bromoadenosine</strong> (11) (1.32 g, 3.82 mmol) and imidazole (2.08 g, 30.5 mmol) in DMF (15 ml) and the solution stirred at room temperature for 24 h. Saturated aqueous NH4CI (35 ml) was added and the mixture extracted with EtOAc (2 x 40 ml). The organic extracts were washed with water (2 x 15 ml), dried (MgS04) and evaporated to provide the title compound; yield 2.16 g (82%).
8-Mercaptoadenosine (10). NaSH (0.8 g, 10 eq) was added to a solution of <strong>[2946-39-6]8-bromoadenosine</strong> (0.5 g, 1.44 mmol) in DMF (7 mL). The mixture was warmed to 100 C. and a few drops of water were added to improve solubility. The mixture was stirred at 100 C. overnight. The solvent was evaporated under high vacuum and the residue was coevaporated repeatedly with MeOH, until the residue turned into a solid. The residue was dissolved in water and neutalized with NaOH. After freeze drying, the product was purified on a silica gel column (CHCl3:MeOH 10:1). The product was obtained as a yellowish powder (100% yield, mp 169-170 C.). 1H-NMR (CD3OD, 200 MHz) 8.09 (s, 1H, H-2), 6.65 (d, J=7 Hz, 1H, H-1'), 5.01 (dd, J=7, 5.5 Hz, 1H, H-2'), 4.39 (dd, J=5.5, 2.5 Hz, 1H, H-3'), 4.13 (q, J=2.5 Hz, 1H, H-4'), 3.87 (dd, J=12.5, 2.5 Hz, 1H, H-5'), 3.71 (dd, J=1.25, 3 Hz, 1H, H-5'); 13C-NMR (CD3OD, 300 MHz) delta 167.88 (C-6), 151.92 (C-2), 148.12 (C-4), 147.88 (C-8), 107.00 (C-5), 88.62 (C-1'), 85.59 (C-4'), 70.70 (C-2'), 70.62 (C-3'), 62.13 (C-5'); MS (CI/NH3): m/z 317 M+NH4+.
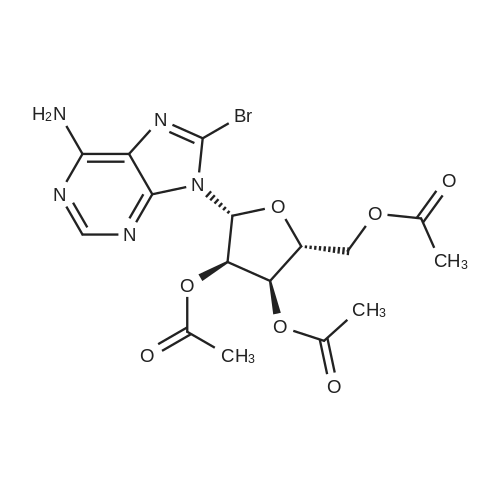
Example 14 2',3',5'-Tris-(O-acetyl)-<strong>[2946-39-6]8-bromoadenosine</strong> (12); Acetic anhydride (5.66 ml, 60 mmol) was added dropwise to a solution of <strong>[2946-39-6]8-bromoadenosine</strong> (11) (3.46 g, 10 mmol) in pyridine (50 ml) followed by DMAP (1 mmol) and the mixture stirred at room temperature for 5 h. The reaction was quenched by addition of methanol (10 ml). The solution was evaporated to dryness at reduced pressure, the residual material dissolved in ethyl acetate (250 ml), the solution shaken with NaHC03 (100 ml), water (3 x 50 ml), dried (MgS04) and the solution concentrated at reduced pressure. The product was a solid; yield 3.21 g, (68%).
62%
With pyridine;
8-[3-N-(5'-Deoxyadenosin-5'-yl)aminoprop-1-yn-1-yl]adenosine (19) Treatment of commercial <strong>[2946-39-6]8-bromo-adenosine</strong> with acetic anhydride (4 eq.) in pyridine gave after purification by flash chromatography (CH2Cl2 /MeOH) 8-bromo-2',3',5'-tri-O-acetyl-adenosine in 62% yield. To a degassed (3 times) solution of the bromo derivative (0.21 g, 0.45 mmol), alkyne 13 (0.20 g, 0.58 mmol) and triethylamine (3 eq.) in THF (10 mL) was added CuI (10% mol) and Pd(PPh3)4 (5% mol). After stirring under Ar at 60C overnight, the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography (CH2Cl2/MeOH) to give the coupling product 18 (0.17 g, 52%). A solution of 18 (0.13 g, 0.18 mmol) in MeOH (4 mL) and 36% aq. ammonia (2 mL) was heated at 50C. After 3 h the reaction was complete and the solvent was evaporated under reduced pressure. Purification by flash chromatography (CH2Cl2/MeOH) afforded deacetylated product (58 mg, 51% yield). This compound (48 mg) was treated with 80% aq. formic acid (1 mL) at 30C. After 7h, the reaction was complete and the reaction mixture was neutralized by addition of aq. ammonia before lyophilisation. The crude product was purified by reverse phase HPLC to give 19 (6 mg, 16%) as a white powder. Rt (5-25% ACN in 20 mM TEAA buffer) 12.3 min; 1H NMR (DMSO-d6) delta: 2.88 (m, 1H, H5'a), 3.01 (m, 1H, H5'a), 3.53 (m, 1H, H5'b), 3.70 (m, 1H, H5'b), 3.76 (d, 2H, NCH2), 3.98 (m, 1H, H4'b), 4.06 (m, 1H, H4'a), 4.17 (m, 1H, H3'a), 4.21 (m, 1H, H3'b), 4.72 (m, 1H, H2'a), 4.99 (m, 1H, H2'b), 5.18 (bs, 2H, OH), 5.40 (bs, 2H, OH), 5.50 (bs, 1H, OH), 5.86 (d, 1H, H1'a, J = 6.1 Hz), 5.96 (d, 1H, H1'b, J = 6.7 Hz), 7.24 (bs, 2H, NH2), 7.57 (bs, 2H, NH2), 8.15 (s, 1H, H2), 8.17 (s, 1H, H2), 8.35 (s, 1H, H8); UV lambdamax 262 nm, 279 nm; 1HRMS (ESI-TOF) m/z calcd for C23H28N11O7 (M+H) 570.2173; found 570.2174.
62%
With pyridine;
S-P-^-iS'-Deoxyadenosin-S'-y^aminoprop-l-yn-l-ylladenosine (19)Treatment of commercial <strong>[2946-39-6]8-bromo-adenosine</strong> with acetic anhydride (4 eq.) in pyridine gave after purification by flash chromatography (CH2CI2 /MeOH) 8- bromo-2',3,,5'-tri-O-acetyl-adenosine in 62% yield. To a degassed (3 times) solution of the bromo derivative (0.21 g, 0.45 mmol), alkyne 13 (0.20 g, 0.58 mmol) and triethylamine (3 eq.) in THF (10 mL) was added Cul (10% mol) and Pd(PPh3)4 (5% mol). After stirring under Ar at 60C overnight, the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography (CH2Cl2 MeOH) to give the coupling product 18 (0.17 g, 52%). A solution of 18 (0.13 g, 0.18 mmol) in MeOH (4 mL) and 36% aq. ammonia (2 mL) was heated at 50C. After 3 h the reaction was complete and the solvent was evaporated under reduced pressure. Purification by flash chromatography (CH2Cl2/MeOH) afforded deacetylated product (58 mg, 51% yield). This compound (48 mg) was treated with 80% aq. formic acid (1 mL) at 30C. After 7h, the reaction was complete and the reaction mixture was neutralized by addition of aq. ammonia before lyophilisation. The crude product was purified by reverse phase HPLC to give 19 (6 mg, 16%) as a white powder. Rt (5-25% ACN in 20 mM TEAA buffer) 12.3 min; NMR (DMSO-< ) delta: 2.88 (m, 1H, H5'a), 3.01 (m, 1H, H5'a), 3.53 (m, 1H, H5'b), 3.70 (m, 1H, H5'b), 3.76 (d, 2H, NCH2), 3.98 (m, 1H, H4'b), 4.06 (m, 1H, H4'a), 4.17 (m, 1H, H3'a), 4.21 (m, 1H5 H3'b)5 4.72 (m, 1H, H2'a), 4.99 (m, 1H, H2'b), 5.18 (bs, 2H, OH), 5.40 (bs, 2H, OH), 5.50 (bs, 1H, OH), 5.86 (d, 1H, HI 'a, J = 6.1 Hz), 5.96 (d, 1H, Hl 'b, J = 6.7 Hz), 7.24 (bs, 2H, NH2), 7.57 (bs, 2H, NH2), 8.15 (s, 1H, H2), 8.17 (s, 1H, H2), 8.35 (s, 1H, H8); UV max 262 nm, 279 nm; 1HRMS (ESI-TOF) m/z calcd for C23H28N?07 (M+H) 570.2173; found 570.2174.
With sodium azide; In N,N-dimethyl-formamide; at 75℃; for 6h;
<strong>[2946-39-6]8-Bromoadenosine</strong> (100 mg) was heated at 75 C for 6 h with 33 mg of NaN3 in 10 mL of dimethylformamide. After dilution with water, the mixture was applied on to a Dowex 50 (H+) column. After washing the column successively with water and 30% ethanol, 8-azidoadenosine was eluted with 1 N NH4OH in 30% ethanol. Rf on silica gel TLC in Solvent B, 0.69. UV: lambdamax in nm 284 at pH 1 and 285 at pH 7 and 13. 8-azidoadenosine (40 mg) was methylated with CH3I, essentially as described previously [20]. The mixture was diluted with water and applied on to a carboxymethyl-cellulose(H+) column. The column was washed successively with 190 mL of water and 190 mL of 30% ethanol, and 8-azido-1-methyladenosine was eluted with 1 N NH4OH in 30% ethanol. Rf on silica gel TLC in Solvent B, 0.13. UV: lambdamax in nm 285 at pH 1 and 7 and 286 at pH 13. 8-azido-1-methyladenosine was hydrolyzed by heating at 75 C for 3 h in 3.6 mL of 0.5 N HCl. After neutralization, Reagent III was purified by HPLC on Cosmosil C18 using 2.5% methanol containing 1% acetic acid (retention time, 16.8 min at a flow rate of 2 mL/min).
2-(6-amino-8-methoxy-purin-9-yl)-5-hydroxymethyl-tetrahydro-furan-3,4-diol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
94%
In methanol; at 85℃; for 2h;
Example 112; Synthesis of 2- .-5-hydroxamethyl-tetrahydro- furan-3, 4-diol; To a solution of <strong>[2946-39-6]8-bromoadenosine</strong> (Aldrich, O. lg, 0.289mmol) in MeOH (25mL) was added sodium methoxide (O. lg, 1. 81mmol) and the mixture was heated to 85C for 2 hours. The reaction was quenched with Dow-X 500 resin (H+), filtered and Dow-X washed with MeOH (15 mL) followed by 7M ammonia in methanol (15mL). The flowthrough was concentrated and purified by column chromatography on silica gel using 20% methanol in methylene as eluent. The appropriate fractions were pooled, concentrated in vacuo to give 81mg (94%) of the title compounds.
2-[6-amino-8-(N'-methyl-hydrazino)-purin-9-yl]-5-hydroxymethyl-tetrahydro-furan-3,4-diol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
100%
In DMF (N,N-dimethyl-formamide); at 85℃; for 3h;
Example 111; Synthesis of 2-E6-Amino-8-(N'-methYl-hydrazino .-purin-9-yl]-5-hydroxymethyl-tetrahydro-furan-3, dro-furan-3,4-diol (185); To a solution of <strong>[2946-39-6]8-bromoadenosine</strong> (Aldrich, 0. 1 g, 0. 289mmol) in DMF was added methyl hydrazine (0. 15mL, 2. 89mmol) and the mixture was heated to 85C for 3 hours. The crude product was purified by column chromatography on silica gel using 2.5% methanol in methylene chloride to wash and the product eluded with 20% methanol. The appropriate fractions were pooled, concentrated in vacuo to give 90mg (100%) of the title compound.
[0102] This example provides characterization data and synthetic guidance for the synthesized compound number 7 shown here: <n="33"/>[0103] Characterization data: 1H NMR (CD3OD, 400 MHz) delta 8.14 (s, IH), 5.95- 5.88 (m, IH), 5.90 (d, / = 4.8 Hz, IH), 5.76-5.72 (m, IH), 5.15 (t, / = 5.2 Hz, IH), 4.41 (t, /= 5.2 Hz, IH), 4.27-4.23 (m, IH), 3.53 (d, / = 6.8 Hz, 2H), 3.36-3.29 (m, IH), 3.16 (dd, /= 9.2, 12.8 Hz, IH), 2.94 (t, / = 7.6 Hz), 2.92-2.88 (m, IH), 2.40 (s, 3H), 1.92-1.86 (m, 2H), 1.07 (t, / = 7.6 Hz, 3H); MS mlz 392.4 (M + H)+.[0104] In preparation of compound 7, the intermediate compound, 8-ethyl adenosine, can be synthesized by the following procedure.[0105] A solution of <strong>[2946-39-6]8-Bromoadenosine</strong> (0.5g, 1.5 mmol) and hexamethyldisilazane (5mL) in 1,4-dioxane (1OmL) was heated overnight at 80 0C. The reaction was cooled to room temperature, concentrated in vacuo, and azeotroped with toluene. The residue was taken up in NMP (3 mL) and tri-w-butylpropenyltin (1.0 g, 3.0 mmol) and Pd(PPh3 )4 (0.175g, 0.15 mmol) were added. The reaction was heated at 190 0C under microwave conditions for 0.5h before cooling to room temperature. The catalyst was filtered through celite washing with EtOAc. The filtrate was partitioned between H2O and EtOAc. The aqueous layer was extracted with EtOAc (2 x 20 mL). The combined organic layers were washed with brine,dried over MgSO4, and concentrated in vacuo. The residue was taken up in MeOHiH2O (4:1) (25mL) and ammonium chloride was added (400 mg). The reaction was heated at 65 0C for 3h. When judged complete by TLC, the reaction was concentrated in vacuo and was subjected to silica gel column chromatography running 0- 25% MeOH:DCM with 0.1% NH4OH to afford 8-propenyl adenosine. The compound from the previous step was taken up in EtOHiH2O (3:1) (4mL) and TFA (5 drops) and hydrogenated under H2 atmosphere in the presence of Pd/C 10% (20% w/w) at room temperature overnight. The catalyst was filtered off and the filtrate evaporated in vacuo. The 8-propyl adenosine was used without further purification.[0106] The 8-propyl adenosine intermediate can be used to prepare Compound 7 by following the general route detailed in Scheme 1.
With bromine; sodium acetate; acetic acid; In water; at 45℃; for 2.5h;
6-Benzylamino-9-(beta-D-ribofuranosyl)purine (469.4 mg; 1.313 mmol) was suspended in 15 ml 1 M AcONa and 15 ml 1 M AcOH. Bromine water (12.7 ml) was added to suspension and mixture was heated for 2.5 h at 45 C. Excess bromine was eliminated by addition of solid NaHSO3 and then the mixture was neutralized by 10% NaOH and evaporated. Residue was shaken out with water and chloroform. Organic layer was separated, dried in MgSO4 and after filtration of desiccant evaporated to dryness. The residue was purified by column chromatography in CHCl3-MeOH-NH4OH (95:5:0.5). Yield 126.5 mg 6-benzylamino-8-bromo-9-(beta-D-ribofuranosyl)purine (22%), 42.2 mg starting material (9%), 205 mg 8-bromoadenosine (45%) and mixture of benzaldehyde and bromobenzaldehyde. Crystallization from CHCl3-hexan; mp: 98-100 C. MS ESI+: 436.2 [M+H+]. For C17H13BrN5O4 calculated 435.0542, found 436.0680 [M+H+]. 1H NMR (400 MHz; CDCl3) delta 3.75 (dd, J=2.7 Hz, 12.7 Hz, H5?), 3.90 (dd, J=2.4 Hz, 12.7 Hz, H5?), 4.20 (d, J=1.8 Hz, H4?), 4.39 (dd, J=1.8 Hz, 5.3 Hz, H3?), 4.81 (bs, -CH2-), 5.08 (dd, J=5.3 Hz, 7.2 Hz, H2?), 6.07 (d, J=7.2 Hz, H1?), 7.24 (m, H4-Ph), 7.31 (m, H3-Ph), 7.38 (m, H2-Ph), 8.19 (s, H2). MS ESI+ (8-bromoadenosine): 346.3 [M+H+]. GC: Rt (benzaldehyde)=321 s. MS EI (benzyldehyde): 105 (100%), 77 (25%). Rt (bromobenzyldehyde)=722 s. MS EI: 185 (100%), 155 (50%), 77 (45%).
8-(10-aminodecylamino)-6-amino-9-β-D-ribofuranosylpurine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In propan-1-ol; at 100℃; for 10h;
General procedure: BAP was reacted with ethyl acrylate to yield ethyl 3-(6-benzylaminopurin-9-yl)propionate [39].This ester with MS, NMR and other properties identical to those previously reported [36] (102 mg),1,10-diaminodecane (163 mg) and toluene (200 L) were heated in a sealed vial at 110 C for 2.5 h.The reaction mixture was dissolved in ethanol (2 mL) and water (4 mL) added. The solution wasacidified with HCl and extracted with ethyl acetate (extracts discarded). The aqueous phase was thenadjusted to pH 11-12 with KOH and extracted again with ethyl acetate. The extracts were washed withwater, concentrated and subjected to preparative TLC on silica gel (solvent D) yielding compound 4a.
With triethylamine; In 1,2-dichloro-ethane; at 33℃; for 18h;
(1) In a three-necked bottle with mechanical stirring, distillation unit and thermometer,Add 10g (0.029mol) of 1,6-amino-8-bromopurine nucleoside,16.8 g (0.088 mol) of p-toluenesulfonyl chloride and 19.4 g (0.191 mol) were added.Triethylamine was used as a solvent of 54.7 g (0.55 mol) of 1,2-dichloroethane.Then, the reaction was heated to 33 C using an electric heating jacket, and the reaction was kept for 18 hours.Using high-performance liquid phase method for central control,The content of 1,6-amino-8-bromopurine nucleoside is 0.61%.The content of 8-bromo-2'-O-p-toluenesulfonyl adenosine is 98.6%.Stop the reaction.(2) After the reaction is completed, the reaction system is cooled to room temperature.Filtration, the filter cake was recrystallized using methanol, filtered, and dried in vacuo.Obtained 13.91 g of 8-bromo-2'-O-p-toluenesulfonyl adenosine as a pale yellow powder.The yield was 95.8%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping