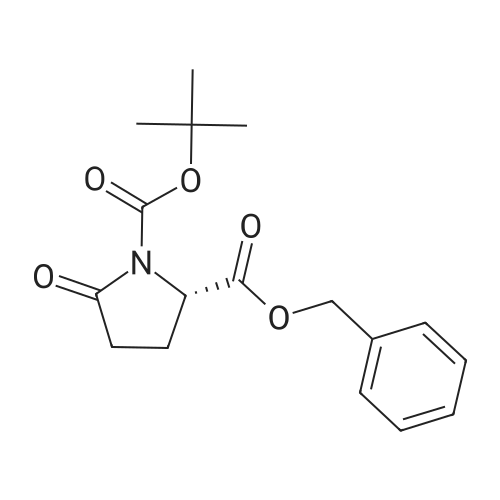
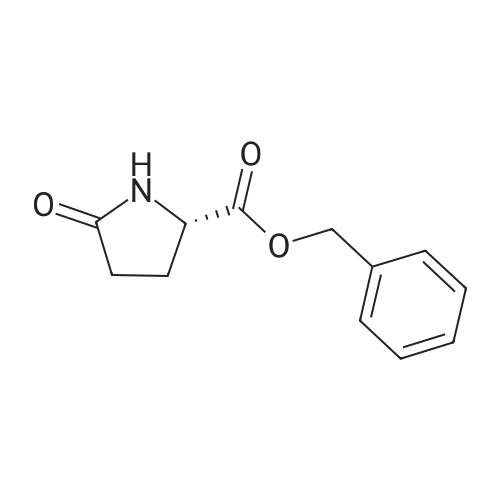
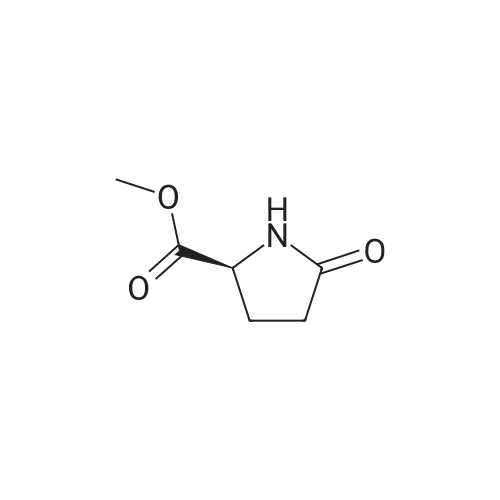
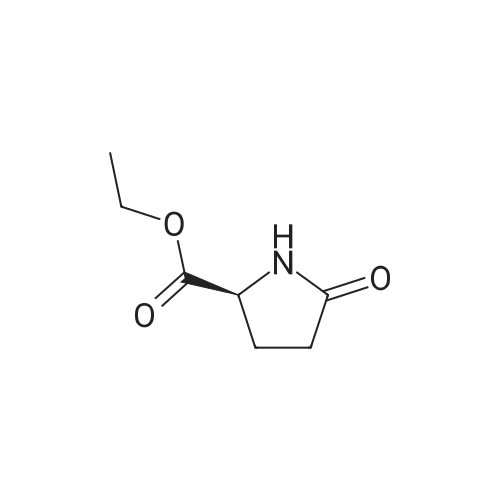
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With lithium hydroxide monohydrate In ethanol; water for 16 h;
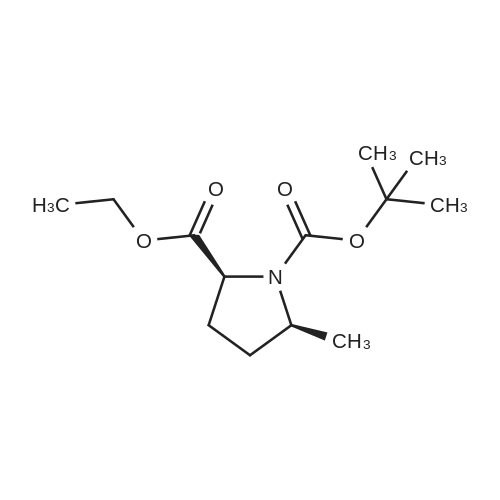
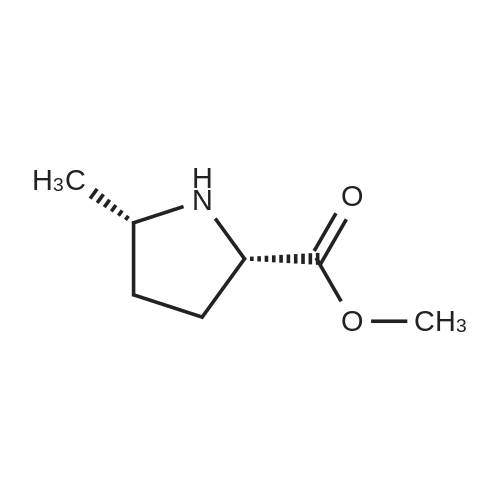
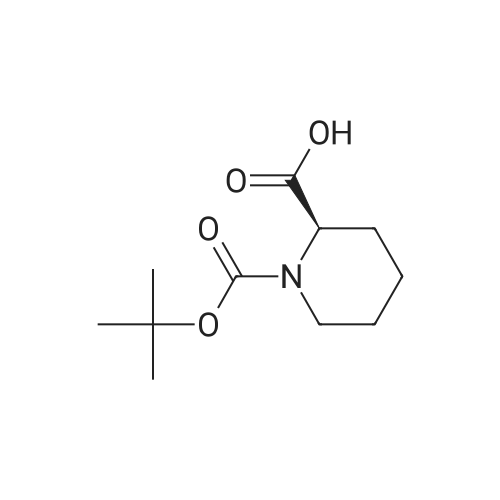
(2S,5S)-1-(Tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid To a solution of (2S,5S)-1-tert-butyl 2-ethyl 5-methylpyrrolidine-1,2-dicarboxylate (6.46 g, 25.1 mmol) in ethanol (20 mL) was added lithium hydroxide mono hydrate (2.11 g, 50.2 mmol) and deionized water (12 mL). The mixture was allowed to stir for 16 hours then partitioned between ethylacetate and a 1:1 mixture of saturated brine and 1N HCl. The aqueous layer was extracted an additional time with ethyl acetate. The organic layers were combined, dried with sodium sulfate and the solvent was removed in vacuo to yield (2S,5S)-1-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid as a white solid (quant.) and was used directly in the following step.
100%
With lithium hydroxide monohydrate; water In ethanol for 16 h;
(2S,5S)-1-(Tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid (Compound 1-6) To a solution of (2S,5S)-1-tert-butyl 2-ethyl 5-methylpyrrolidine-1,2-dicarboxylate (6.46 g, 25.1 mmol) in ethanol (20 mL) was added lithium hydroxide monohydrate (2.11 g, 50.2 mmol) and deionized water (12 mL). The mixture was allowed to stir for 16 hours then partitioned between ethylacetate and a 1:1 mixture of saturated brine and 1 N HCl. The aqueous layer was extracted an additional time with ethyl acetate. The organic layers were combined, dried with sodium sulfate and the solvent was removed in vacuo to yield (2S,5S)-1-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid as a white solid (quant.) and was used directly in the following step.
98.5%
With lithium hydroxide monohydrate; water In ethanol at 25℃; for 12 h;
[001651 To a solution of compound 1-5 (9.11 g, 35.45 mmol) in EtOH (80 mL) was added a solution of Lithium hydroxide hydrate (2.5 g, 59.67 mmol) in 20 mL of H20 at 25 °C. The mixture was stirred at 25 °C for 12 hours. After the reaction was completed, the mixture was extracted with EtOAc (150 mL), then the EtOAc phase was discarded. The aqueous phase was adjusted to pH 2 with 6percent hydrochloric acid, then the resulting mixture was extracted with EtOAc (80 mLx3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated /n vacuo to give the title compound as a white solid (8.0 g, 98.5percent).The compound was characterized by the following spectroscopic data:MS (ESI, pos.ion) m/z: 230.3 [M+Hfb; and‘H NMR (400 MHz, CD3OD): 4.24 (m, 1H), 3.56 (m, 1H), 1.75-1.97 (m, 2H), 1.45-1.69 (m, 2H), 1.40 (s, 9H), 1.32-1.34 (m, 3H) ppm.
98.5%
With lithium hydroxide In ethanol; water at 25℃; for 12 h;
Step 5) the Preparation of Compound 1-6 (0231) To a solution of compound 1-5 (9.11 g, 35.45 mmol) in EtOH (80 mL) was added a solution of Lithium hydroxide hydrate (2.5 g, 59.67 mmol) in 20 mL of H2O at 25° C. The mixture was stirred at 25° C. for 12 hours. After the reaction was completed, the mixture was extracted with EtOAc (150 mL), then the EtOAc phase was discarded. The aqueous phase was adjusted to pH 2 with 6percent hydrochloric acid, then the resulting mixture was extracted with EtOAc (80 mL×3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to give the title compound as a white solid (8.0 g, 98.5percent). The compound was characterized by the following spectroscopic data: (0232) MS (ESI, pos. ion) m/z: 230.3 [M+H]+; and (0233) 1H NMR (400 MHz, CD3OD): δ 4.24 (m, 1H), 3.56 (m, 1H), 1.75-1.97 (m, 2H), 1.45-1.69 (m, 2H), 1.40 (s, 9H), 1.32-1.34 (m, 3H) ppm.
9.1 g
With lithium hydroxide monohydrate; water In ethanol for 16 h;
In the 12g ii-5 prepared in the previous step, add 30ml of ethanol and stir to dissolve.2 g of lithium hydroxide monohydrate and 12 ml of deionized water are added. The mixture was stirred for 16 hours,Add 100ml of saturated sodium chloride solution and acidify with 1N hydrochloric acid;Then add 100ml of ethyl acetate to extract;It is extracted once more with 50 ml of ethyl acetate. The ethyl acetate extracts were combined and dried over anhydrous sodium sulfate overnight. The solid was filtered off and the filtrate was evaporated to dryness under reduced pressure.(9.1 g) of a white solid of (2S,5S)-N-(tert-butoxycarbonyl)-5-methyl-pyrrolidine-2-carboxylic acid (II) was obtained.
9.1 g
With lithium hydroxide monohydrate In ethanol; water for 16 h;
Step 5: In 12 g of ii-5 prepared in the previous step, add 30 ml of ethanol, stir to dissolve, add 2 g of lithium hydroxide monohydrate and12ml of deionized water. The mixture was stirred for 16 hours,Add 100ml of saturated sodium chloride solution and acidify with 1N hydrochloric acid; Then add 100ml of ethyl acetate to extract;It is extracted once more with 50 ml of ethyl acetate.The ethyl acetate extracts were combined and dried over anhydrous sodium sulfate overnight.The solid was filtered off and the filtrate was evaporated to dryness under reduced pressure.9.1 g of white solid II was obtained.
Reference:
[1] Patent: US2014/178336, 2014, A1, . Location in patent: Paragraph 0252
[2] Patent: US2015/361087, 2015, A1, . Location in patent: Paragraph 0311; 0316
[3] Patent: WO2015/110048, 2015, A1, . Location in patent: Paragraph 00165
[4] Patent: US2017/44140, 2017, A1, . Location in patent: Paragraph 0218; 0231; 0232; 0233
[5] Journal of Medicinal Chemistry, 2006, vol. 49, # 12, p. 3520 - 3535
[6] Journal of Medicinal Chemistry, 2006, vol. 49, # 21, p. 6416 - 6420
[7] Patent: WO2004/26822, 2004, A2, . Location in patent: Page 91
[8] Patent: US2013/115194, 2013, A1, . Location in patent: Paragraph 0426
[9] Patent: WO2013/75029, 2013, A1, . Location in patent: Page/Page column 131; 133
[10] Patent: US2013/309196, 2013, A1, . Location in patent: Paragraph 0348
[11] Patent: CN107540679, 2018, A, . Location in patent: Paragraph 0011; 0016
[12] Patent: CN107556324, 2018, A, . Location in patent: Paragraph 0010; 0011; 0016
With sodium hydroxide In water; acetonitrile at 20℃; for 1 h;
General procedure: Trifluoroacetic acid (10ml) was added to a solution of compound4a(4.7g, 0.014mol) in dichloromethane (30ml). The reaction mixture was stirred for 3h at room temperature. The solvent was removed in vacuo, and the residue was azeotroped with toluene to afford the crude product. To a solution of the product in methanol (50ml) was added 10percent Pd/C (500mg, 50percent wetted, type AD). The resulting mixture was stirred under a hydrogen atmosphere (1atm) for 16h at room temperature. The mixture was filtered to remove the catalyst, and the filtrate was concentrated in vacuo to afford the crude product. To a solution of the product in acetonitrile (60ml) with water (10ml) was added di-tert-butyl dicarbonate (4.58g, 21mmol) and 1N aqueous sodium hydroxide solution (35ml, 35mmol). The resulting solution was stirred for 1h at room temperature. After removing the solvent in vacuo, the residue was diluted with chloroform, and extracted with water. The aqueous layer was diluted with saturated aqueous NH4Cl, and the solvent was removed in vacuo. The residue was diluted with 10percent MeOH/CHCl3solution, and dried over magnesium sulfate. The mixture was filtered, and the solvent was removed in vacuo to afford the crude product. Purification by flash silica gel chromatography with CHCl3/MeOH (15:1, v/v) provided the title compound (2.14g, 67percent yield from4a);1H NMR (400MHz, CDCl3)δ: 1.25 (3H, d,J=6.3Hz), 1.48 (9H, s), 1.63–1.68 (1H, m), 2.00–2.09 (2H, m), 2.26–2.33 (1H, m), 3.91–3.97 (1H, m), 4.30–4.34 (1H, m); ESI/MS:m/z=252 (M+Na).
Reference:
[1] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 14, p. 4319 - 4331
With lithium hydroxide monohydrate; In ethanol; water; for 16h;
(2S,5S)-1-(Tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid To a solution of (2S,5S)-1-tert-butyl 2-ethyl 5-methylpyrrolidine-1,2-dicarboxylate (6.46 g, 25.1 mmol) in ethanol (20 mL) was added lithium hydroxide mono hydrate (2.11 g, 50.2 mmol) and deionized water (12 mL). The mixture was allowed to stir for 16 hours then partitioned between ethylacetate and a 1:1 mixture of saturated brine and 1N HCl. The aqueous layer was extracted an additional time with ethyl acetate. The organic layers were combined, dried with sodium sulfate and the solvent was removed in vacuo to yield (2S,5S)-1-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid as a white solid (quant.) and was used directly in the following step.
100%
With lithium hydroxide monohydrate; water; In ethanol; for 16h;
(2S,5S)-1-(Tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid (Compound 1-6) To a solution of (2S,5S)-1-tert-butyl 2-ethyl 5-methylpyrrolidine-1,2-dicarboxylate (6.46 g, 25.1 mmol) in ethanol (20 mL) was added lithium hydroxide monohydrate (2.11 g, 50.2 mmol) and deionized water (12 mL). The mixture was allowed to stir for 16 hours then partitioned between ethylacetate and a 1:1 mixture of saturated brine and 1 N HCl. The aqueous layer was extracted an additional time with ethyl acetate. The organic layers were combined, dried with sodium sulfate and the solvent was removed in vacuo to yield (2S,5S)-1-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid as a white solid (quant.) and was used directly in the following step.
98.5%
With lithium hydroxide monohydrate; water; In ethanol; at 25℃; for 12h;
[001651 To a solution of compound 1-5 (9.11 g, 35.45 mmol) in EtOH (80 mL) was added a solution of Lithium hydroxide hydrate (2.5 g, 59.67 mmol) in 20 mL of H20 at 25 °C. The mixture was stirred at 25 °C for 12 hours. After the reaction was completed, the mixture was extracted with EtOAc (150 mL), then the EtOAc phase was discarded. The aqueous phase was adjusted to pH 2 with 6percent hydrochloric acid, then the resulting mixture was extracted with EtOAc (80 mLx3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated /n vacuo to give the title compound as a white solid (8.0 g, 98.5percent).The compound was characterized by the following spectroscopic data:MS (ESI, pos.ion) m/z: 230.3 [M+Hfb; and?H NMR (400 MHz, CD3OD): 4.24 (m, 1H), 3.56 (m, 1H), 1.75-1.97 (m, 2H), 1.45-1.69 (m, 2H), 1.40 (s, 9H), 1.32-1.34 (m, 3H) ppm.
98.5%
With lithium hydroxide; In ethanol; water; at 25℃; for 12h;
Step 5) the Preparation of Compound 1-6 (0231) To a solution of compound 1-5 (9.11 g, 35.45 mmol) in EtOH (80 mL) was added a solution of Lithium hydroxide hydrate (2.5 g, 59.67 mmol) in 20 mL of H2O at 25° C. The mixture was stirred at 25° C. for 12 hours. After the reaction was completed, the mixture was extracted with EtOAc (150 mL), then the EtOAc phase was discarded. The aqueous phase was adjusted to pH 2 with 6percent hydrochloric acid, then the resulting mixture was extracted with EtOAc (80 mL×3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to give the title compound as a white solid (8.0 g, 98.5percent). The compound was characterized by the following spectroscopic data: (0232) MS (ESI, pos. ion) m/z: 230.3 [M+H]+; and (0233) 1H NMR (400 MHz, CD3OD): delta 4.24 (m, 1H), 3.56 (m, 1H), 1.75-1.97 (m, 2H), 1.45-1.69 (m, 2H), 1.40 (s, 9H), 1.32-1.34 (m, 3H) ppm.
Example 84A (3.69 g, 14.34 mmol) in 15 mL [OF ETOH] was treated with 14.3 mL of 1.7 N [LIOH] solution at room temperature. After 4 h, the mixture was concentrated, acidified with 1N [HC1] and then extracted with EtOAc (3X). The combined organic extracts were dried with [NA2S04,] and then concentrated to give the crude acid. MS (ESI) [M/Z] 228 (M-H)-.
(d) (2S,5S)-5-Methyl-pyrrolidine-1,2-dicarboxylic acid 1-tert-butyl ester A mixture of the product of the previous step (2 g, 7.77 mmol) and lithium hydroxide (466 mg, 23.3 mmol) in 3:1 methanol:water (24 mL) was stirred at RT overnight, and concentrated. The residue was dissolved in water and extracted with EtOAc (2*20 mL). The aqueous layer was adjusted to pH 2 with 1N HCl and extracted with 3:1 DCM:methanol (3*24 mL) to give the title intermediate (1.7 g) as a white crystal.
With lithium hydroxide monohydrate; water; In ethanol; for 16h;
(2S,5S)-l-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid To a solution of (2S,5S)-l -tert-butyl 2-ethyl 5-methylpyrrolidine-l,2-dicarboxylate (6.46 g, 25.1 mmol) in ethanol (20 mL) was added lithium hydroxide mono hydrate (2.11 g, 50.2 mmol) and deionized water (12mL). The mixture was allowed to stir for 16 hours then partitioned between ethylacetate and a 1 : 1 mixture of saturated brine and IN HC1. The aqueous layer was extracted an additional time with ethyl acetate. The organic layers were combined, dried with sodium sulfate and the solvent was removed in vacuo to yield (2S,5S)-l -(tert-butoxycarbonyl)-5-methylpyrrolidine-2- carboxylic acid as a white solid (quant.) and was used directly in the following step.
To a solution of (2S,5S)-1-tert-butyl 2-ethyl 5-methylpyrrolidine-1,2-dicarboxylate (6.46 g, 25.1 mmol) in ethanol (20 mL) was added lithium hydroxide mono hydrate (2.11 g, 50.2 mmol) and deionized water (12 mL). The mixture was allowed to stir for 16 hours then partitioned between ethylacetate and a 1:1 mixture of saturated brine and 1N HCl. The aqueous layer was extracted an additional time with ethyl acetate. The organic layers were combined, dried with sodium sulfate and the solvent was removed in vacuo to yield (2S,5S)-1-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid as a white solid (quant.) and was used directly in the following step.
9.1 g
With lithium hydroxide monohydrate; water; In ethanol; for 16h;
In the 12g ii-5 prepared in the previous step, add 30ml of ethanol and stir to dissolve.2 g of lithium hydroxide monohydrate and 12 ml of deionized water are added. The mixture was stirred for 16 hours,Add 100ml of saturated sodium chloride solution and acidify with 1N hydrochloric acid;Then add 100ml of ethyl acetate to extract;It is extracted once more with 50 ml of ethyl acetate. The ethyl acetate extracts were combined and dried over anhydrous sodium sulfate overnight. The solid was filtered off and the filtrate was evaporated to dryness under reduced pressure.(9.1 g) of a white solid of (2S,5S)-N-(tert-butoxycarbonyl)-5-methyl-pyrrolidine-2-carboxylic acid (II) was obtained.
9.1 g
With lithium hydroxide monohydrate; In ethanol; water; for 16h;
Step 5: In 12 g of ii-5 prepared in the previous step, add 30 ml of ethanol, stir to dissolve, add 2 g of lithium hydroxide monohydrate and12ml of deionized water. The mixture was stirred for 16 hours,Add 100ml of saturated sodium chloride solution and acidify with 1N hydrochloric acid; Then add 100ml of ethyl acetate to extract;It is extracted once more with 50 ml of ethyl acetate.The ethyl acetate extracts were combined and dried over anhydrous sodium sulfate overnight.The solid was filtered off and the filtrate was evaporated to dryness under reduced pressure.9.1 g of white solid II was obtained.
With hydrogenchloride; ammonia; triethylamine; In tetrahydrofuran; 1,4-dioxane; ethyl acetate;
Example 84C (2S,5S)-2-Carbamoyl-5-methyl-pyrrolidine-1-carboxylic acid tert-butyl ester Example 84B (2.055 g, 8.96 mmol) and Et3N (2.24 mL, 1.8 equiv.) were mixed in 15 mL of THF and then cooled to 0° C. Isobutyl chloroformate (1.51 mL, 1.3 equiv.) was added. After stirring for 35 min, 0.5 M NH3 in dioxane (35.8 mL, 2 equiv.) was added. After stirring at 0° C. for 3 h, the mixture was warmed to room temperature and stirred overnight. The volatiles were evaporated, and 1N HCl was added. The mixture was extracted with EtOAc (3*). The combined organic extracts were dried with Na2SO4 and then concentrated. The crude product was purified by chromatography (silica gel, 50percent then 75-80percent EtOAc/hexane) to give the desired amide. MS (ESI) m/z 229 (M+H)+.
Example 84B (2. [055G,] 8.96 mmol) and Et3N (2.24 mL, 1.8 equiv. ) were mixed in 15 mL of THF and then cooled to [0 °C.] Isobutyl [CHLOROFORMATE] (1.51 mL, 1.3 equiv. ) was added. After stirring for 35 min, 0.5 M NH3 in dioxane [(35.] 8 mL, 2 equiv. ) was added. After stirring at [0 °C] for 3 h, the mixture was warmed to room temperature and stirred overnight. The volatiles were evaporated, and 1N [HC1] was added. The mixture was extracted with EtOAc (3X). The combined organic extracts were dried with [NA2S04] and then concentrated. The crude product was purified by chromatography (silica gel, 50percent then 75-80percent EtOAc/hexane) to give the desired amide. MS (ESI) m/z 229 (M+H) [+.]
With O-(benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate; triethylamine; In DMF (N,N-dimethyl-formamide);pH 6 - 7;
Example 3F 2S (2S-CARBAMOYL-4, 4-DIFLUORO-PYRROLIDINE-1-CARBONYL)-5S-METHYL-PYRROLIDINE-1- carboxylic acid ter-butyl ester The compound from Example 3E (303 mg, 0.93 mmol), 5S-methyl- pyrrolidine-1, 2S-dicarboxylic acid 1-TERT-BUTYL ester (leq, prepared as described in Example 5) AND 2-(LH-BENZOKIAZOLE-1-YL)-L, 1, 3, 3-tetramethyluronium tetrafluoroborate (TBTU, 1.4 eq. ) were mixed in 2 mL of DMF, then Et3N was added until the pH of the mixture reached 6-7 (wet pH paper). The mixture was stirred overnight and purified by reverse-phase HPLC to provide the titled compound. (170 mg, 56percent).
Example 84B (2S,5S)-5-Methyl-pyrrolidine-1,2-dicarboxylic acid 1-tert-butyl ester Example 84A (3.69 g, 14.34 mmol) in 15 mL of EtOH was treated with 14.3 mL of 1.7 N LiOH solution at room temperature. After 4 h, the mixture was concentrated, acidified with 1N HCl and then extracted with EtOAc (3*). The combined organic extracts were dried with Na2SO4, and then concentrated to give the crude acid. MS (ESI) m/z 228 (M-H)-.
Example 84B (2S,5S)-5-Methyl-pyrrolidine-1,2-dicarboxylic acid 1-tert-butyl ester Example 84A (3.69 g, 14.34 mmol) in 15 mL of EtOH was treated with 14.3 mL of 1.7 N LiOH solution at room temperature. After 4 h, the mixture was concentrated, acidified with 1N HCl and then extracted with EtOAc (3*). The combined organic extracts were dried with Na2SO4, and then concentrated to give the crude acid. MS (ESI) m/z 228 (M+H)-.
13
[ 334769-80-1 ]
[ 99-73-0 ]
[ 1250939-12-8 ]
Yield
Reaction Conditions
Operation in experiment
52%
With triethylamine; In dichloromethane; at 0 - 25℃; for 3.5h;
[001691 To a cooled 0°C solution of compound 1-6 (5.0 g, 21.8 mmol) and compound 1-6-2 (7.28 g, 26.17 mmol) in DCM (140 mL) was added TEA (4.54 mL, 32.7 mmol) dropwise. The mixture was stirred at 25 °C for 3.5 hours. After the reaction was completed, the mixture was quenched with H20 (80 mL) and extracted with DCM (100 mLx3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (PE/EtOAc (v/v) = 5/1) to give the title compound as a white solid (4.73 g, 52percent). The compound was characterized by the following spectroscopic data:MS (ESI, pos.ion) m/z: 426.3 [M+Hfb; and?H NMR (400 MHz, CDC13): 7.78-7.75 (m, 2H), 7.65-7.63 (m, 2H), 5.53-5.15 (m, 2H), 4.49-4.39 (m, 1H), 3.59-3.54 (m, 1H), 2.31-2.21 (m, 2H), 2.12-2.01 (m, 1H), 1.98-1.85 (m, 1H), 1.45 (s, 9H), 1.12-1.13 (d,J= 6.4 Hz, 3H) ppm.
52%
With triethylamine; In dichloromethane; at 0 - 25℃; for 3.5h;
Step 9) the Preparation of Compound 1-12 (0243) To a cooled 0° C. solution of compound 1-6 (5.0 g, 21.8 mmol) and compound 1-6-2 (7.28 g, 26.17 mmol) in DCM (140 mL) was added TEA (4.54 mL, 32.7 mmol) dropwise. The mixture was stirred at 25° C. for 3.5 hours. After the reaction was completed, the mixture was quenched with H2O (80 mL) and extracted with DCM (100 mL×3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (PE/EtOAc (v/v)=5/1) to give the title compound as a white solid (4.73 g, 52percent). The compound was characterized by the following spectroscopic data: (0244) MS (ESI, pos. ion) m/z: 426.3 [M+H]+; and (0245) 1H NMR (400 MHz, CDCl3): delta 7.78-7.75 (m, 2H), 7.65-7.63 (m, 2H), 5.53-5.15 (m, 2H), 4.49-4.39 (m, 1H), 3.59-3.54 (m, 1H), 2.31-2.21 (m, 2H), 2.12-2.01 (m, 1H), 1.98-1.85 (m, 1H), 1.45 (s, 9H), 1.12-1.13 (d, J=6.4 Hz, 3H) ppm.
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 20℃; for 5h;
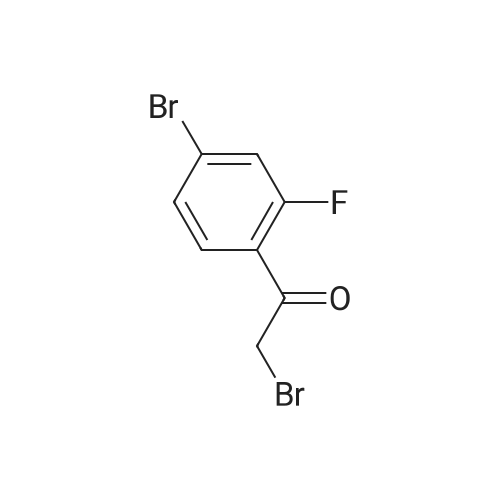
Example M10, Step cTo a solution of acid MlOb (2.16 g, 9.42 mmol) and 2-bromo-l-(4- bromophenyl)ethanone (2.62 g, 9.42 mmol) in acetonitrile (50 mL) was added slowly diisopropylethylamine (1.645 mL, 9.42 mmol), and the reaction mixture was stirred at room temperature for 5 hr. Solvent was removed in vacuo, and the residue was partitioned between ethyl acetate and water (1 : 1, 100 mL). The organic layer was washed with sat. aHC03, dried ( a2S04) and concentrated in vacuo to afford ketoester MlOc as white solid (3.9g), which was used in the next step without further purification. XH NMR (400 MHz , DMSO-d6, delta = 2.5 ppm): 7.92 (d, J = 8.3, 2H), 7.78 (d, J = 8.5, 2H), 5.6-5.4 (m, 2H), 4.35 (m, 1H), 3.85 (m, 1H), 2.25 (m, 1H), 2.05 (m, 2H), 1.60 (m, 1H), 1.5/1.4 (two overlapping br s, 9H), 1.18 (d, J = 6.6, 3H). LC/MS: Anal. Calcd. for [M+Na Ci9H2481BrNaN05: 450.07; found: 450.00.
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 20℃; for 16h;
To a solution of 2-bromo- 1 -(4-bromophenyl)ethanone (2.425 g, 8.72 mmol) and (2S,5S)-l-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid (2 g, 8.72 mmol) in acetonitrile (50 mL) was added DIEA (1.524 mL, 8.72 mmol), and the mixture was stirred at room temperature for 16 hrs. Solvent was removed in vacuo and the residue was partitioned between EtOAc (40 mL) and water (30 mL). The organic phase was washed with sat. NaHCCh and brine, and dried with a2S04. Removal of the volatile component in vacuo afforded Example 4, step d (1.74 g) as light yellow solid, which was used without further purification. 1H NMR (DMSO-d6, delta = 2.5 ppm, 400 MHz): 7.95-7.90 (m, 2H), 7.81 (m, 1H), 7.79 (m, 1H), 5.63-5.44 (m, 2H), 4.36 (m, 1H), 3.99 (m, 1H), 2.27 (m, 1H), 2.09 (m, 2H), 1.63 (m, 1H), 1.41- 1.37 (two singlet, 9H), 1.19 (m, 3H). LC/MS: Anal. Calcd. for [M+Na]+ Ci9H24BrNNa05: 448.07; found: 448.06.
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 20℃; for 16h;
Exam le QC-18.1, step dTo a solution of 2-bromo- 1 -(4-bromophenyl)ethanone (2.425 g, 8.72 mmol) and (2S,5S)-l-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid (2 g, 8.72 mmol) in acetonitrile (50 mL) was added DIEA (1.524 mL, 8.72 mmol), and the mixture was stirred at room temperature for 16 hrs. Solvent was removed in vacuo and the residue was partitioned between EtOAc (40 mL) and water (30 mL). The organic phase was washed with sat. aHC03 and brine, and dried with Na2S04. Removal of the volatile component in vacuo afforded Example 18.1, step d (1.74 g) as light yellow solid, which was used without further purification. lH NMR (DMSO- d6, delta = 2.5 ppm, 400 MHz): 7.95-7.90 (m, 2H), 7.81 (m, 1H), 7.79 (m, 1H), 5.63-5.44 (m, 2H), 4.36 (m, 1H), 3.99 (m, 1H), 2.27 (m, 1H), 2.09 (m, 2H), 1.63 (m, 1H), 1.41- 1.37 (two singlet, 9H), 1.19 (m, 3H). LC/MS: Anal. Calcd. for [M+NafCi9H24Br a05: 448.07; found: 448.06.
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 20℃; for 2h;Inert atmosphere;
(a) (2S,5S)-5-Methyl-pyrrolidine-1,2-dicarboxylic acid 2-[2-(4-bromo-phenyl)-2-oxo-ethyl]ester 1-tert-butyl ester A mixture of the product of Preparation 41 (1.7 g, 7.41 mmol), 2-bromo-1-(4-bromo-phenyl)-ethanone (2.08 g, 7.49 mmol) and DIPEA (2.87 g, 22.23 mmol) in ACN (50 mL) was stirred under nitrogen at RT for 2 h. The reaction mixture was concentrated under reduced pressure to give the title intermediate (3.16 g).
In acetonitrile; at 20℃; for 5h;Inert atmosphere;
General procedure: 2, 4'-Dibromoacetophenone (24 g, 87 mmol) was added under N2 at room temperature to a solution of compound BB2-d (154 g, 87 mmol) in MeCN (100 ml) followed by slow addition of DIEA (34 g, 261 mmol). The mixture was stirred at rt for 5 h then concentrated in vacuo and the resulting residue was diluted with EtOAc, washed with 1 N HCI (2x50 ml), saturated NaHC03 (2x50 ml) and brine (2x50 ml), dried over anhydrous Na2S04, filtered and concentrated. The residue was purified by column chromatography (p.ether/EtOAc 10: 1) which gave the title compound (27 g, 84percent). MS (ESI): 369 [M+H]+.
1.74 g
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 20℃; for 16h;
To a solution of 2-bromo- 1 -(4-bromophenyl)ethanone (2.425 g, 8.72 mmol) and (2S,5S)-l-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid (2 g, 8.72 mmol) in acetonitrile (50 mL) was added DIEA (1.524 mL, 8.72 mmol), and the mixture was stirred at room temperature for 16 h. Solvent was removed in vacuo and the residue was partitioned between EtOAc (40 mL) and water (30 mL). The organic phase was washed with sat. aHC03 and brine, and dried with Na2S04. Removal of the volatile component in vacuo afforded Example 18.1, step d (1.74 g) as light yellow solid, which was used without further purification. 1H NMR (DMSO-d6, delta = 2.5 ppm, 400 MHz): 7.95-7.90 (m, 2H), 7.81 (m, 1H), 7.79 (m, 1H), 5.63-5.44 (m, 2H), 4.36 (m, 1H), 3.99 (m, 1H), 2.27 (m, 1H), 2.09 (m, 2H), 1.63 (m, 1H), 1.41-1.37 (two singlet, 9H), 1.19 (m, 3H). LC/MS: Anal. Calcd. for [M+Naf Ci9H24BrNNa05: 448.07; found: 448.06.
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 20℃;
To a solution of acid MlOb (2.16 g, 9.42 mmol) and 2-bromo-l-(4- bromophenyl)ethanone (2.62 g, 9.42 mmol) in acetonitrile (50 mL) was added slowly diisopropylethylamine (1.645 mL, 9.42 mmol), and the reaction mixture was stirred at room temperature for 5 hr. Solvent was removed in vacuo, and the residue was partitioned between ethyl acetate and water (1 :1, 100 mL). The organic layer was washed with sat. NaHCO3, dried (Na2SO4) and concentrated in vacuo to afford ketoester MlOc as white solid (3.9g), which was used in the next step without further purification. 1H NMR (400 MHz , DMSO-d6} delta - 2.5 ppm): 7.92 (d, J = 8.3, 2H), 7.78 (d, J = 8.5, 2H), 5.6-5.4 (m, 2H), 4.35 (m, IH), 3.85 (m, IH), 2.25 (m, IH), 2.05 (m, 2H), 1.60 (m, IH), 1.5/1.4 (two overlapping br s>; 9H), 1.18 (d, J - 6.6, 3H). LC/MS: Anal. Calcd. for [M+Naf Cj9H2481BrNaNO5: 450.07; found: 450.00.
(2S,5S)-1-tert-butyl 2-methyl 5-methylpyrrolidine-1,2-dicarboxylate[ No CAS ]
[ 334769-80-1 ]
Yield
Reaction Conditions
Operation in experiment
79%
1 M aq LiOH (12.3 ml, 12.3 mmol) was added to a solution of BB21-d (3 g, 12.3 mmol) in THF (10 ml) and the resulting mixture was stirred overnight. The mixture was adjusted to pH 4 with 1 N HCI and extracted with EtOAc. The organic phase was concentrated in vacuo which gave the title compound (2.2 g, 79percent). LC-MS (ESI): 230 [M+H]+.
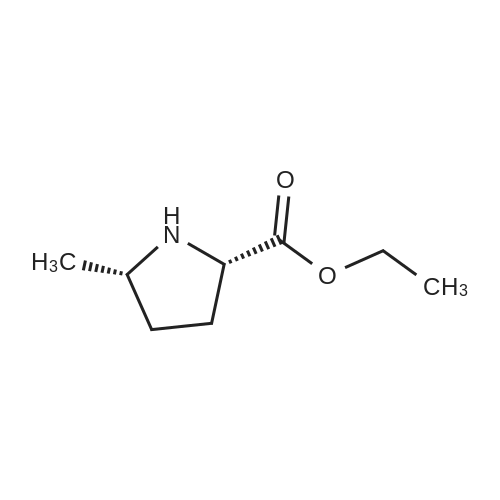
Exam le M10, Step bA solution of lithium hydroxide (0.23g, 9.62 mmol) in water (5 mL) was added dropwise to a solution of ester MlOa (1.8g, 7.4 mmol) in ethanol (10 mL), and stirred at room temperature for 17 hr. Most of the solvent was evaporated, and the residue was diluted with water, 1 N HCl was added dropwise to bring it to pH 3. It was extracted with ethyl acetate (20 mL, 4x), dried (Na2S04) and evaporated in vacuo to afford a colorless oil, which yielded crystals when dissolved inEtOAc/hexanes solvent system and allowed to stand at room temperature. The white solid was filtrated and dried in vacuo (1.42g). XH NMR (CDC13, 400 MHz, d = 7.24 ppm): 4.35 (m, 1H), 3.95 (m, 1H), 2.35 (m, 1H), 2.05 (m, 2H), 1.70 (m, 1H), 1.50 (br s, 9H), 1.25 (d, J = 7.1, 3H).
Example MlO, Step b; A solution of lithium hydroxide (0.23g, 9.62 mmol) in water (5 mL) was added dropwise to a solution of ester MlOa (1.8g, 7.4 mmol) in ethanol (10 mL), and stirred at room temperature for 17 hr. Most of the solvent was evaporated, and the residue was diluted with water, 1 N HCl was added dropwise to bring it to pH 3. It was extracted with ethyl acetate (20 mL, 4x), dried (Na2SO4) and evaporated in vacuo to afford a colorless oil, which yielded crystals when dissolved in EtOAc/hexanes solvent system and allowed to stand at room temperature. The white solid was filtrated and dried in vacuo (1.42g). 1H NMR (CDCl3, 400 MHz, d - 7.24 ppm): 4.35 (m, IH), 3.95 (m, IH), 235 (m, IH), 2.05 (m, 2H), 1.70 (m, IH), 1.50 (br s, 9H), 1.25 (d, J = 7.1, 3H).
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 20℃; for 3h;
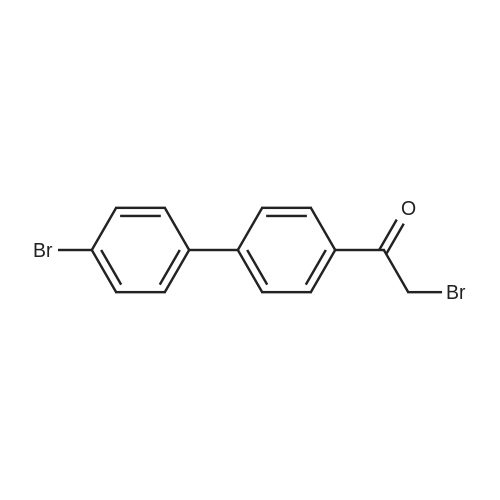
To a mixture of 2-bromo-1 -[4-(4-bromophenyl)phenyl]ethanone (735.3 mg, 2mmol), (2S,5S)-1 -tert-butoxycarbonyl-5-methyl-pyrrolidine-2-carboxylic acid (500 mg, 2.18 mmol) in acetonitrile (10 ml) (suspension) is added DIPEA (380 muIota_, 2.18 mmol). The reaction mixture is stirred at rt for 3 hours, diluted with EtOAc and H20. The organic phase is washed with brine, dried over Na2S04, filtered and concentrated. The residue is purified by flash column chromatography on silica gel (0 to 30percent EtOAc in Hexanes) to afford 2-[2-[4-(4- bromophenyl)phenyl]-2-oxo-ethyl]-1 -tert-butyl-(2S,5S)-5-methylpyrrolidine-1 ,2- dicarboxylate (0.8 g, 76.7percent) as a white solid.
EEDQ (1.67 g, 6.75 mmol) was added to a solution of Example J.9g2 (1.6 g,6.75 mmol) and M.4a (1.55 g, 6.75 mmol) in DCM (100 mL) and stirred for 6h.(Note: The dianiline was not completely soluble). The reaction mixture was diluted with DCM (1 vol) and washed with half sat'd NaHC03 soln. Concentration gave a solid (2.5 g). LC/MS (Cond. J2): RT = 3.07 min. LC/MS Anal. Calcd. for[M+H]+ C2iH27BrN303: 448.13; found 448.11. The crude solid (2.5 g, 5.58 mmol) was taken up in AcOH (200 mL) and stirred for 18 h at 60 °C. Concentration under high vacuum removed the solvent. The residue was taken up in DCM, washed with sat'd aHC03 soln, and concentrated. The residue was charged (DCM) to a 80 g Thompson silica gel cartridge and gradient elution was performed from 15percent to 100percent B over 750 mL. (A/B Hex/EtOAc) to give Example J.9g (2.6 g). XH NMR (MeOD, 500 MHz, delta): 8.36-8.35 (m, 2H), 8.0 (d, J = 9Hz, 1H), 7.91 (dd, J = 9, 2 Hz, 1H), 7.87 (d, J = 9 Hz, 1H), 5.31-5.28 (m, 1H), 4.17 (br. s, 1H), 2.59-2.56 (m, 1H), 2.39-2.31 (m, 2H) 1.86-1.83 (m, 1H), 1.52-1.19 (m, 12H). LC/MS (Cond. J2): RT = 2.57 min.LC/MS Anal. Calcd. for [M+H]+ 430.12; found 430.09.
With borane; In tetrahydrofuran; at 0 - 25℃; for 2h;
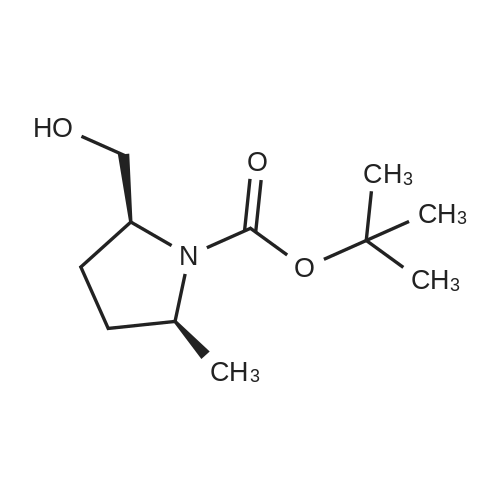
[001661 To a cooled 0 °C solution of compound 1-6 (2.24 g, 9.78 mmol) in THF (20 mL) was added a solution of borane in THF (14.6 mL, 1 mol/L). The mixture was stirred at 25 °C for 2 hours, and then to the mixture was added MeOH (8.0 mL). The THF was removed /n vacuo and the residue was dissolved in DCM (30 mL). The resulting mixture was washed with water (20 mLx3). The separated organic phase was dried over anhydrous Na2504 and concentrated /n vacuo. The residue was purified by silica gel column chromatography (PE/EtOAc (v/v) = 3/1) to give the title compound as colorless oil (1.98 g, 94.3percent). The compound was characterized by the following spectroscopic data:MS (ESI, pos.ion) m/z: 216.2 [M+Hfb; and?H NMR (400 MHz, CDC13): 3.90-3.91 (br, 2H), 3.62-3.65 (m, 2H), 3.50-3.54 (m, 1H), 1.91-1.93 (m, 2H), 1.51-1.60 (m, 2H), 1.44 (s, 9H), 1.13-1.14 (d,J= 6.2 Hz, 3H) ppm.
93%
To a solution of <strong>[334769-80-1](2S,5S)-1-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid</strong> (5.91 g, 25.8 mmol) in tetrahydrofuran at 0° C., was added borane in dimethylsulfide (1.0 M, 3.4 mL, 34 mmol) dropwise. The reaction was stirred for 4 hours at 0° C. then 18 hours at room temperature. The mixture was then cooled to 0° C. and methanol (70 mL) was added dropwise. The reaction was warmed to room temperature and the solvents were removed in vacuo. The residue was taken up in dichloromethane (200 mL) and extracted with saturated sodium bicarbonate. The organic layer was dried with sodium sulfate and the solvent was removed in vacuo to yield (2S,5S)-tert-butyl 2-(hydroxymethyl)-5-methylpyrrolidine-1-carboxylate as a clear oil (5.15 g, 23.9 mmol, 93percent) and was used directly in the following step.
Example 17, Step hTo a solution of (2S,5S)-l-(tert-butoxycarbonyl)-5-methylpyrrolidine-2- carboxylic acid (3.5 g, 15.27 mmol) in THF (10 mL) was added BH3.DMS (1.595 mL, 16.79 mmol) at 0 °C and then refluxed at 80 °C for 12 h. The reaction mixture was cooled to 0 °C and added slowly MeOH (10 mL). After stirring for 10 minutes the volatile components were removed under reduced pressure. The resulting residue was dissolved in EtOAc (50 mL), washed with 10percent NaHC03 (25 mL), water (25 mL), brine (10 mL), dried over Na2S04 and concentrated in vacuum. The crude was purified by flash chromatography (Silica gel 230-400, 30-40percent EtO Ac/petroleum ether) to obtain 17h (3.1 g) as a colorless liquid. LC/MS (Condition 75): Rt = 1.71 min. XH NMR (CDC13> delta = 7.26 ppm, 400 MHz): delta 4.95 (br s, 1H), 3.98-3.88 (m, 2H), 3.71-3.63 (m, 1H), 3.57-3.50 (m, 1H), 2.03-1.89 (m, 2H), 1.72-1.50 (m, 2H), 1.47 (s, 9H), 1.16 (d, J = 6.0, 3H). LC/MS: Anal. Calcd. for [M+H-Boc]+ C6H14NO: 116.10; found 116.2.
With dimethylsulfide borane complex; In tetrahydrofuran; at 0 - 20℃; for 22h;
(2S,5S)-tert-butyl 2-(hydroxymethyl)-5-methylpyrrolidine-l-carboxylate To a solution of (2S,5S)-l -(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid (5.91 g, 25.8 mmol) in tetrahydrofuran at 0 °C, was added borane in dimethylsulfide (1.0 M, 3.4 mL, 34 mmol) dropwise. The reaction was stirred for 4 hours at 0 °C then 18 hours at room temperature. The mixture was then cooled to 0 °C and methanol (70 mL) was added dropwise. The reaction was warmed to room temperature and the solvents were removed in vacuo. The residue was taken up in dichloromethane (200 mL) and extracted with saturated sodium bicarbonate. The organic layer was dried with sodium sulfate and the solvent was removed in vacuo to yield (2S,5S)-tert-butyl 2- (hydroxymethyl)-5-methylpyrrolidine-l -carboxylate as a clear oil (5.15 g, 23.9 mmol, 93percent) and was used directly in the following step.
With borane-THF; In tetrahydrofuran; at 0 - 25℃; for 2h;
Step 6) the Preparation of Compound 1-7 (0234) To a cooled 0° C. solution of compound 1-6 (2.24 g, 9.78 mmol) in THF (20 mL) was added a solution of borane in THF (14.6 mL, 1 mol/L). The mixture was stirred at 25° C. for 2 hours, and then to the mixture was added MeOH (8.0 mL). The THF was removed in vacuo and the residue was dissolved in DCM (30 mL). The resulting mixture was washed with water (20 mL×3). The separated organic phase was dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (PE/EtOAc (v/v)=3/1) to give the title compound as colorless oil (1.98 g, 94.3percent). The compound was characterized by the following spectroscopic data: (0235) MS (ESI, pos. ion) m/z: 216.2 [M+H]+; and (0236) 1H NMR (400 MHz, CDCl3): delta 3.90-3.91 (br, 2H), 3.62-3.65 (m, 2H), 3.50-3.54 (m, 1H), 1.91-1.93 (m, 2H), 1.51-1.60 (m, 2H), 1.44 (s, 9H), 1.13-1.14 (d, J=6.2 Hz, 3H) ppm
With N-ethyl-N,N-diisopropylamine; HATU; In N,N-dimethyl-formamide; at 0 - 20℃; for 1.5h;
Example 17, Step eTo a stirred solution of 2-amino-l-(4-bromophenyl)ethanone hydrochloride (5 g, 19.96 mmol) in DMF (50 mL) was added (2S,5S)-l-(tert-butoxycarbonyl)-5- methylpyrrolidine-2-carboxylic acid (4.80 g, 20.96 mmol) at 0 °C. Then HATU (7.74 g, 20.36 mmol) was added to the reaction mixture followed by DIPEA (10.46 mL, 59.9 mmol). The reaction mixture was allowed to warm to room temperature and stirred for 1.5 h. Water (100 mL) was added to the reaction mixture and extracted with EtOAc (2 x 100 mL). The organic layer was washed with brine (50 mL), dried over a2S04 and concentrated under reduced pressure. The crude product was purified by flash chromatography (Silica gel 60-120, 1.8percent MeOH /DCM) to yield 17e (7.9 g) as an off white solid. LC/MS (Condition 15): Rt = 1.99 min. XH NMR (CDCls, delta = 7.26 ppm, 400 MHz): delta 7.84 (d, J = 8.8, 2H), 7.65 (d, J = 8.8, 2H), 4.84-4.62 (m, 2H), 4.39 (br s, 1H), 3.94 (br s, 1H), 2.23 (br s, 1H), 2.10-2.02 (m, 2H), 1.64-1.54 (m, 1H), 1.48/1.47 (s, 9H), 1.39 (d, J = 6.0, 3H). LC/MS: Anal. Calcd. for [M-H]" Ci9H24BrN204: 424.32; found 425.0.
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 0 - 20℃; for 2h;
Example 15, Step eA solution of crude dibromide 15d (350 mg, 0.829 mmol) in ACN (5 mL) was cooled to 0 °C. Then (2S,5S)-l-(tert-butoxycarbonyl)-5-methylpyrrolidine-2- carboxylic acid (418 mg, 1.824 mmol) was added followed by drop-wise addition of DIPEA (0.579 mL, 3.32 mmol). The reaction mixture was allowed to warm to room temperature and stirred for 2 h. The reaction mixture was diluted with EtOAc (30 mL) and washed with saturated NH4C1 (20 mL), 10percent NaHC03 (20 mL), water (20 mL) and brine (10 mL). The organic layer was dried over Nu3/4804 and concentrated under reduced pressure. The crude was purified by Combiflash Isco (Silicycle, 40 g, silica, EtOAc: petroleum ether, 35:65) to afford diketoester 15e (300 mg) as a yellow solid. LC/MS (Condition 14): Rt = 2.36 min. 3/4 NMR (CDC13, delta = 7.26 ppm, 400 MHz): delta 8.01-7.99 (m, 2H), 7.89 (d, J = 8.4, 1H), 7.79-7.77 (m, 2H), 7.62 (d, J = 8.4, 1H), 5.60- 5.05 (m, 4H), 4.55-4.47 (m, 1H), 4.45-4.38 (m, 1H), 4.09-4.01 (m, 1H), 3.98-3.92 (m, 1H), 3.52 (br s, 4H), 2.38-2.29 (m, 4H), 2.17-2.04 (m, 2H), 1.80-1.69 (m, 2H), 1.48/1.45 (s, 18 H), 1.33 (br s, 6H). LC/MS: Anal. Calcd. for [M-H]" C4oH49N2Oio: 717.35; found 717.6.
With N-ethyl-N,N-diisopropylamine;potassium iodide; In acetonitrile; at 0 - 20℃; for 2h;
Example 16, Step eTo a solution of crude 16d (350 mg, 1.008 mmol) and (2S,5S)-l-(tert- butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid (462 mg, 2.016 mmol) in ACN (50 mL) was added KI (36.8 mg, 0.222 mmol) followed by DIPEA (0.704 mL, 4.03 mmol) at 0°C. Then the reaction mixture was stirred at RT for 2 h. The reaction mixture was diluted with EtOAc (50 mL) and washed with saturated NH4C1 (50 mL), 10percent NaHC03 (50 mL), brine (50 mL), dried over a2S04 and concentrated under reduced pressure. The crude was dissolved in MeOH (5 mL) and precipitated with diethyl ether:petroleum ether, 50:45 mL). The resulting precipitate was redissolved in MeOH (5 mL) and precipitated with diethyl ether:petroleum ether, 50:45 mL). This process was repeated for one more time. The precipitate was dried under high vacuum to obtain 16e (700 mg) as a yellow solid. LC/MS (Condition 10): Rt = 2.55 min. XH NMR (CDC13> delta = 7.26 ppm, 400 MHz): delta 8.14-8.12 (m, 2H), 7.85-7.82 (m, 2H), 7.70-7.65 (m, 1H), 7.59 (s, 1H), 7.59-7.56 (m, 1H), 7.08-7.04 (m, 1H), 5.89- 5.26 (m, 4H), 4.58-4.40 (m, 2H), 4.10-3.72 (m, 2H), 2.41-2.24 (m, 4H), 2.15-2.03 (m, 2H), 1.78-1.60 (m, 2H), 1.50 (br s, 18H), 1.40-1.31 (m, 6H). LC/MS: Anal. Calcd. For [M+H]+ C39H49N4O10: 733.34; found 733.4.
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 20℃; for 64h;
To a solution of Example 4, step a (2.58 g, 8.72 mmol) and (2S,5S)-l-(tert- butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid (2.00 g, 8.72 mmol) in acetonitrile (50 mL) was added DIEA (2.285 mL, 13.08 mmol), and the mixture was stirred at room temperature for 64 hrs. Solvent was removed in vacuo and the residue was partitioned between EtOAc (40 mL) and water (30 mL). The organic layer was washed with sat. aHC03 and brine, dried with a2S04 and evaporated in vacuo to afford Example 4, step b (3.8 g) as yellow solid. 1H NMR (CDC13, 400 MHz): 7.87 (m, 1H), 7.44 (m, 2H), 5.42-5.09 (m, 2H), 4.53-4.40 (m, 1H), 4.10-3.95 (m, 1H), 2.31 (m, 2H), 2.09 (m, 1H), 1.75 (m, 1H), 1.49-1.46 (two singlet, 9H), 1.33 (m, 3H). LC/MS: Anal. Calcd. for [M+Na]+ Ci9H24BrNNa05: 466.06; found: 466.03.
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 20℃; for 64h;
Exam le QC-18.1, step bTo a solution of Example 18.1, step a (2.58 g, 8.72 mmol) and (2S,5S)-l-(tert- butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid (2.00 g, 8.72 mmol) in acetonitrile (50 mL) was added DIEA (2.285 mL, 13.08 mmol), and the mixture was stirred at room temperature for 64 hrs. Solvent was removed in vacuo and the residue was partitioned between EtOAc (40 mL) and water (30 mL). The organic layer was washed with sat. aHC03 and brine, dried with Na2S04 and evaporated in vacuo to afford Example 18.1, step b (3.8 g) as yellow solid. XH NMR (CDC13, 400 MHz): 7.87 (m, 1H), 7.44 (m, 2H), 5.42-5.09 (m, 2H), 4.53-4.40 (m, 1H), 4.10-3.95 (m, 1H), 2.31 (m, 2H), 2.09 (m, 1H), 1.75 (m, 1H), 1.49-1.46 (two singlet, 9H), 1.33 (m, 3H). LC/MS: Anal. Calcd. for [M+Naf 466.06; found: 466.03.
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 0 - 20℃; for 2.16667h;
To a solution of crude Example 3, step d (5.1 g, 12 mmol) in acetonitrile (75 mL) was added (2S,5S)-l-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid (Example 1, step a) (5.70 g, 24.9 mmol) followed by DIPEA (8.69 mL, 49.7 mmol) at 0 °C. After 10 minutes, the temperature was raised to RT and stirred for 2 h. Then the reaction mixture was diluted with EtO Ac (100 mL) and washed with 10percent NaHCO3 (50 mL), brine (50 mL), dried over a2S04, filtered and concentrated under reduced pressure. The residue was purified by combiflash (Silicycle, S1O2, 25- 30percent EtOAc/petroleum ether) to afford Example 3, step e (5.8 g) as a pale yellow oil. NMR (CDCI3, delta = 7.26 ppm, 400 MHz): delta 8.00 (app bd, 2H), 7.71 (app d, 3H), 7.53-7.51 (m, 2H), 5.61-5.34 (m, 2H), 5.29-5.04 (m, 2H), 4.51-4.36 (m, 2H), 4.09- 3.91 (m, 2H), 2.59 (s, 3H), 2.35-2.21 (m, 4H), 2.15-2.04 (m, 2H), 1.80-1.63 (m, 2H), 1.47/1.44 (s, 18H), 1.35-1.27 (m, 6H). LC/MS: Anal. Calcd. for [Mu-EtaGammaC39H49 2Oio: 705.35; found 705.30.
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 50℃; for 18h;
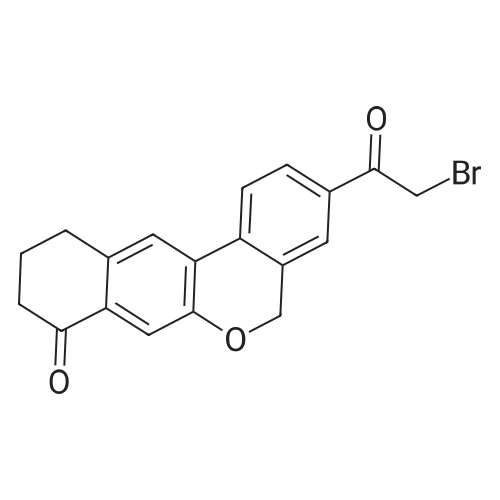
To a solution of 9-bromo-3-chloro-10,l l -dihydro-5H-dibenzo[c,g]chromen-8(9H)-one (1.41 g, 3.88 mmol) in MeCN (17 mL) was added (2S,5S)-l-(tert-butoxycarbonyl)-5-methylpyrrolidine-2- carboxylic acid (980 mg, 4.27 mmol) and DIPEA (1.49 mL, 8.54 mmol). After stirring for 18 h at 50 °C, the solution was diluted with EtOAc and washed successively with IN HCI, saturated aqueous NaHC03 and brine. The organics were dried over Na2S04, filtered and concentrated under reduced pressure. The crude residue was purified by silica column chromatography (10percent to 30percent EtOAc/hexanes) to afford (2S,5S)-l-tert-butyl 2-(3-chloro-8-oxo-8,9,10,l l-tetrahydro-5H- dibenzo[c,g]chromen-9-yl) 5-methylpyrrolidine-l,2-dicarboxylate (1.63 g, 81percent>).
81%
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 50℃; for 18h;
To a solution of 9-bromo-3-chloro-10,11-dihydro-5H-dibenzo[c,g]chromen-8(9H)-one (1.41 g, 3.88 mmol) in MeCN (17 mL) was added <strong>[334769-80-1](2S,5S)-1-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid</strong> (980 mg, 4.27 mmol) and DIPEA (1.49 mL, 8.54 mmol). After stirring for 18 h at 50° C., the solution was diluted with EtOAc and washed successively with 1N HCl, saturated aqueous NaHCO3 and brine. The organics were dried over Na2SO4, filtered and concentrated under reduced pressure. The crude residue was purified by silica column chromatography (10percent to 30percent EtOAc/hexanes) to afford (2S,5S)-1-tert-butyl 2-(3-chloro-8-oxo-8,9,10,11-tetrahydro-5H-dibenzo[c,g]chromen-9-yl) 5-methylpyrrolidine-1,2-dicarboxylate (1.63 g, 81percent).
5.6 g
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 50℃; for 18h;
The first step: 4.2g (11.6mmol) IV, add 60ml acetonitrile, stir to dissolve, add 2.94g in order(12. 8 mmol) II and 4. 5 ml (25.6 mmol) DIPEA. Stir 18h at 50°C, add ethyl acetate, use lN HC1, then saturateWash with NaHC03 and saline. The organic phase was dried over anhydrous Na2SO4 overnight, filtered and concentrated under reduced pressure; isolated by silica gel column chromatography,Elution with a mixture of ethyl acetate:petroleum ether (2:8), the desired fractions were collected and evaporated to dryness under reduced pressure to give (2S,5S)-N-(tert-butyl)Oxycarbonyl)-2-(3-chloro-8-oxo-8,9,10,11-tetrahydro-5H-dibenzo[c,g]chromene-9-yl)-5-methylpyrrolidine -2_ Carboxylate (v_l) 5.6 g.
With triethylamine; In acetonitrile; at 50℃; for 15h;
To a solution of 3-(2-bromoacetyl)-10,l l -dihydro-5H-dibenzo[c,g]chromen-8(9H)-one in MeCN (30 mL) was added (2S,5S)-l -(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid (1.2 g, 3.23 mmol) and triethyl amine (0.48 mL, 3.55 mmol) and the solution was heated to 50 °C. After stirring for 15 h, the solution was cooled to rt, and diluted with EtOAc and washed successively with saturated aqueous NaHCC>3 and brine. The organics were dried over MgSO i, filtered and concentrated under reduced pressure. The crude residue was purified by silica column chromatography (20percent to 50percent> EtOAc/hexanes) to afford (2S,5S)-1 -tert-butyl 2-(2-oxo-2-(8-oxo- 8,9,10, 11 -tetrahydro-5H-dibenzo[c,g] chromen-3 -yl)ethyl) 5-methylpyrrolidine-l ,2-dicarboxylate (1.09 g, 65percent).
65%
With triethylamine; In acetonitrile; at 50℃; for 15h;
To a solution of 3-(2-bromoacetyl)-10,11-dihydro-5H-dibenzo[c,g]chromen-8(9H)-one in MeCN (30 mL) was added <strong>[334769-80-1](2S,5S)-1-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid</strong> (1.2 g, 3.23 mmol) and triethyl amine (0.48 mL, 3.55 mmol) and the solution was heated to 50° C. After stirring for 15 h, the solution was cooled to rt, and diluted with EtOAc and washed successively with saturated aqueous NaHCO3 and brine. The organics were dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue was purified by silica column chromatography (20percent to 50percent EtOAc/hexanes) to afford (2S,5S)-1-tert-butyl 2-(2-oxo-2-(8-oxo-8,9,10,11-tetrahydro-5H-dibenzo[c,g]chromen-3-yl)ethyl) 5-methylpyrrolidine-1,2-dicarboxylate (1.09 g, 65percent).
With caesium carbonate; In acetone; at 40℃; for 16h;
(2S,5S)-2-(2-(9-bromo-8-oxo-8,9,10,11-tetrahydro-5H-dibenzo[c,g]chromen-3-yl)-2-oxoethyl) 1-tert-butyl 5-methylpyrrolidine-1,2-dicarboxylate (700 mg, 1.17 mmol) was treated with a solution of <strong>[334769-80-1](2S,5S)-1-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid</strong> (375 mg, 1.64 mmol) in acetone (6 mL) and Cs2CO3 (267 mg, 0.82 mmol). The stirred reaction mixture was heated to 40° C. for 16 h, then cooled to RT and diluted with CH2Cl2 and extracted 3×. The organic phase was washed with brine, then dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue was purified by silica column chromatography (40percent to 100percent EtOAc/hexanes) to afford (2S,5S)-2-(2-(9-((2S,5S)-1-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carbonyloxy)-8-oxo-8,9,10,11-tetrahydro-5H-dibenzo[c,g]chromen-3-yl)-2-oxoethyl) 1-tert-butyl 5-methylpyrrolidine-1,2-dicarboxylate (464 mg, 53percent).
464 mg
With caesium carbonate; In acetone; at 40℃; for 16h;
2S,5S)-2-(2-(9-((2S,5S)-l-(teri-butoxycarbonyl)-5-methylpyrrolidine-2-carbonyloxy)-8-oxo- 8,9,10,ll-tetrahydro-5H-dibenzo[c,g]chromen-3-yl)-2-oxoethyl) 1-tert-butyl 5- methylpyrrolidine-l,2-dicarboxylate (2S,5S)-2-(2-(9-bromo-8-oxo-8,9,10,l l -tetrahydro-5H-dibenzo[c,g]chromen-3-yl)-2- oxoethyl) 1-tert-butyl 5-methylpyrrolidine-l,2-dicarboxylate (700 mg, 1.17 mmol) was treated with a solution of (2S,5S)-l -(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid (375 mg, 1.64 mmol) in acetone (6 mL) and Cs2C03 (267 mg, 0.82 mmol). The stirred reaction mixture was heated to 40 °C for 16 h, then cooled to RT and diluted with CH2C12 and extracted 3X. The organic phase was washed with brine, then dried over MgS04, filtered and concentrated under reduced pressure. The crude residue was purified by silica column chromatography (40percent> to 100percent> EtOAc/hexanes) to afford (2S,5S)-2-(2-(9-((2S,5S)-l-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carbonyloxy)-8-oxo- 8,9,10,1 l-tetrahydro-5H-dibenzo[c,g] chromen-3 -yl)-2-oxoethyl) 1 -tert-butyl 5-methylpyrrolidine-l, 2- dicarboxylate (464 mg, 53percent>).
(2S,5S)-2-(2-(9-bromo-8-oxo-8,9,10,11-tetrahydro-5H-dibenzo[c,g]chromen-3-yl)-2-oxoethyl) 1-tert-butyl 5-methylpyrrolidine-1,2-dicarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84%
With potassium carbonate; In dichloromethane; at 20℃;
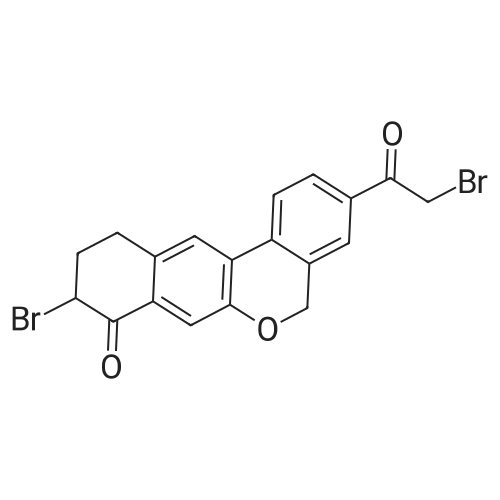
9-bromo-3-(2-bromoacetyl)-10,l l-dihydro-5H-dibenzo[c,g]chromen-8(9H)-one (1.43 g, 3.17 mmol) was treated with a solution of (2S,5S)-l-(tert-butoxycarbonyl)-5-methylpyrrolidine-2- carboxylic acid (800 mg, 3.49 mmol) in dichloromethane (14 mL) and K2CO3 (658 mg, 1.18 mmol). The stirred reaction mixture was stirred at RT and diluted with CH2CI2 and extracted 3X. The organic phase was washed with brine, then dried over MgS04, filtered and concentrated under reduced pressure to afford ((2S,5S)-2-(2-(9-bromo-8-oxo-8,9,10,l l-tetrahydro-5H-dibenzo[c,g]chromen-3-yl)- 2-oxoethyl) 1 -tert-butyl 5-methylpyrrolidine-l,2-dicarboxylate (1.61 g, 84percent).
With sodium hydroxide; In water; acetonitrile; at 20℃; for 1h;
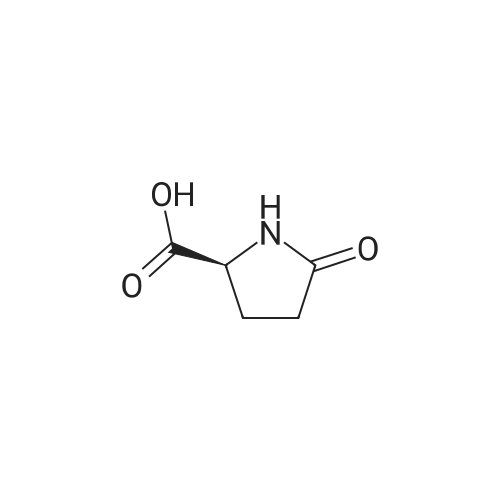
General procedure: Trifluoroacetic acid (10ml) was added to a solution of compound4a(4.7g, 0.014mol) in dichloromethane (30ml). The reaction mixture was stirred for 3h at room temperature. The solvent was removed in vacuo, and the residue was azeotroped with toluene to afford the crude product. To a solution of the product in methanol (50ml) was added 10percent Pd/C (500mg, 50percent wetted, type AD). The resulting mixture was stirred under a hydrogen atmosphere (1atm) for 16h at room temperature. The mixture was filtered to remove the catalyst, and the filtrate was concentrated in vacuo to afford the crude product. To a solution of the product in acetonitrile (60ml) with water (10ml) was added di-tert-butyl dicarbonate (4.58g, 21mmol) and 1N aqueous sodium hydroxide solution (35ml, 35mmol). The resulting solution was stirred for 1h at room temperature. After removing the solvent in vacuo, the residue was diluted with chloroform, and extracted with water. The aqueous layer was diluted with saturated aqueous NH4Cl, and the solvent was removed in vacuo. The residue was diluted with 10percent MeOH/CHCl3solution, and dried over magnesium sulfate. The mixture was filtered, and the solvent was removed in vacuo to afford the crude product. Purification by flash silica gel chromatography with CHCl3/MeOH (15:1, v/v) provided the title compound (2.14g, 67percent yield from4a);1H NMR (400MHz, CDCl3)delta: 1.25 (3H, d,J=6.3Hz), 1.48 (9H, s), 1.63?1.68 (1H, m), 2.00?2.09 (2H, m), 2.26?2.33 (1H, m), 3.91?3.97 (1H, m), 4.30?4.34 (1H, m); ESI/MS:m/z=252 (M+Na).
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; triethylamine; In dichloromethane; at 20℃; for 16h;
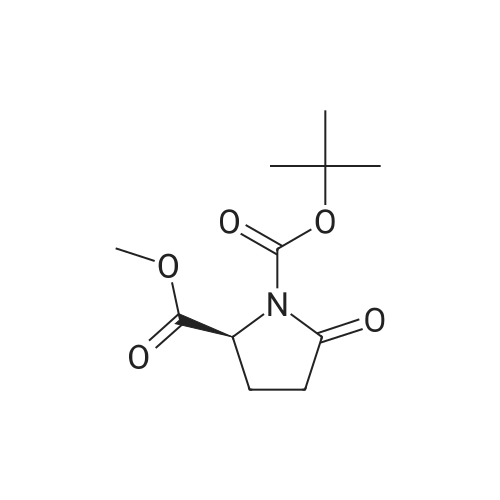
Triethylamine (280mul, 2.02mmol) was added to a solution of compound5a(232mg, 1.01mmol) in dichloromethane (6ml), followed by dimethylamine hydrochloride (124mg, 1.52mmol), HOBt (14mg, 0.10mmol) and EDC/HCl (233mg, 1.21mmol). The solution was stirred for 16h at room temperature. The solvent was removed in vacuo to afford crude product. The residue was purified by flash silica gel chromatography with CHCl3/MeOH (50:1, v/v) to give the colorless oil (161mg, 62percent yield);1H NMR (400MHz, CDCl3)delta: 1.35 (3H, d,J=6.1Hz), 1.40 and 1.46 (9H, each s), 1.66?1.75 (1H, m), 1.87?1.93 (1H, m), 2.01?2.12 (2H, m), 2.97 (3H, s), 3.07 and 3.11 (3H, s), 3.91?3.95 and 4.02?4.07 (1H, m), 4.53?4.58 and 4.68?4.72 (1H, m); ESI/MS:m/z=157 (M-Boc).
With potassium carbonate; In dichloromethane; at 20℃;
9-bromo-3-(2-bromoacetyl)-10,11-dihydro-5H-dibenzo[c,g]chromen-8(9H)-one (1.43 g, 3.17 mmol) was treated with a solution of <strong>[334769-80-1](2S,5S)-1-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carboxylic acid</strong> (800 mg, 3.49 mmol) in dichloromethane (14 mL) and K2CO3 (658 mg, 1.18 mmol). The stirred reaction mixture was stirred at RT and diluted with CH2Cl2 and extracted 3×. The organic phase was washed with brine, then dried over MgSO4, filtered and concentrated under reduced pressure to afford ((2S,5S)-2-(2-(9-bromo-8-oxo-8,9,10,11-tetrahydro-5H-dibenzo[c,g]chromen-3-yl)-2-oxoethyl) 1-tert-butyl 5-methylpyrrolidine-1,2-dicarboxylate (1.61 g, 84percent).
(2S,5S)-2-(2-(9-((2S,5S)-1-(tert-butoxycarbonyl)-5-methylpyrrolidine-2-carbonyloxy)-8-oxo-8,9,10,11-tetrahydro-5H-dibenzo[c,g]chromen-3-yl)-2-oxoethyl) 1-tert-butyl 5-methylpyrrolidine-1,2-dicarboxylate[ No CAS ]
tert-butyl (2S,5S)-2-(9-{2-[(2S,5S)-1-(tert-butoxycarbonyl)-4-(methoxymethyl)pyrrolidin-2-yl]-1H-imidazol-5-yl}-1,4,5,11-tetrahydroisochromeno[4′,3′:6,7]naphtho[1,2-d]imidazol-2-yl)-5-methylpyrrolidine-1-carboxylate[ No CAS ]






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping