|
|
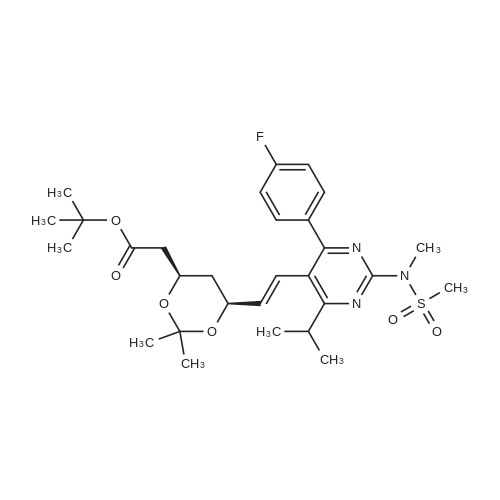
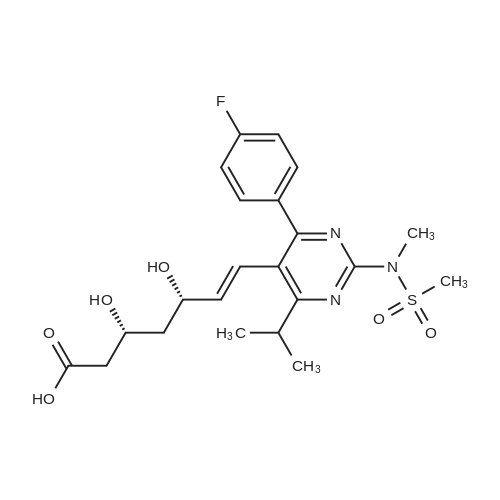
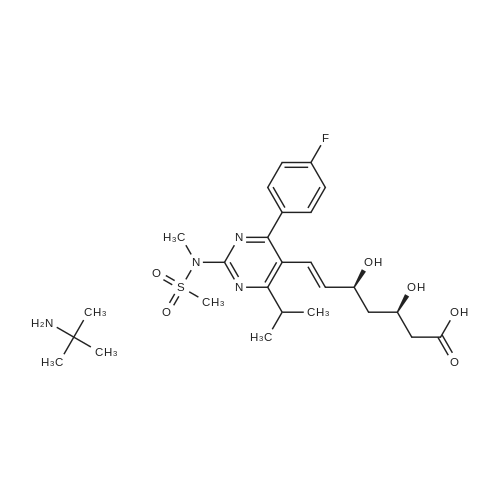
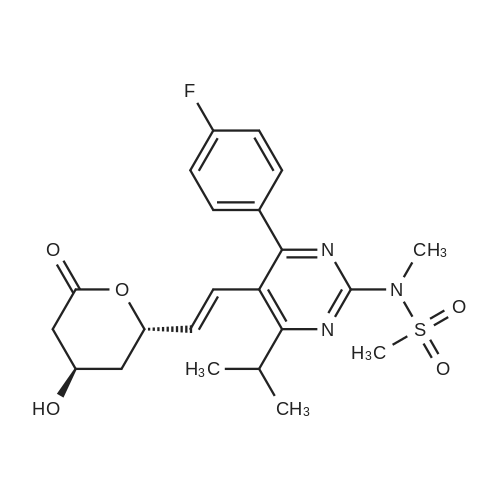
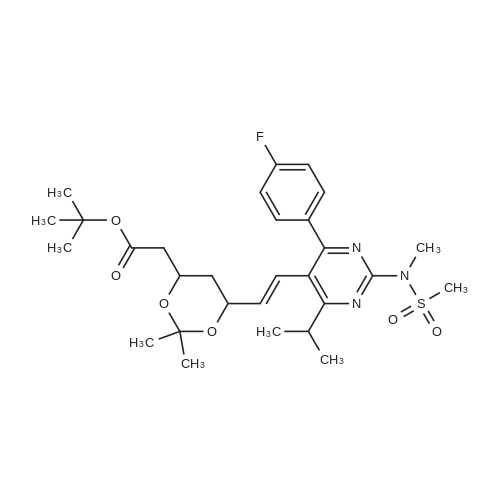
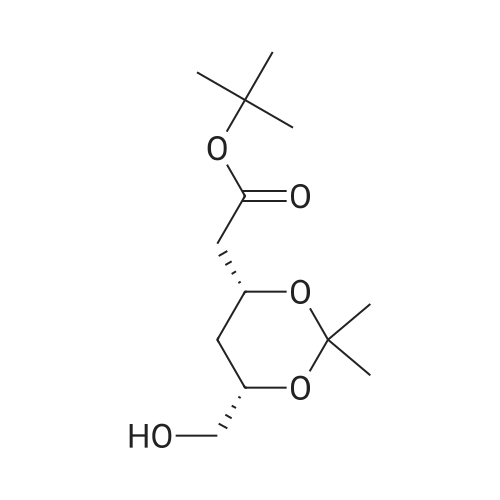
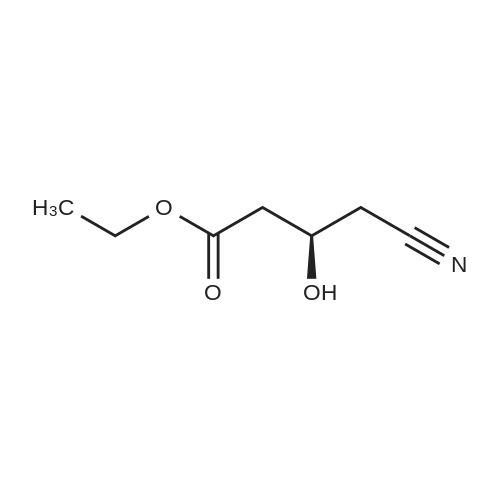
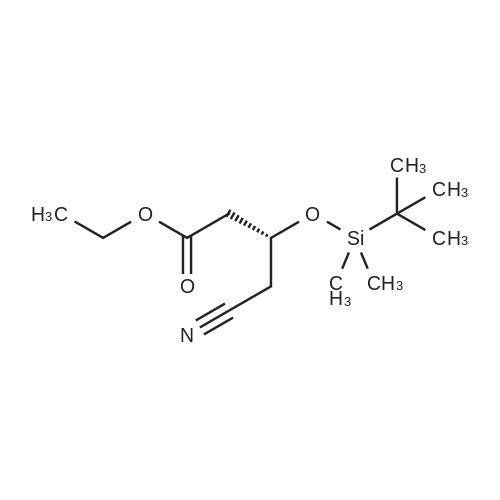
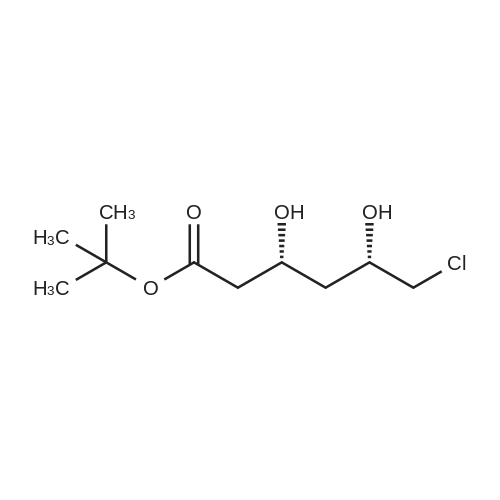
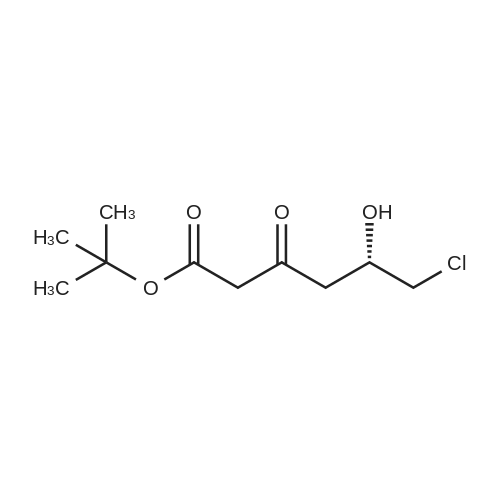
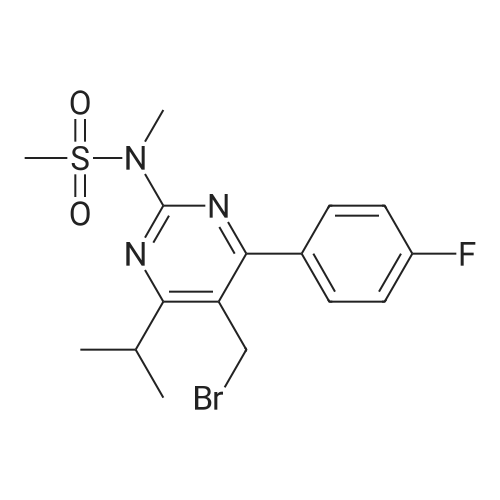
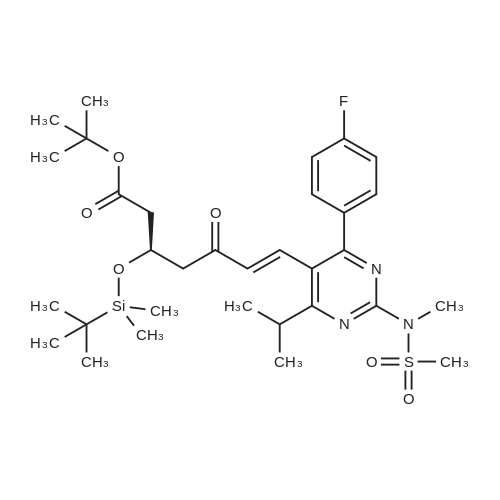
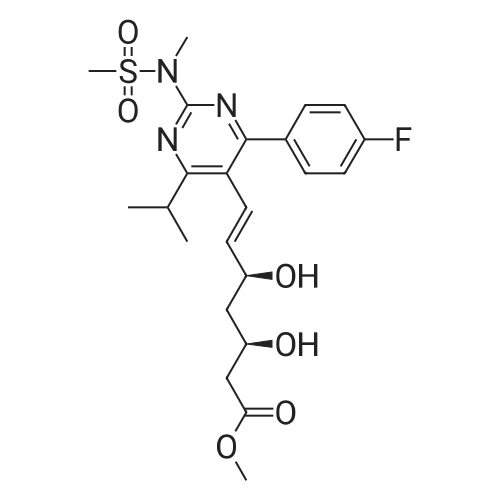
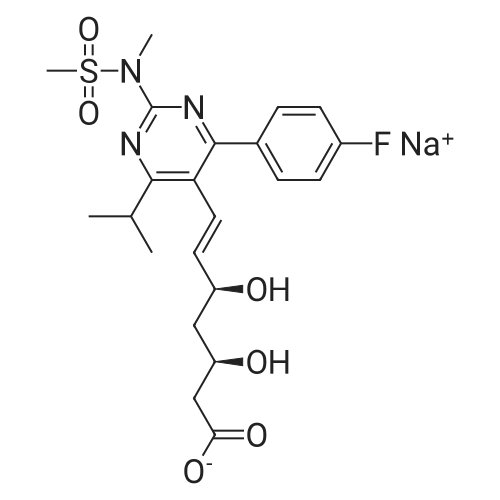
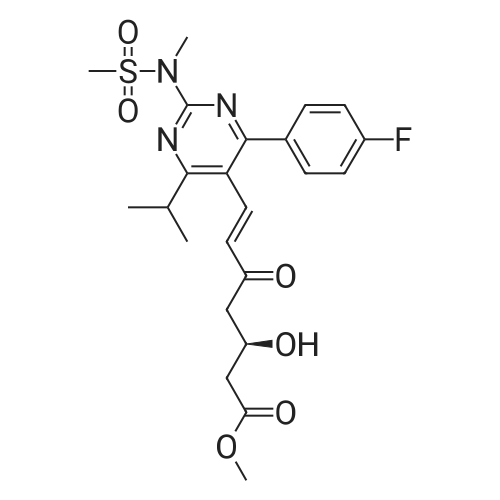
Example 7; General procedure for preparing isolated ammonium salts of rosuvastatin; 5 g of terf-butyl ester of rosuvastatin 1.75 ml of 8 M NaOH 25 ml of demineralized water 10 ml of tetrahydrofuran EPO <DP n="19"/>The reactants and the solvents are stirred from 50 to 55C for 1 hour. The solution formed is then allowed to cool to room temperature and washed with 50 ml methylcyclohexane yielding 33 ml of aqueous solution of sodium salt of rosuvastatin.To 33 ml of sodium rosuvastatinate solution prepared in the above described experiment is added 1.3 ml 85% phosphoric acid, previously dissolved in 5 ml of water. Reaction mixture is extracted with 40 ml of /so-butyl acetate. Organic layer is separated off and dried with 5 g of anhydrous magnesium sulphate. Drying agent is filtered off and washed with 10 ml /so-butyl acetate obtaining 52 ml of filtrate containing rosuvastatinic acid, which is divided into smaller portions for preparing various ammonium salts.To 5 ml of the obtained solution 1.5 equivalents of appropriate amine and 5 ml tert-butyl methyl ether are added. Rosuvastatin substituted ammonium salt is filtered off and dried on filter. The following solid salts are prepared:cyclohexylammonium salt of rosuvastatin: 0.45 g, 99.71% area by HPLC;1H-NMR: (CD3OD): 1.10 - 1.45 (12H,m), 1.31 (6H,d, J=7Hz), 1.48 - 1.56 (1H,m), 1.62 - 1.72 (1 H,m), 1.80 - 1.87 (2H,m), 1.97 - 2.03 (2H,m), 2.25 (1H,dd, J1=MHz, J2=7.6Hz), 2.34 (1H,dd, J1=MHz1 J2=4.9Hz), 2.98 - 3.09 (1 H,m), 3.51 (1H,h, J=7Hz), 3.52 (3H,s), 3.54 (3H,s), 3.92 - 3.97 (1H,m), 4.33 - 4.40 (1H,m), 5.56 (1 H,dd, J1=^Hz, J2=6Hz), 6.62 (1H,dd, J1=^Hz, J2=1.2Hz), 7.14 - 7.22 (2H,m), 7.69 - 7.75 (1H,m);dicyclohexylammonim salt of rosuvastatin: 0.35 g, 99.82% area;1H-NMR: (CD3OD): 1.12 - 1.76 (20H,m), 1.29 (d, J=7Hz), 1.48 - 1.56 (1H,m), 1.62 - 1.72 (1H,m), 1.83 - 1.92 (4H,m), 2.01 - 2.09(4H,m), 2.25 (1H,dd, J1=HHz, J2=7,6Hz), 2.34 (1H,dd, J1=MHz, J2=4.9Hz), 3.07 - 3.17 (2H,m), 3.51 (1H,h, J=7Hz), 3.52 (3H,s), 3.54 (3H,s), 3.92 - 3.97 (1H,m), 4.33 - 4.40 (1H,m), 5.56 (1H,dd, J1=IeHz, J2=6Hz), 6.62 (1H,dd, J,=16Hz, J2=1.2Hz), 7.14 - 7.22 (2H,m), 7.69 - 7,75 (2H,m);pyrrolidinium salt of rosuvastatin: 0.28 g, 99.71% area,1H-NMR: (CD3OD): 1.29 (6H,d, J=7Hz), 1.48 - 1.56 (1H,m), 1.62 - 1.72 (1H,m), 1.96 - 2.01 (4H,m), 2.25 (1H,dd, J1=MHz, J2=7,6Hz), 2.34 (1H,dd, J1=MHz, J2=4,9Hz), 3.20 - 3.25 (4H,m), 3,51 (1H,h, J=7Hz), 3.52 (3H,s), 3.54 (3H,s), 3.92 - 3.97 (1 H,m), 4.33 - 4.40 EPO <DP n="20"/>(1 H,m), 5.56 (1H,dd, J1=IeHz, J2=6Hz), 6.62 (1 H,dd, J1=IeHz, J2=1 ,2Hz), 7.14 - 7.22 (2H,m), 7.69 - 7.75 (2H,m);piperidinium salt of rosuvastatin: 0.28 g, 99.77% area;1H-NMR: (CD3OD): 1.29 (6H,d, J=7Hz), 1.48 - 1.56 (1H,m), 1.62 - 1.81 (7H,m), 2.25 (1 H,dd, J1=MHz, J2=7.6Hz), 2,34 (1 H,dd, J1=MHz, J2=4,9Hz), 3.09 - 3.13 (4H,m), 3.51 (1 H,h, J=7Hz), 3.52 (3H,s), 3.54 (3H,s), 3.92 - 3.97 (1H,m), 4.33 - 4.40 (1 H,m), 5.56 (1 H,dd, J1=IeHz, J2=6Hz), 6.62 (1H,dd, J1=^Hz, J2=1.2Hz), 7.14 - 7,22 (1H,m), 7.69 - 7.75 (2H,m);morpholinium salt of rosuvastatin: 0.30 g, 99.51% area;1H-NMR: (CD3OD): 1.29 (6H,d, J=7Hz), 1.49 - 1.57 (1 H,m), 1.62 - 1.72 (1H,m), 2.25 (1 H,dd, J1=MHz, J2=7.6Hz), 2.34 (1 H,dd, J1=HHz, J2=4.9Hz), 3.12 - 3.16 (4H,m), 3.51 (1 H,h, J=7Hz), 3.52 (3H,s), 3.53 (3H,s), 3.81 - 3.,85 (4H,m), 3.92 - 4.00 (1H,m), 4.33 - 4.40 (1H,m), 5.57 (1H,dd, J1=IeHz, J2=6Hz), 6.62 (1H,dd, J1=IeHz, J2=1.2Hz), 7.14 - 7.22 (2H,m), 7.69 - 7.75 (1H,m);1-adamantylammonium salt of rosuvastatin: 0.66 g, 99.75% area;1H-NMR: (CD3OD): 1.29 (6H,d, J=7Hz), 1.48 - 1.56 (1H,m), 1.62 - 1.85 (16H,m), 2.15 (3H,s (broad)), 2.25 (1 H,dd, J1=HHz, J2=7,6Hz), 2.34 (1H,dd, J1=UHz, J2=4.9Hz), 3.51 (1 H,h, J=7Hz), 3.52 (3H,s), 3.54 (3H,s), 3.92 - 3.97 (1H,m), 4.33 - 4.40 (1 H,m), 5.56 (1H,dd, J1=IeHz, J2=6Hz), 6.62 (1 H,dd, J1=^Hz, J2=1.2Hz), 7.14 - 7.22 (2H,m), 7.69 - 7.75 (1 H,m).; Example 8; Preparation of N-cyclohexylammonium salt of rosuvastatin; 1O g tert-butyl ester of rosuvastatin3.5 ml 8 M NaOH50 ml demineralized water20 ml tetrahydrofuran EPO <DP n="21"/>The reactants and the solvents are stirred from 50 to 55C for 1 hour. The solution formed is then allowed to cool to room temperature and washed with 100 ml methylcyclohexane yielding 66 ml of aqueous solution of sodium rosuvastatinate.To 33 ml of the obtained solution is added 1.3 ml 85% phosphoric acid in 5 ml demineralized water. Rosuvastatinic acid is extracted with 40 ml /so-propyl acetate. 4.7 g of anhydrous magnesium sulphate and 0.5 g charcoal is added to organic phase and suspension is stirred for 45 min. Magnesium sulphate and charcoal are filtered off yielding 41 ml of filtrate.16 ml of the filtrate is separated and treated by addition of 0.5 ml of cyclohexylamine in 8 ml of /so-propyl acetate during stirring and rosuvastatin cyclohexylammonium salt precipitate instantaneously as white solid. It is separated by filtration, precipitate is washed on the filter with 10 ml of /so-propyl acetate and dried on the filter yielding 1.34 g of the de... |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping