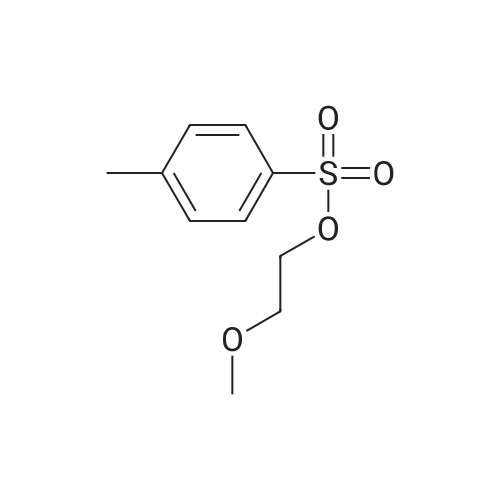
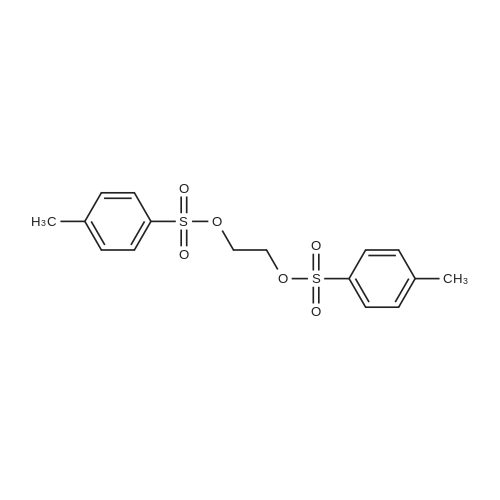
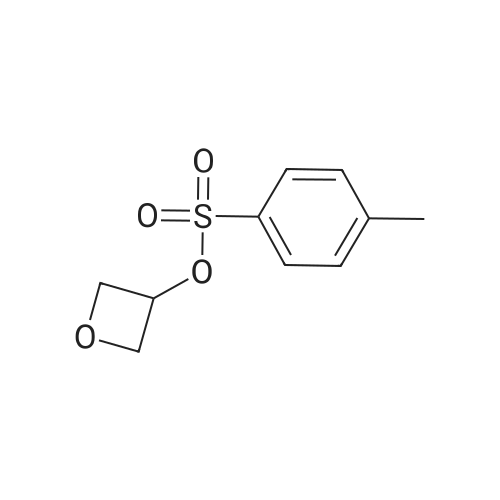
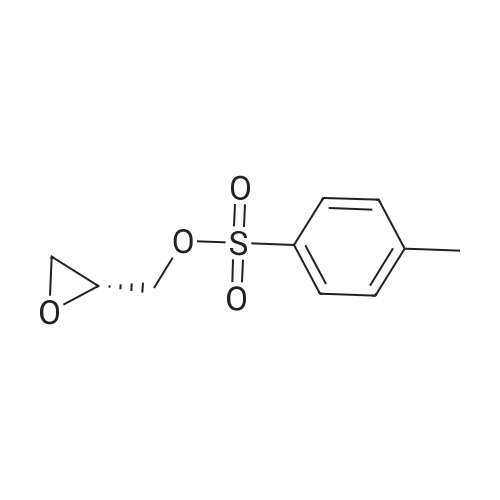
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
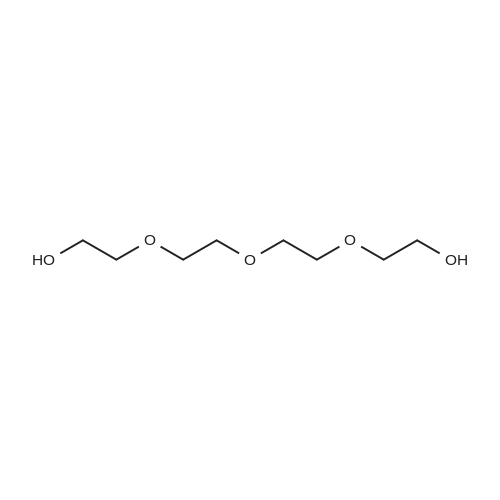
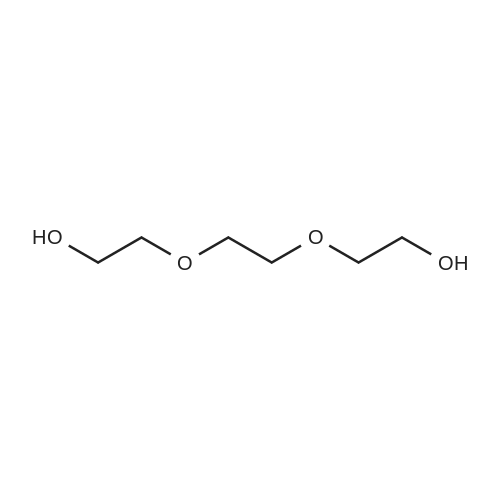
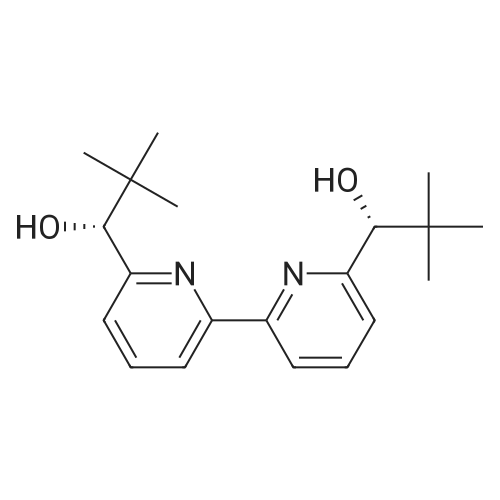
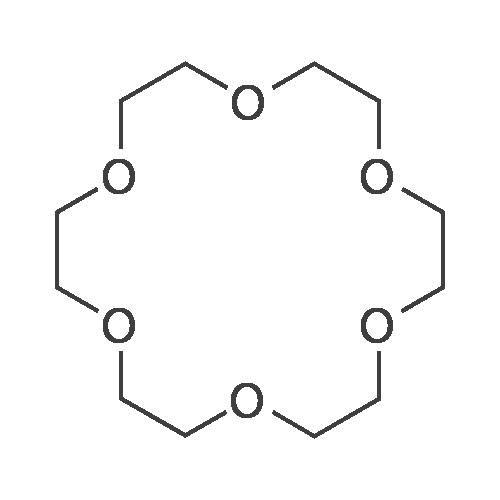
A 3 L, three-neck round bottom flask was charged with a magnetic stir bar, 1.50 L tBuOH, andKOtBu (30.7 g, 274 mmol). The mixture was warmed to 40°C stirred for 30 min to dissolve theKOtBu, and then tetraethylene glycol di(toluene-p-sulphonate) (46 g, 91.5 mmol, in 100 mLdioxane) and diethanolamine (9.6 g, 91.3 mmol, in 100 mL tBuOH) were added dropwise(simultaneously from two different dropping funnels) over the course of 2 h. Note: Slow additionof the solutions is crucial; the slower the addition, the higher the yield After addition, the reactionmixture was stirred for 1h and allowed to cool. The reaction mixture was then filtered twicethrough a Büchner funnel and the solvent was removed on a rotary evaporator. Deionized water(100 mL) was added to the brown, sticky residue and the resulting solution was first extracted withhexane (1×60 mL, hexane phase discarded), followed by CH2Cl2 (5×60 mL). The CH2Cl2 phaseswere collected, dried over MgSO4, and solvent was removed on a rotary evaporator. The resultingdark brown residue was distilled through a bulb-to-bulb distillation under high static vacuum usinga heat gun to yield the product as a colorless liquid (8.4 g, 35percent). The NMR spectra are consistentwith published data. We have found that the tBuOH used in this preparation can be distilled andre-used for subsequent azacrown syntheses despite the presence of small amounts of dioxane(<5percent).
Reference:
[1] Bulletin of the Chemical Society of Japan, 1983, vol. 56, # 1, p. 212 - 218
[2] Polyhedron, 2018, vol. 141, p. 385 - 392
[3] Phosphorus, Sulfur and Silicon and the Related Elements, 2006, vol. 181, # 1, p. 219 - 225
[4] Journal of the Chemical Society, Chemical Communications, 1981, # 10, p. 471 - 472
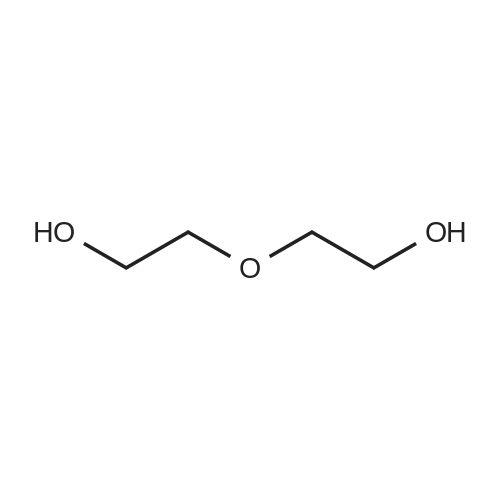
2
[ 37860-51-8 ]
[ 33941-15-0 ]
Reference:
[1] Chemische Berichte, 1984, vol. 117, # 1, p. 234 - 245
[2] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1979, p. 357 - 371
[3] Russian Journal of Organic Chemistry, 2012, vol. 48, # 10, p. 1345 - 1352[4] Zh. Org. Khim., 2012, vol. 48, # 10, p. 1350 - 1357,8
3
[ 112-60-7 ]
[ 98-59-9 ]
[ 37860-51-8 ]
[ 77544-60-6 ]
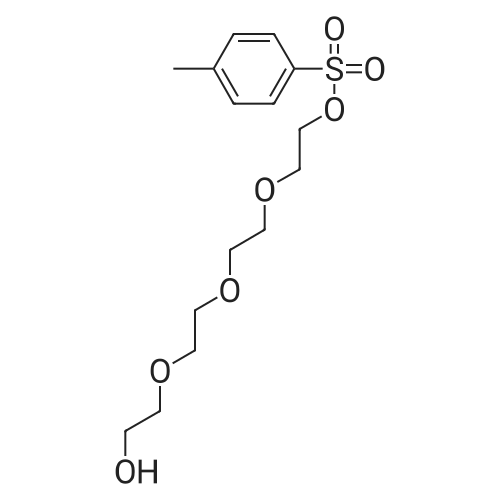
Yield
Reaction Conditions
Operation in experiment
35.9%
With triethylamine In acetonitrile at 0 - 25℃; for 1 h; Cooling with ice
20.0 g of triethylene glycol and 10.2 g of triethylamine were charged into a reaction flask, 300 ml of acetonitrile was added thereto, and the mixture was cooled in an ice bathTo 0-5 ° C, 19.0gTsCl dissolved in 100ml acetonitrile slowly dripping into the solution, 1h drops finished, finished after the drop to 20-25Deg.] C for 12-14 h, the solvent was removed by rotary column chromatography, and eluted with n-hexane: ethyl acetate volume ratio= 4: 1 and 1.5: 1 gradient elution, in n-hexane: ethyl acetate volume ratio = 4: 1 eluentTo give 12.9 g of an oily product (compound thirteen) in a yield of 35.9percent in n-hexane: ethyl acetate volume ratio = 1.5: 1To give 5.4 g of an oil (Compound 14) in a yield of 10.4percent.
Reference:
[1] Organic Letters, 2002, vol. 4, # 14, p. 2329 - 2332
[2] Journal of Organic Chemistry USSR (English Translation), 1990, vol. 26, # 11, p. 2094 - 2100[3] Zhurnal Organicheskoi Khimii, 1990, vol. 26, # 11, p. 2425 - 2433
[4] Patent: CN105541736, 2016, A, . Location in patent: Paragraph 0044; 0052
[5] Journal of the Chemical Society, Perkin Transactions 2, 2001, # 9, p. 1573 - 1584
[6] Patent: WO2009/108484, 2009, A1, . Location in patent: Page/Page column 22; 23
[7] Patent: US2012/4423, 2012, A1, . Location in patent: Page/Page column 10
4
[ 112-60-7 ]
[ 98-59-9 ]
[ 37860-51-8 ]
Yield
Reaction Conditions
Operation in experiment
98%
With potassium hydroxide In dichloromethane at 0 - 10℃; for 3 h;
General procedure: Compounds 2a–c were prepared by a modified procedure known from the literature1. Oligo ethylene glycol(37.7 mmol) and p-toluenesulfonyl chloride (14.4 g, 75.4 mmol, 2 equiv) were dissolved in dichloromethane (36 mL)and this solution was cooled to 0 °C. During permanent stirring, milled potassium hydroxide (17 g, 302 mmol, 8equiv) was added and the solution was then stirred for 3 hours while the temperature was kept from 0 to 10 °C. This reaction was monitored by TLC (UV detection). After warming up to room temperature the reaction mixture was diluted by chloroform and extracted with water (3 × 1:1). Chloroform extracts were collected, dried over magnesium sulfate overnight, filtered and solvents were evaporated under reduced pressure.
98%
With potassium hydroxide In dichloromethane for 1.5 h; Cooling with ice
The 10mL (0.068mol) of tetraethylene glycol,300ml dichloromethane,25.8g (0.136mol)MethylBenzenesulfonyl chloride,Added to a 500ml round-bottomed flask,Under ice-cooling,Was slowly added 30.4g (0.544mol) of potassium hydroxide,1.5h the reaction is substantially complete.The reaction solution was washed three times with water (3 × 100ml),The organic layer was collected,Dried over anhydrous sodium sulfate.filter,Spin dry solvent,To give the crude product as an oil.Column chromatography (petroleum ether: ethyl acetate = 1:4) to give a pale yellow oily liquid 34g.The yield was 98percent.
95%
With 1H-imidazole; triethylamine In dichloromethane at 0 - 20℃;
0-Ditosylate Tetraethylne Glycol (32). Compound 2 (5.00 mL, 28.95 mmol) was added dropwise to a stirred solution of /Moluenesulfonyl chloride (12.15 g, 63.73 mmol), imidazole (0.04 g, 0.65 mmol) and Et3N (16.15 mL, 108.30 mmol) in CH2Cl2 (30 mL) at 0 0C. The reaction mixture was allowed to stir at room temperature (12 h), and then Et2O (80 mL) was added and the reaction washed with H2O (5 x 60 mL). The organic layer was dried (Na2SO4) and evaporated in vacuo, and then the crude product was purified by column chromatography (SiO2; 3/1 EtOAc/hexanes) to yield 13.81 g (95percent) of 32 as a clear oil (Busch et al., 2002): Rf = 0.65 (3/1 EtOAc/hexanes); 1H NMR (CDCl3) δ 2.44 (s, 2 CH3), 3.53-3.59 (m, 2 OCH2CH2O), 3.68 (t, J = 4.8 Hz, OCH2CH2OTs), 4.15 (t, J = 4.8 Hz, OCH2CH2OTs), 7.34 (d, J= 8.4 Hz, 4 ArH), 7.79 (d, J= 8.4 Hz, 4 ArH); 13C NMR (CDCl3) δ 21.7 (CH3), 68.8 , 69.4, 70.6, 70.8 (OCH2CH2O, OCH2CH2OTs), 128.1, 130.1, 133.1, 145.0 (C6H4).
89%
Stage #1: With potassium hydroxide In dichloromethane at 0 - 20℃; Stage #2: at 20℃; for 10 h;
General procedure: KOH (20 g, 356.5 mmol for 9; and 10 g, 181.5 mmol for 10–11) was added to 300 mL of a CH2Cl2 solution containing the oligoethylene glycol (10 g,94 mmol for 9, 66.6 mmol for 10, and 51.5 mmol for 11) at 0 °C. After stirring for 30 min at room temperature, TsCl (45 g, 236 mmol for 9–11) was added and the resulting solution was stirred for 10 h at room temperature. The resulting solution was filtered and then washed with aq. K2CO3. The organic layer was dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to silicagel column chromatography (EtOAc:Hex = 1:1) to yield 35 g of 9 (90percent), 27 g of 10 (88percent), and 23 g of 11 (89percent).
74.4%
With triethylamine In dichloromethane at 0 - 20℃;
To a solution of 2-{2-[2-(2-hydroxyethoxy)ethoxy]ethoxy}ethan-l-ol (13 g, 67.1 mmol), Et3N (13 mL) in DCM (100 mL) was added TsCl (25.5 g, 134.2 mmol) in portions at 0°C. The resulting mixture was allowed to stir at room temperature overnight. TLC showed the reaction completed. The mixture was partitioned between DCM and H20. The organic phase was washed with brine, dried over magnesium sulfate and evaporated to dryness. The crude product was purified by silica gel chromatography using with 10-20percent EtOAc in hexane as eluent to afford the desired compound (25.0 g, 74.4percent).
73%
With triethylamine In dichloromethane at 25℃; for 12 h;
Tetraethylene glycol (3.88 g, 20 mmol) and triethylamine (TEA) (8.0 mL) were dissolved in dichloromethane (60 mL). Then, tosyl-chloride (9.50 g, 50 mmol) was added in one portion. The resulting mixture was stirred at 25 oC for 12 h. After washing with KHSO4 (1 M, 40 mL) and NaHCO3 (5percent, 40 mL), respectively and drying over Na2SO4, the crude product was obtained by evaporation and subsequently purification by column chromatography over silica gel (dichloromethane) to obtain the target product 7 as a colorless oil (7.5 g, 14.6 mmol, 73percent).
73%
With potassium hydroxide In tetrahydrofuran; water at 0 - 20℃; for 18 h; Inert atmosphere; Glovebox
A 1 L round bottom flask, charged with a magnetic stir bar, THF (400 mL), ptoluenesulfonylchloride(165.6 g, 0.868 mol) and tetraethylene glycol (56.4 g, 0.290 mol). Theflask was cooled to 0°C and a solution of KOH (104.8 g in 100 mL water, 1.87 mol) was addeddropwise through a dropping funnel over the course of 3 h. After addition, the mixture was allowedto stand at ambient temperature for 18 h. At this stage, the reaction mixture had generated a whiteprecipitate. The reaction was poured into 600 mL of water/CH2Cl2 mixture (70/30). The two-phasesolvent system was separated, and the aqueous phase was extracted four times with 100 mLCH2Cl2. Note: Before extraction the aqueous phase is denser than CH2Cl2. The organic phaseswere combined, dried over MgSO4 and solvent was evaporated under reduced pressure to yield acolorless oil. (53.3 g, 73percent). The NMR spectra are consistent with published data.
58%
With triethylamine In dichloromethane at 0 - 20℃; for 6 h;
Under room temperature, the methyl sulfonyl chloride (29.5g, 155mmol) by adding 2,2 ' - ((oxygen dihydrogenmethylenebisphosphonate (ethane -2,1-diyl)) double (oxy)) b ethanol 20a (10.0g, 51 . 5mmol) in dichloromethane (150 ml) solution, in 0 °C next, add triethylamine (32.6 ml, 232mmol), stirring 5 minutes, to the reaction room temperature for 6 hours. Added to the reaction solution 100 ml water quenching reaction, separation, of sequentially separated organic phase is washed with water (100 ml), saturated salt water washing (100 ml), then dried with anhydrous sodium sulfate, filtered, filtrate concentrated. The resulting residue is purified by silica gel column chromatography [petroleum ether/ethyl acetate (v/v)=10/1], to obtain title compound 20b (15g, pale yellow liquid), yield: 58.0percent.
43%
With dmap In dichloromethane at 5 - 20℃;
Into a 250-mL 3-necked round-bottom flask, was placed a solution of tetraethylene glycol (10 g, 51 55 mmol, 1 00 equiv) in DCM (100 mL) This was followed by the addition of a solution of 4-methylbenzene-l-sulfonyl chloride (21 4 g, 112 63 mmol, 2 20 equiv) in DCM (50 mL) dropwise with stirring at 5°C To this was added N,N-dimethylpyridin-4-amine (15 7 g, 128 69 mmol, 2 50 equiv) The resulting solution was stirred for 2 h at room temperature at which time it was diluted with 100 mL of water The resulting solution was extracted with 3x100 mL of DCM and the organic layers combined The resulting mixture was washed with 1x100 mL of brine and then concentrated under vacuum The residue was applied onto a silica gel column and eluted with ethyl acetate/petroleum ether (1 2) to afford H g (43percent) of the title compound as white oil.
21.4 g
at 0 - 10℃; for 2 h;
Example 4 19.1 g paratoluensulfonyl chloride and 40 mL pyridine are added in 250 mL three-neck flask, and cooled to 0° C. 9.7 g HO-PEG(n=4)-OH is mixed uniformly with 20 mL pyridine, and then dripped in the three-neck flash, and the temperature is controlled between 0 and 10° C. Stirring and reaction is continued at this temperature for two hours. TLC monitors that the reaction is complete. 300 mL cold water and 60 mL concentrated hydrochloric acid are added in the reaction liquid and slowly stirred for half an hour, then the reaction liquid is transferred into a 500 mL separating funnel, acetic ether is added to extract twice (300 mL+200 mL). The organic layers are merged and washed by water to neutrality, dried by anhydrous sodium sulfate for two hours. A rotary evaporator evaporates out solvent to obtain 21.4 g viscous liquid, which will be directly used for next reaction.
Reference:
[1] Bioorganic and Medicinal Chemistry, 2007, vol. 15, # 14, p. 4841 - 4856
[2] Inorganica Chimica Acta, 2011, vol. 365, # 1, p. 38 - 48
[3] Journal of Medicinal Chemistry, 2016, vol. 59, # 17, p. 7840 - 7855
[4] Journal of Organic Chemistry, 1999, vol. 64, # 18, p. 6870 - 6873
[5] Journal of the American Chemical Society, 2008, vol. 130, # 33, p. 10882 - 10883
[6] Journal of Organic Chemistry, 1992, vol. 57, # 24, p. 6678 - 6680
[7] Beilstein Journal of Organic Chemistry, 2016, vol. 12, p. 349 - 352
[8] Patent: CN105384745, 2016, A, . Location in patent: Paragraph 0084; 0085; 0086
[9] Journal of Organic Chemistry, 1999, vol. 64, # 3, p. 721 - 725
[10] Phosphorus, Sulfur and Silicon and the Related Elements, 2006, vol. 181, # 1, p. 219 - 225
[11] Russian Journal of Organic Chemistry, 2012, vol. 48, # 10, p. 1345 - 1352[12] Zh. Org. Khim., 2012, vol. 48, # 10, p. 1350 - 1357,8
[13] Chemical Communications, 2013, vol. 49, # 81, p. 9311 - 9313
[14] Organic and Biomolecular Chemistry, 2017, vol. 15, # 17, p. 3681 - 3705
[15] Collection of Czechoslovak Chemical Communications, 1987, vol. 52, # 8, p. 2057 - 2060
[16] Patent: WO2010/14236, 2010, A2, . Location in patent: Page/Page column 34
[17] Chemistry - A European Journal, 2014, vol. 20, # 40, p. 12894 - 12900
[18] Langmuir, 2015, vol. 31, # 49, p. 13410 - 13419
[19] Chemical Communications, 2018, vol. 54, # 10, p. 1249 - 1252
[20] Organic Letters, 2012, vol. 14, # 18, p. 4866 - 4869
[21] Bulletin of the Chemical Society of Japan, 1990, vol. 63, # 4, p. 1260 - 1262
[22] Journal of Organic Chemistry, 1983, vol. 48, # 25, p. 4864 - 4869
[23] Bulletin of the Chemical Society of Japan, 1990, vol. 63, # 4, p. 1260 - 1262
[24] Chemical Communications, 2016, vol. 52, # 45, p. 7310 - 7313
[25] Journal of the American Chemical Society, 1994, vol. 116, # 8, p. 3192 - 3196
[26] Tetrahedron, 2005, vol. 61, # 33, p. 7924 - 7930
[27] Journal of Physical Organic Chemistry, 2009, vol. 22, # 1, p. 1 - 8
[28] Tetrahedron, 1999, vol. 55, # 5, p. 1491 - 1504
[29] Tetrahedron, 2007, vol. 63, # 23, p. 5083 - 5087
[30] Organic Letters, 2011, vol. 13, # 22, p. 6006 - 6009
[31] Journal of Porphyrins and Phthalocyanines, 2013, vol. 17, # 1-2, p. 104 - 117
[32] Chinese Journal of Chemistry, 2013, vol. 31, # 5, p. 607 - 611
[33] Bulletin of the Korean Chemical Society, 2015, vol. 36, # 6, p. 1654 - 1660
[34] Journal of Organic Chemistry, 1995, vol. 60, # 24, p. 7984 - 7996
[35] ChemMedChem, 2010, vol. 5, # 5, p. 777 - 789
[36] Tetrahedron Asymmetry, 2005, vol. 16, # 12, p. 2119 - 2124
[37] Journal of the American Chemical Society, 2011, vol. 133, # 8, p. 2749 - 2759
[38] Journal of Organic Chemistry, 1984, vol. 49, p. 1408 - 1412
[39] Patent: WO2014/9429, 2014, A1, . Location in patent: Sheet 13/23
[40] Tetrahedron Letters, 1995, vol. 36, # 25, p. 4377 - 4380
[41] Canadian Journal of Chemistry, 1997, vol. 75, # 11, p. 1472 - 1482
[42] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1994, # 4, p. 447 - 460
[43] Organic and Biomolecular Chemistry, 2006, vol. 4, # 11, p. 2082 - 2087
[44] Journal of Medicinal Chemistry, 2012, vol. 55, # 7, p. 2981 - 2993
[45] Organometallics, 2014, vol. 33, # 16, p. 4323 - 4335
[46] Chemical Communications, 2015, vol. 51, # 8, p. 1524 - 1527
[47] Journal of Heterocyclic Chemistry, 1998, vol. 35, # 1, p. 209 - 215
[48] MedChemComm, 2013, vol. 4, # 10, p. 1400 - 1404
[49] Chemical Communications, 2013, vol. 49, # 86, p. 10097 - 10099
[50] Chemistry - A European Journal, 2012, vol. 18, # 52, p. 16689 - 16697
[51] Chemical Communications, 2013, vol. 49, # 22, p. 2195 - 2197
[52] Synlett, 2013, vol. 24, # 12, p. 1523 - 1528
[53] Patent: WO2017/30814, 2017, A1, . Location in patent: Paragraph 00240
[54] Molecular Crystals and Liquid Crystals Science and Technology Section A: Molecular Crystals and Liquid Crystals, 2001, vol. 365, p. 427 - 437
[55] Chinese Chemical Letters, 2016, vol. 27, # 11, p. 1655 - 1660
[56] Polyhedron, 2018, vol. 141, p. 385 - 392
[57] Organic Letters, 2010, vol. 12, # 13, p. 3050 - 3053
[58] Bioorganic and Medicinal Chemistry, 1999, vol. 7, # 9, p. 1881 - 1890
[59] Journal of Organic Chemistry, 1999, vol. 64, # 14, p. 5156 - 5161
[60] Patent: CN105461762, 2016, A, . Location in patent: Paragraph 0661; 0663; 0664; 0665; 0666
[61] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1985, p. 607 - 624
[62] Macromolecules, 2004, vol. 37, # 13, p. 4761 - 4769
[63] Journal of the American Chemical Society, 2012, vol. 134, # 1, p. 83 - 86
[64] Patent: WO2010/78449, 2010, A2, . Location in patent: Page/Page column 279
[65] Liebigs Annalen der Chemie, 1983, # 5, p. 770 - 801
[66] Journal of Organic Chemistry, 1994, vol. 59, # 8, p. 2186 - 2196
[67] Journal of Heterocyclic Chemistry, 1994, vol. 31, # 4, p. 1047 - 1052
[68] Zeitschrift fuer Naturforschung, B: Chemical Sciences, 2002, vol. 57, # 1, p. 107 - 112
[69] Tetrahedron, 2003, vol. 59, # 50, p. 9939 - 9950
[70] Organic and Biomolecular Chemistry, 2005, vol. 3, # 12, p. 2255 - 2261
[71] Tetrahedron Letters, 2006, vol. 47, # 48, p. 8563 - 8566
[72] Journal of Organic Chemistry, 2006, vol. 71, # 26, p. 9884 - 9886
[73] Journal of the American Chemical Society, 2010, vol. 132, # 2, p. 656 - 666
[74] Chemistry Letters, 2010, vol. 39, # 2, p. 100 - 101
[75] Journal of Molecular Structure, 2010, vol. 982, # 1-3, p. 162 - 168
[76] Synthesis, 2012, vol. 44, # 5, p. 717 - 722
[77] Chemical Communications, 2012, vol. 48, # 45, p. 5650 - 5652
[78] Dalton Transactions, 2012, vol. 41, # 29, p. 8767 - 8769
[79] Langmuir, 2012, vol. 28, # 33, p. 12357 - 12363
[80] Journal of Materials Chemistry, 2012, vol. 22, # 33, p. 16927 - 16932
[81] Chemical Communications, 2013, vol. 49, # 38, p. 3982 - 3984
[82] Chinese Chemical Letters, 2014, vol. 25, # 12, p. 1643 - 1647
[83] Letters in Organic Chemistry, 2015, vol. 12, # 2, p. 85 - 90
[84] Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 7, p. 1488 - 1494
[85] Patent: US2016/82117, 2016, A1, . Location in patent: Paragraph 0052
[86] Tetrahedron, 2016, vol. 72, # 38, p. 5744 - 5748
[87] Tetrahedron, 2014, vol. 70, # 50, p. 9545 - 9553
[88] Patent: KR101508710, 2015, B1, . Location in patent: Paragraph 0023-0032
[89] Journal of Medicinal Chemistry, 2017, vol. 60, # 7, p. 2890 - 2907
[90] New Journal of Chemistry, 2018, vol. 42, # 14, p. 11324 - 11333
[91] Tetrahedron, 2018, vol. 74, # 37, p. 4777 - 4789
[92] Journal of Heterocyclic Chemistry, 2018, vol. 55, # 9, p. 2172 - 2177
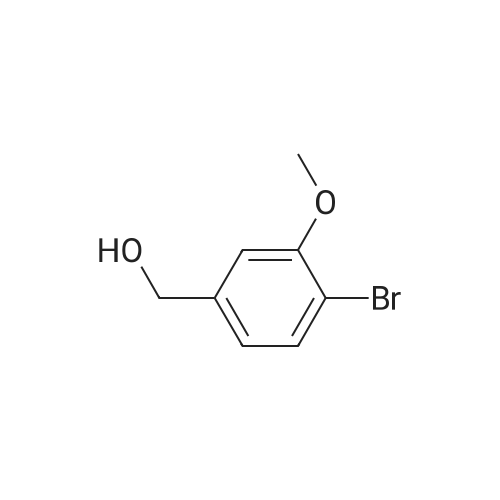
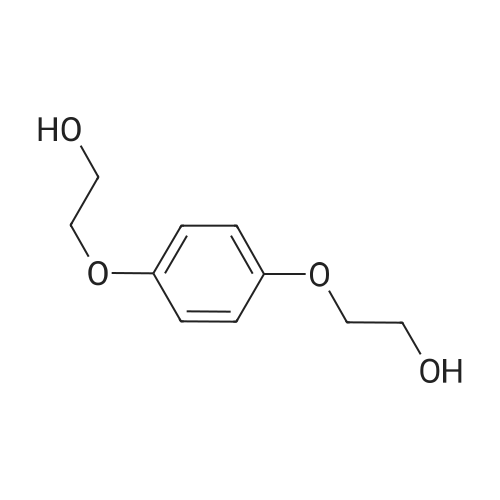
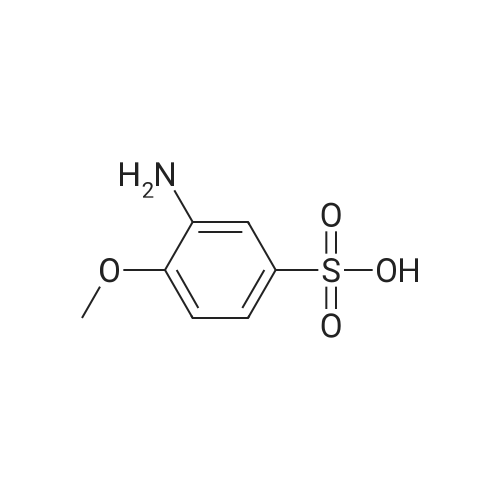
5
[ 98-59-9 ]
[ 112-27-6 ]
[ 37860-51-8 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2014, vol. 12, # 9, p. 1430 - 1439
6
[ 37860-51-8 ]
[ 101187-40-0 ]
Reference:
[1] Journal of the American Chemical Society, 2012, vol. 134, # 1, p. 83 - 86
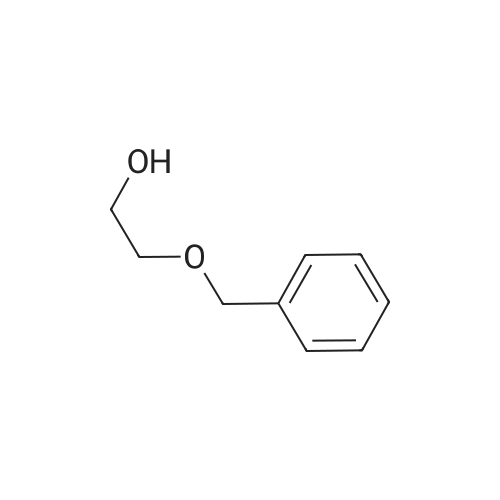
Stage #1: 2-Benzyloxyethanol With sodium hydride In tetrahydrofuran for 1h; Cooling with ice;
Stage #2: tetraethylene glycol di(p-toluenesulfonate) In tetrahydrofuran at 25℃;
1 Synthesis of compound 8:
General procedure: Monobenzyl glycol (40 mmol) was dissolved in anhydrous THF (250 mL), and sodium hydride (60%, 4 g, 100 mmol) was added in portions under ice bath.After stirring for 1 hour, a solution of compound 7 (4.58 g, 10 mmol) in THF was added dropwise.The mixture was stirred at room temperature overnight. The reaction was quenched with ice water and concentrated to remove THF.The residue was dissolved in ethyl acetate (200 mL) and washed thoroughly with water.The organic phase was concentrated and purified by column chromatography (DCM/MeOH=50:1) to give the intermediate dibenzyl diol.Then, dibenzyl ethylene glycol was dissolved in an appropriate amount of MeOH, and debenzylation reaction was performed under H2 to obtain compound 8. The yield is 63-76%.
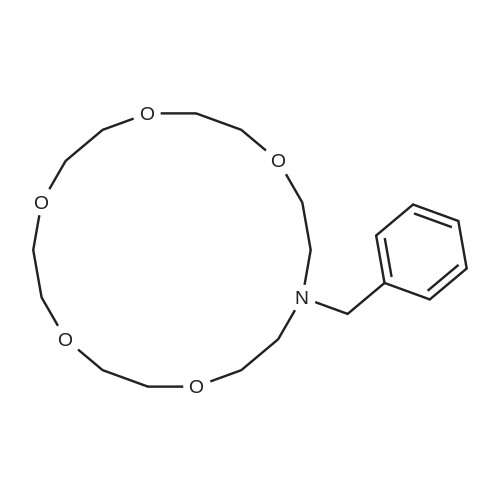
With sodium hydroxide; tetra-(n-butyl)ammonium iodide In toluene for 16h; Heating;
52%
Stage #1: benzene-1,2-diol With sodium hydroxide; tetra-(n-butyl)ammonium iodide In water; toluene at 50 - 60℃; for 0.5h;
Stage #2: tetraethylene glycol di(p-toluenesulfonate) In water; toluene for 16h; Reflux;
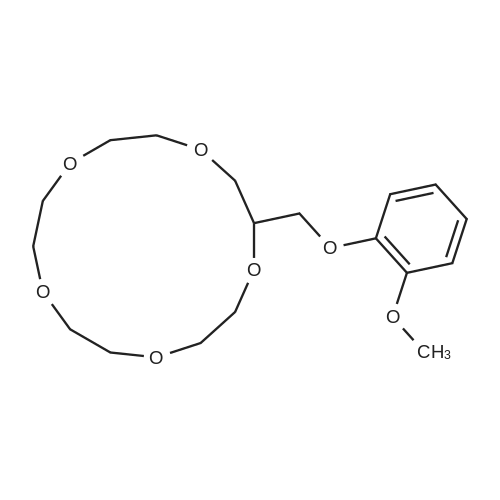
With potassium <i>tert</i>-butylate In 1,4-dioxane; <i>tert</i>-butyl alcohol at 40℃; for 2h;
35%
With potassium <i>tert</i>-butylate; <i>tert</i>-butyl alcohol at 40℃; for 1h; Inert atmosphere; Glovebox;
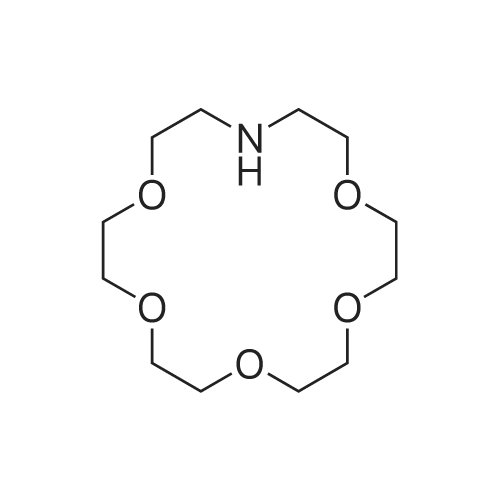
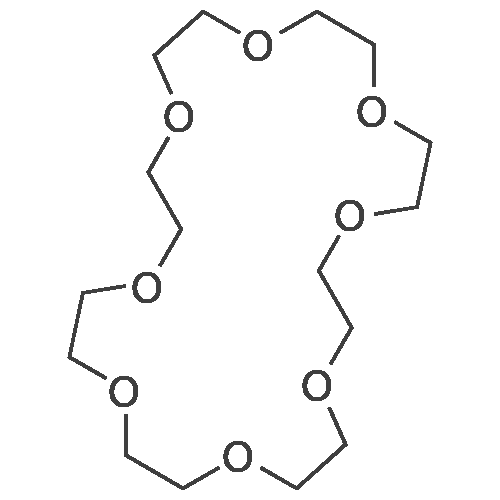
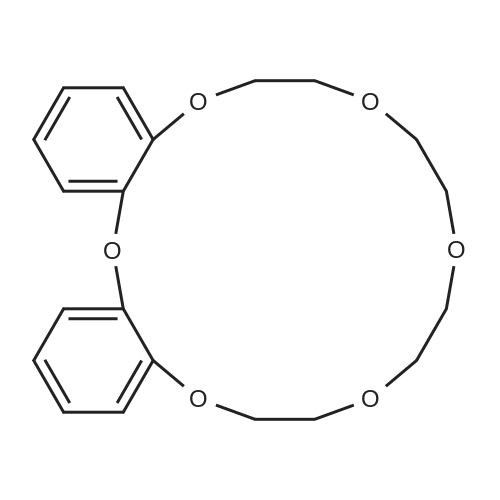
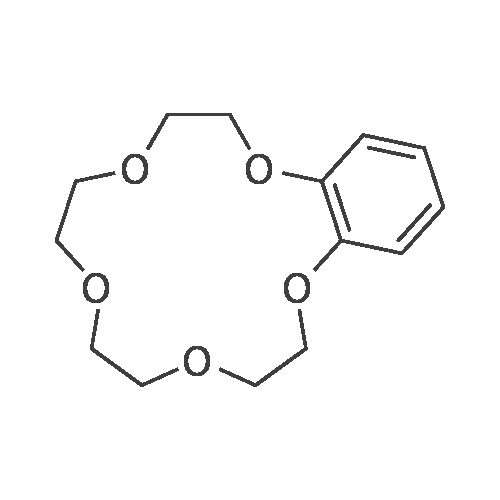
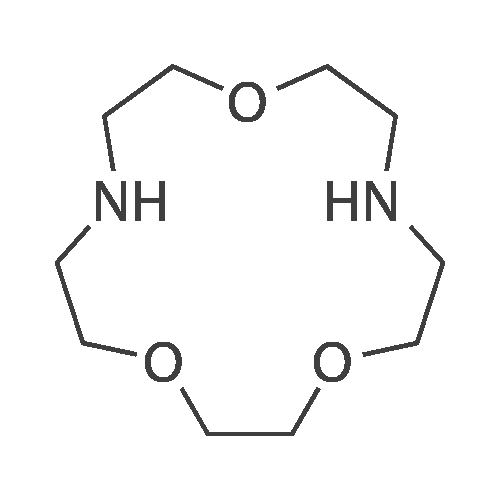
Synthesis of 1-Aza-18-Crown-6 (1b)
A 3 L, three-neck round bottom flask was charged with a magnetic stir bar, 1.50 L tBuOH, andKOtBu (30.7 g, 274 mmol). The mixture was warmed to 40°C stirred for 30 min to dissolve theKOtBu, and then tetraethylene glycol di(toluene-p-sulphonate) (46 g, 91.5 mmol, in 100 mLdioxane) and diethanolamine (9.6 g, 91.3 mmol, in 100 mL tBuOH) were added dropwise(simultaneously from two different dropping funnels) over the course of 2 h. Note: Slow additionof the solutions is crucial; the slower the addition, the higher the yield After addition, the reactionmixture was stirred for 1h and allowed to cool. The reaction mixture was then filtered twicethrough a Büchner funnel and the solvent was removed on a rotary evaporator. Deionized water(100 mL) was added to the brown, sticky residue and the resulting solution was first extracted withhexane (1×60 mL, hexane phase discarded), followed by CH2Cl2 (5×60 mL). The CH2Cl2 phaseswere collected, dried over MgSO4, and solvent was removed on a rotary evaporator. The resultingdark brown residue was distilled through a bulb-to-bulb distillation under high static vacuum usinga heat gun to yield the product as a colorless liquid (8.4 g, 35%). The NMR spectra are consistentwith published data. We have found that the tBuOH used in this preparation can be distilled andre-used for subsequent azacrown syntheses despite the presence of small amounts of dioxane(<5%).
30%
With potassium <i>tert</i>-butylate In 1,4-dioxane; <i>tert</i>-butyl alcohol for 12h;
24%
With sodium t-butanolate In 1,4-dioxane; <i>tert</i>-butyl alcohol at 20 - 40℃; for 1h;
With potassium <i>tert</i>-butylate In <i>tert</i>-butyl alcohol at 40℃; Yield given;
With sodium hexafluorophosphate(V); anhydrous sodium carbonate In acetonitrile for 72h; Inert atmosphere; Reflux;
40%
With caesium fluoride In acetonitrile
26%
With anhydrous sodium carbonate In acetonitrile at 60℃; for 144h;
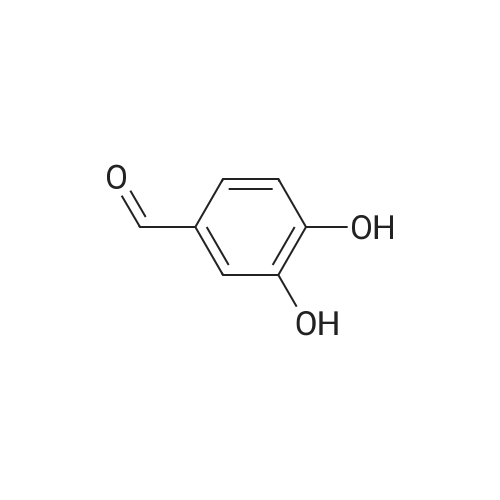
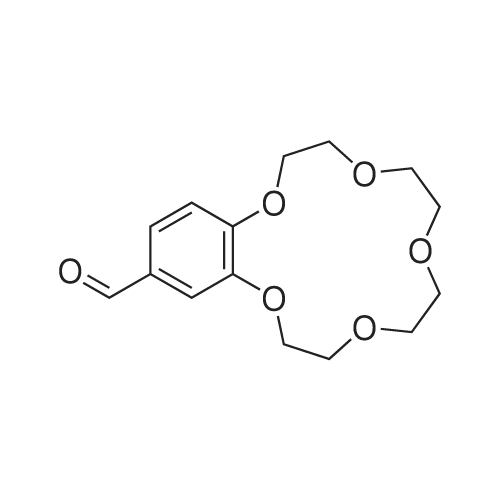
6 Synthesis of aldehyde-benz-15-crown-5-ether
Powdered sodium carbonate (26g, 0.245 mol) was added to a stirred solution of benzaldehyde (13.05g, 0.094 mol) in acetonitrile and ditosylate from example 3 (47.5g, 0.094 mol) . The reaction mixture was stirred at 60 °C for 6 days. Then insoluble inorganic part was filtered off and the solvent was removed under reduced pressure affording colorless oil. The oil was triturated with water (80 mL) containing 6.2 mL of concentrated hydrochloric acid. The mixture was extracted with Et2O/EtOAc (1:2) (4x120 mL) . The organic extracts were combined, and solvents were removed under reduced pressure to yield a viscous oil. The oil was purified by flash chromatography on silica gel (Combiflash, dichloromethane/ isopropanol : 2-3%) to afford white solid. Yield 7.2 g (26%) . iH NMR (CDCI3) , 5 9.82 (s, 1H) , 7.43 (dd, 1H, J = 8.0 Hz, J = 2.0 Hz) , 7.38 (d, 1H, J = 2.0 Hz) , 6.93 (d, 1H, J = 8.0 Hz) , 4.21 - 4.17 (m, 4H) , 3.94 - 3.90 (m, 4H) , 3.79 - 3.73 (m, 8H) , 3.46 (brs, 2H) ppm.
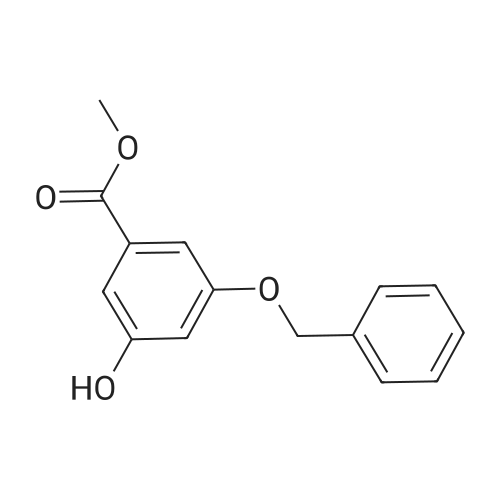
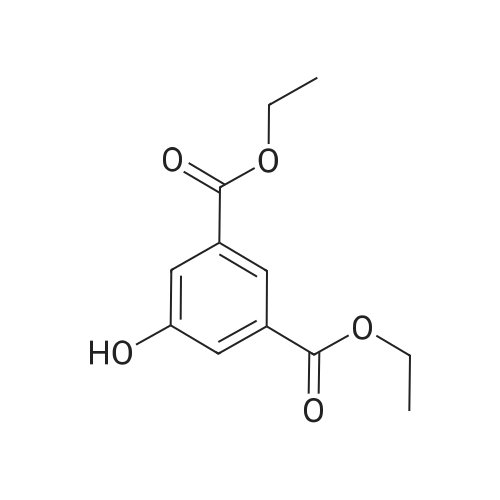
tetra(ethylene glycole) di(3-(methoxycarbonyl)phenyl) ether[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
With potassium carbonate In N,N-dimethyl-formamide at 70℃; for 16h;
4.5. Tetraethylene glycole di(3-methoxycarbonyl phenyl) ether (6a)
Tetraethylene glycol di(p-toluenesulfonate) (0.1 mL, 0.25 mmol) was added dropwise to a solution of methyl 3-hydroxybenzoate (229 mg, 1.5 mmol) and K2CO3 (415 mg, 3.0 mmol) in DMF (4.6 mL). After being stirred at 70 °C for 16 h the mixture was treated with a saturated solution of NaHCO3 and subsequently extracted with EtOAc. The organic layer was washed with brine and dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash chromatography (hexane/EtOAc 1:1) to give 6a in 90% yield (104 mg). EI-MS: m/z 463 (M+); 1H NMR: (CDCl3, 360 MHz) δ (ppm): 3.67-3.75 (m, 8H), 3.84-3.89 (m, 4H), 3.90 (s, 6H), 4.14-4.19 (m, 4H,), 7.09-7.13 (m, 2H), 7.32 (t, J = 8.0 Hz, 2H,); 7.55-7.58 (m, 2H), 7.60-7.64 (m, 2H); 13C NMR: (CDCl3, 90 MHz) δ (ppm): 52.1, 67.7, 69.7, 70.7, 70.9, 114.8, 120.1, 122.2, 129.4, 131.4, 158.8, 167.0; IR: (NaCl) ν (cm-1): 2875, 1723, 1445, 1290, 1230, 1106, 756.
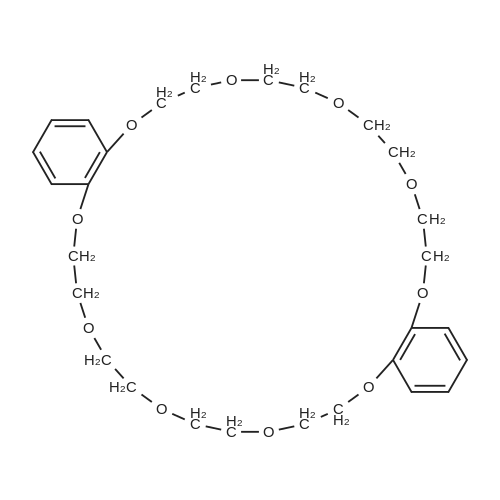
Stage #1: hydroquinone With sodium hydroxide In water; butan-1-ol for 0.5h; Reflux; Inert atmosphere;
Stage #2: tetraethylene glycol di(p-toluenesulfonate) With caesium carbonate In 1,4-dioxane; butan-1-ol for 20h; Reflux;
4.3 Bis(p-phenylene)-34-crown-10 (2)
A solution of hydroquinone (11.1 g, 100 mmol), NaOH (9.10 g, 228 mmol), water (10 mL) and n-butanol (450 mL) was stirred at reflux for 30 min under N2. Tetra(ethylene glycol) ditosylate (50.2 g, 100 mmol) and Cs2CO3 (32.6 g, 100 mmol) in 500 mL of a 3:2 (v:v) mixture of 1,4-dioxane:n-butanol were added. The mixture was refluxed with stirring for 20 h. The cooled mixture was filtered and the filtrate was evaporated to afford a dark solid, which was dissolved in CH2Cl2 (100 mL) and passed through a short silica gel column with ethyl acetate. The eluent solution was evaporated to dryness and subjected to column chromatography on alumina using diethyl ether as an eluent; a colored oil eluted first and then the desired product, which was recrystallized from 3:2 (v:v) toluene:n-hexane as clear, colorless crystals that were dried overnight in vacuo, 3.35 g (12.5%, lit. yield 8% [24] by a similar one-step process), mp 95.0-96.8 °C (lit. mp 93.5-94.0 °C [24], 87-88 °C [25]).
150 mg
With sodium hydroxide In isopropyl alcohol for 24h; Heating;
Multi-step reaction with 3 steps
1.1: NaH / dimethylformamide / 2 h / 50 °C
2.1: 1.34 g / AcOH / tetrahydrofuran; H2O / 4 h / 20 °C
3.1: PPh3 / pyridine / 2 h / 20 °C
3.2: conc. NH4OH / pyridine / 2 h / 20 °C
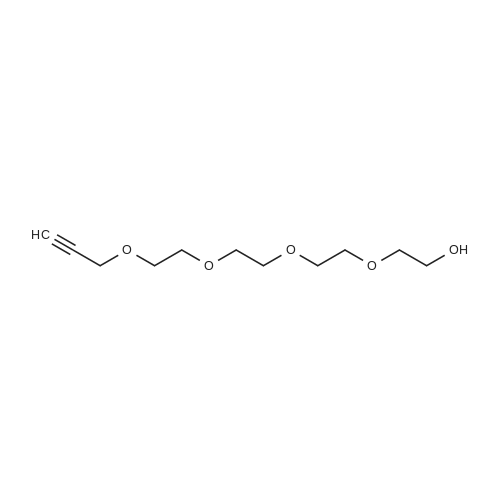
j00767j 1-Bromo-2-(2-(2-(2-bromoethoxy)ethoxy)ethoxy)ethane (2):
1007681 A solution of ((oxybis(ethane-2, 1-diyl))bis(oxy))bis(ethane-2, 1-diyl) bis(4- methylbenzenesulfonate) (10 g, 197.6 mmol) in acetone (100 mL) was charged with lithium bromide (7 g, 79.5 mmol) and heated to 75 °C for 12 h. The reaction mixture was concentrated in vacuo resulting in the crude compound which was purified by chromatography on silica gel, eluting with 0-20% ethyl acetate in n-hexane to give 5 g, 79% yield, of the title compound as a yellow liquid. ‘H NMR (400 MHz, CDC13): ö=3.82(t,J=6.51 Hz, 8H), 3.48 (t,J= 6.51 Hz,8H).
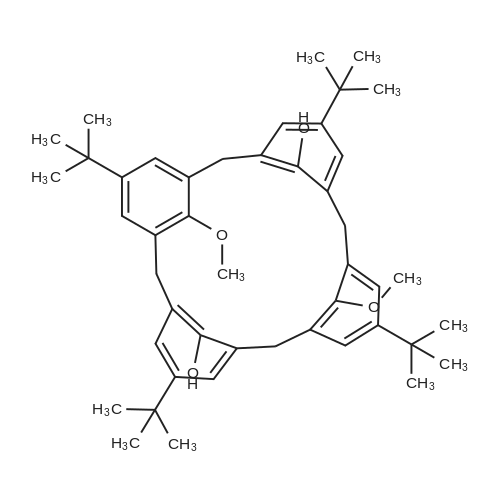
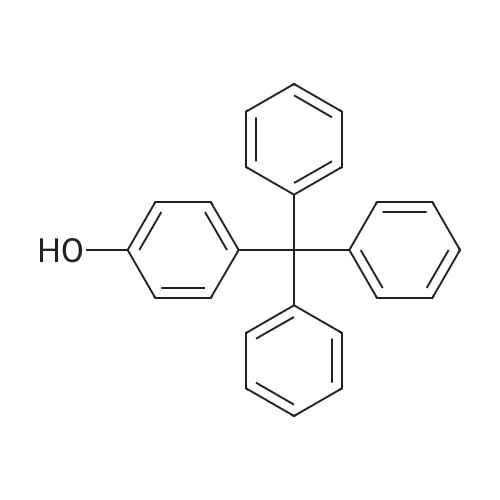
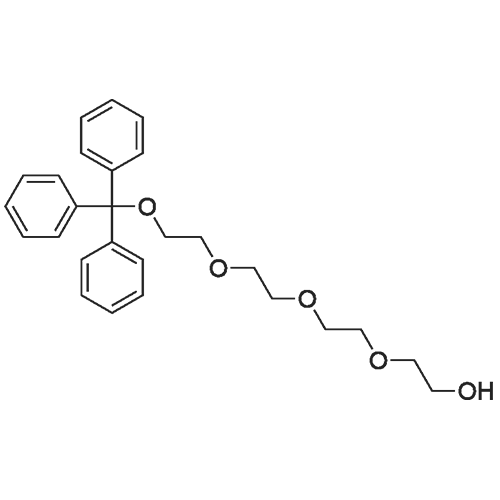
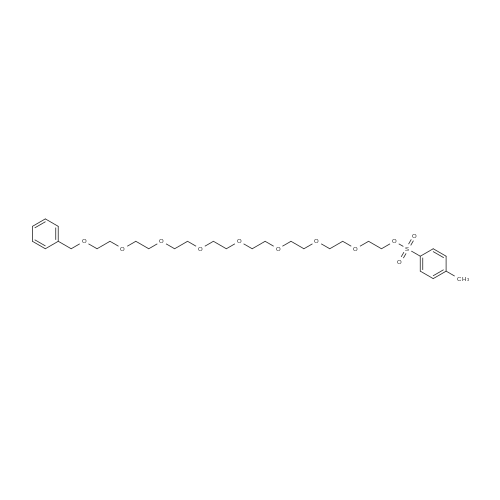
1,1,1,39,39,39-hexaphenyl-2,5,8,11,14,17,20,23,26,29,32,35,38-tridecaoxanonatriacontane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
Stage #1: tetraethylene glycol di(p-toluenesulfonate); 1,1,1-triphenyl-2,5,8,11-tetraoxatridecan-13-ol With sodium hydride In tetrahydrofuran at 25℃; for 2h;
Stage #2: In tetrahydrofuran at 0 - 40℃; Inert atmosphere;
81%
Stage #1: 1,1,1-triphenyl-2,5,8,11-tetraoxatridecan-13-ol With sodium hydride In tetrahydrofuran; hexane; isopropyl alcohol; mineral oil at 20℃; for 4h; Inert atmosphere;
Stage #2: tetraethylene glycol di(p-toluenesulfonate) In tetrahydrofuran; hexane; isopropyl alcohol; mineral oil at 0 - 40℃; Inert atmosphere;
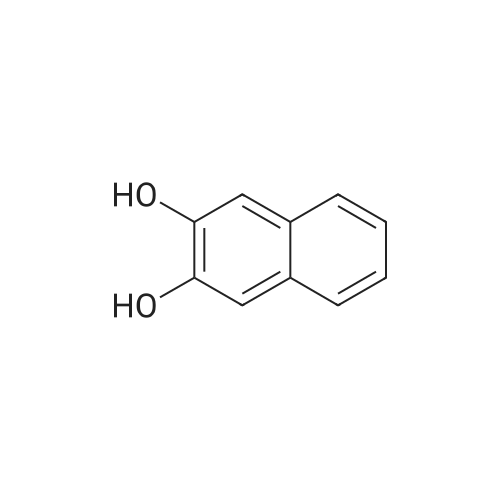
Stage #1: 2,3-naphthalenediol With tetrabutylammomium bromide; sodium hydroxide In benzene at 60℃; for 0.5h;
Stage #2: tetraethylene glycol di(p-toluenesulfonate) In benzene for 15h; Heating;
A suspension of NaH (1.8 g, 60percent in paraffin, 45 mmol) in THF (50 mL) was cooled to 0 °C. Addition of a cooled solution of 41 (3.66 g, 17 mmol) in THF (30 mL) was followed by stirring for 15 min. After adding tetraethylene glycol di-p-tosylate (1.12 mL, 2.8 mmol) the mixture was warmed to room temperature and stirred for 4 h. Addition of water was followed by extraction with EtOAc, drying (Na2SO4) and evaporation of the solvent. The residue was purified by flash-chromatography (hexane/EtOAc 6:1 to 1:1) to give 47 in 74percent yield (1.24 mg). EI-MS: m/z 592 (M+); 1H NMR: (CDCl3, 360 MHz) delta (ppm): 3.67-3.69 (m, 8H), 3.70-3.73 (m, 8H), 3.79 (s, 6H), 4.58 (s, 4H), 6.69 (dd, J1 = 8.5 Hz, J2 = 3.0 Hz, 1H), 7.08 (d, J = 3.0 Hz, 1H), 7.38 (d, J = 9.0 Hz, 1H). 13C NMR: (CDCl3, 150 MHz) delta (ppm): 55.5, 70.2, 70.6, 70.7, 70.8, 72.3, 112.6, 114.3, 114.7, 133.0, 138.7, 159.1; IR: (NaCl) nu (cm-1): 2868, 1595, 1469, 1354, 1296, 1111, 876, 810, 754.
Stage #1: 3,6,9,12-tetraoxapentadec-14-yn-1-ol With sodium hydride In tetrahydrofuran; mineral oil for 1h;
Stage #2: tetraethylene glycol di(p-toluenesulfonate) In tetrahydrofuran; mineral oil at 20℃; for 30h;
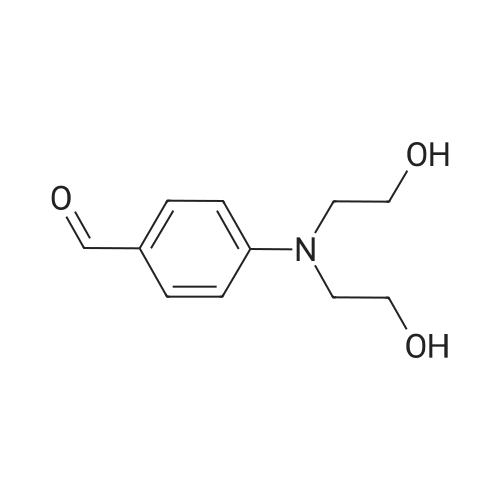
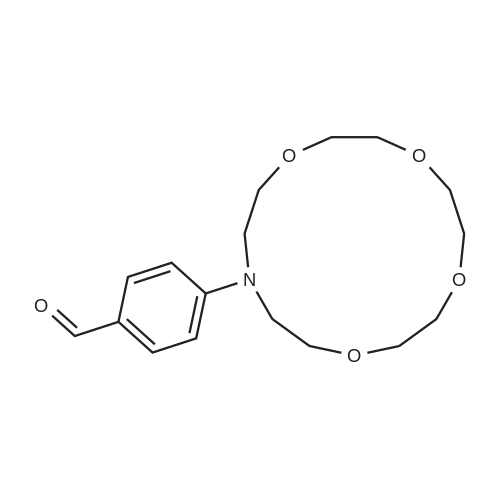
Stage #1: 4-(bis(2-hydroxyethyl)amino)benzaldehyde With sodium hydride In tetrahydrofuran; mineral oil for 0.5h; Inert atmosphere; Reflux;
Stage #2: tetraethylene glycol di(p-toluenesulfonate) In tetrahydrofuran; mineral oil at 70℃; for 48h; Inert atmosphere;
Stage #1: 4-(bis(2-hydroxyethyl)amino)benzaldehyde With sodium hydride In tetrahydrofuran for 2h; Inert atmosphere; Reflux;
Stage #2: tetraethylene glycol di(p-toluenesulfonate) In tetrahydrofuran for 96h; Inert atmosphere; Reflux;
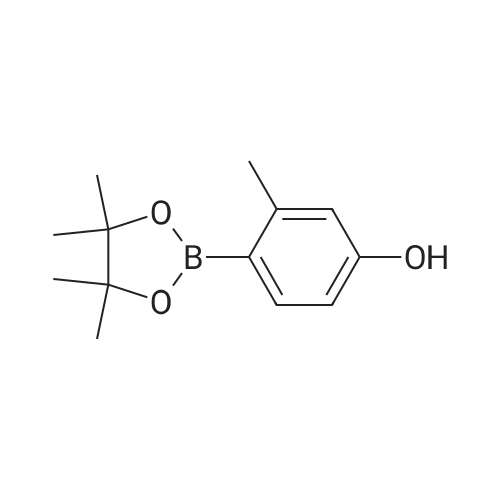
2-(2-(2-(2-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)ethoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
58%
With potassium carbonate In acetonitrile at 85℃; for 16h;
1007741 2-(2-(2-(2-(3-Methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenoxy)ethoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (3):
1007751 A solution of ((oxybis(ethane-2, 1 -diyl))bis(oxy))bis(ethane-2, 1 -diyl) bis(4- methylbenzenesulfonate) (1.34 g, 2.66 mmol) in acetonitrile (50 mL) was charged with potassium carbonate (1.1 g, 8.00 mmol), 3 -methyl-4-(4,4,5,5-tetramethyl- 1,3 ,2-dioxaborolan-2- yl)phenol (499 mg, 2.13 mmol) and heated to 85 °C for 16 h. The reaction mixture was cooledto room temperature, filtered through a pad of celite and concentrated in vacuo resulting in thecrude compound which was purified by chromatography on silica gel, eluting with 10-15%ethyl acetate in n-hexane to give 700 mg (58% yield) of the title compound as pale brown solid.‘H NMR (400 MHz, DMSO-d6): ö = 7.78 (d, J= 7.45 Hz, 2H), 7.55 (d, J= 7.89 Hz, 1H), 7.47(dd, J= 2.63, 7.89 Hz, 2H), 6.67 -6.76 (m, 2H), 4.04-4.14 (m, 4H), 3.67-3.75 (m, 2H), 3.54- 3.59 (m, 4H), 3.47 - 3.52 (m, 2H), 3.44 (br. s, 3H), 3.42 (br. s, 3H), 2.39 -2.43 (m, 4H), 1.28(s, 12H); MS (ES): m/z = 581.23 [M+H2O] LCMS: tR = 3.47 mm.
2,4,6-tris[(13"-tosyl-1",4",7",10",13"-pentaoxadecan-1"-yl)-4'-benzen-1'-yl]-1,3,5-triazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
21%
With caesium carbonate In acetonitrile for 12h; Reflux;
4.6 General procedure for the synthesis of podands 5-7
General procedure: To a solution of ditosylated oligoethyleneglycol (1.4mmol) in acetonitrile (15mL), Cs2CO3 (0.7mmol) were added in small portions, then the suspension was heated to reflux and compound 1 (0.14mmol) in MeCN (5mL) were added dropwise and the mixture was refluxed for 12h. After cooling at rt, the solvent was evaporated, the resulting solid was washed with water (30mL) and extracted with CHCl3 (3×20mL). The combined organic layers were dried over MgSO4 and then the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography on silicagel (using a gradient elution system of ethylacetate: petroleum ether 1:1 to 2:1) to yield the desired podands 5, 6 and 7, respectively.
The reaction mixture of tetraethyleneglycol ditosylate (2.51 g, 5 mmol),13 4-tertoctylphenol(1.03 g, 5 mmol) and NaH (0.36 g, 15 mmol) in10 mL of anhydrous tetrahydrofuran (THF) was refluxed for6 h. The reaction was quenched with aqueous 2-N NaOH(4 mL). The quenched reaction mixture was refluxed for4 h, cooled to room temperature, and carefully neutralized withaqueous 10% HCl. The reaction mixture was extracted withethyl acetate (3 × 100 mL), washed with saturated aqueousNaHCO3 solution (50 mL), brine (100 mL), and dried withanhydrous MgSO4. Evaporation of the filtrate under reducedpressure gave pale yellow crude product. Purification of thecrude product by column chromatography (Hexane:EA =3:1) afforded the M4EG (1.81 g, 82%) as colorless liquid.
Stage #1: dasatanib With potassium carbonate In acetonitrile at 20℃; for 2h;
Stage #2: tetraethylene glycol di(p-toluenesulfonate) In acetonitrile Reflux;
4 Example 4 Synthesis of Double End-Substituted Tetraethylene Glycol-Dasatinib (DSTN-42)
Example 4 Synthesis of Double End-Substituted Tetraethylene Glycol-Dasatinib (DSTN-42) 487 g dasatinib and 166 mg potassium carbonate are added into a 100 mL three-neck flask filled with 20 mL acetonitrile, and stirred at a room temperature for two hours. 10 mL acetonitrile solution in which 258 mg TsO-PEG(n=4)-OTs is dissolved is added to the reaction flask, and reflex reacts overnight. TLC monitors that the reaction is complete. Column separation is carried out to obtain 308 mg white solid with a yield rate 54.5%. m/z [MH]+1133. 1H-NMR (DMSO-d6): 2.23 (s, 6H), 2.49 (s, 6H), 2.51 (m, 8H), 3.53 (m, 32H), 6.04 (s, 2H), 7.27 (m, 4H), 7.40 (m, 2H), 8.22 (s, 2H), 9.88 (s, 2H), 11.48 (s, 2H).
With potassium hydroxide; In tetrahydrofuran; at 20℃; for 1h;
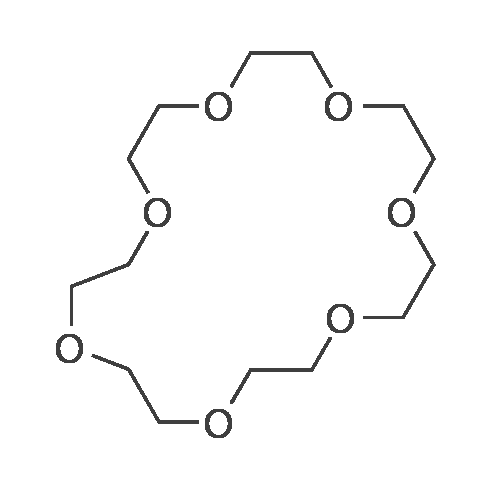
1 part by mole of tetraethylene glycol bis-p-toluenesulfonate, 1.5 parts by mole of diethylene glycol, 2 parts by mole of potassium hydroxide, 2 parts by mole of tetrahydrofuran, and reacted at 20 C for 1 hour in an industrial microwave reactor. After completion of the reaction, the mixture was separated by desalting, extracted, and the solvent was evaporated, and the crude product was distilled under reduced pressure to give 18-crown ether-6 in a yield of 45%.
With sodium hydride In tetrahydrofuran at 0 - 20℃; for 36h;
2.1.3 2.1.3: Synthesis of 3,6,9,12,15,18,21,24-octaoxaheptacos-26-yn-1-yl 4-methylbenzenesulfonate (10')
In an oven dried RBF, monobenzyltetraethylene glycol (4.8 g, 1 eq), ditosyltetraethylene glycol (14 g, 2 eq) was dissolved with stirring in THF at 0°C. Immediately, NaH (2 g, 2 eq) was added to flask in portions and then stirred for 36 hours at RT. Upon completion, NaH was quenched by addition of water at 0°C. and extracted with DCM. The combined organic layer was dried over Na2SO4 and concentrated under vacuum to get crude product which was purified using silica gel column chromatography using MeOH/ DCM as eluent to get pale yellow liquid. (5 g, 42%); 1H NMR (400 MHz, CDCl3) δH 7.77 (d, J=8 Hz, 2H), 7.32 d, J=8 Hz, 2H), 4.18 (d, J=2.4 Hz, 2H), 4.14 (t, J=3.2 Hz, 2H), 3.69-3.66 (m, 6H), 3.65-3.61 (m, 20H), 3.57 (s, 4H), 2.42 (m, 4H); 13C NMR (100 MHz, CDCl3) δC 144.78, 132.79, 129.76, 127.87, 70.60, 70.42, 69.20, 68.56, 21.56; MALDI-ToF (M+K): 601.11.
Stage #1: tetraethylene glycol di(p-toluenesulfonate); 1-phenyl-2,5,8,11-tetraoxatridecan-13-ol With sodium hydride In tetrahydrofuran at 0 - 20℃; for 12h;
Stage #2: p-toluenesulfonyl chloride With dmap; triethylamine at 20℃; for 12h;
1.1.1 1.1.1: Synthesis of 2-(2-(2-(benzyloxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (10)
A mixture of ditosyltetraethylene glycol (42 g, 1.2 eq) and monobenzyltetraethylene glycol (14 g, 0.7 eq) was dissolved under stirring at 0°C. in THF. Then NaH (4.0 g, 1.2 eq) was added in portions. The reaction was stirred for 12 hours at RT. Upon completion of reaction excess of NaH was quenched by drop wise addition of water and extracted with DCM thrice. The combined organic layer was dried over Na2SO4 and concentrated under vacuum. To the obtained residue, tosyl chloride and DMAP was added in an oven dried RBF. Then Triethyl amine was added and allowed to react for 12 hours at RT under stirring. Upon completion, water was added to the reaction and extracted with DCM thrice. The combined organic layer was dried over Na2SO4 and concentrated under vacuum to get crude product which was purified using silica gel column chromatography using MeOH/DCM as eluent to get pale yellow liquid. Yield (3.6 g, 26%) 1H NMR (400 MHz, CDCl3): δH 7.78 (d, J=8 Hz, 2H), 7.33-7.26 (m, 7H), 4.56 (s, 2H), 4.15 (t, J=4.8 Hz, 2H), 3.69-3.55 (m, 32H), 2.44 (s, 3H). 13C NMR (100 MHz, CDCl3): δc 144.95, 133.06, 129.95, 127.97, 77.16, 70.83, 70.64, 69.38, 68.78, 21.75. MALDI-TOF MS: (M+K) 653.48.
With caesium carbonate In N,N-dimethyl-formamide at 20℃; for 48h;
Synthesis of the precursor TPC
To a solution of Crizotinib (2.24 g,5 mmol, 1.0 eq) in DMF (50 mL) was added ((oxybis(ethane-2,1-diyl))bis(oxy))bis(ethane-2,1-diyl) bis(4-methylbenzenesulfonate) (2.5 g,5 mmol, 1.0 eq) and Cs2CO3 (1.6 g, 5 mmol, 1.0 eq) at room temperature. The solution was stirred at RT for 2 days. The reaction was diluted with EA, then dropwised to saturated NaHCO3, extracted with EA. The combined organic phase was washed with brine, dried over Na2SO4 and concentrated. The residue was purified by Pre-HPLC to give the compound of TPC (788 mg, 20% yield) as white solid. 1H NMR(400 MHz, MeOD) δ: 7.75-7.80 (m, 3H), 7.56 (s, 1H), 7.50 (s, 1H),7.27-7.34 (m, 3H), 7.03-7.07 (m, 1H), 6.87 (s, 1H), 6.06-6.08 (m, 1H),4.77 (s, 1H), 4.12-4.17 (m, 3H), 3.63-3.70 (m, 12H), 3.09 (d,J = 12 Hz, 2H), 2.64-2.67 (m, 2H), 2.44 (s, 3H), 2.01-2.24 (m, 6H),1.85-1.86 (d, J = 6.4 Hz, 3H). LCMS:Rt: 2.05 min; MS m/z (ESI):780.3[M + H]
Stage #1: 1,1,1-triphenyl-2,5,8,11-tetraoxatridecan-13-ol With sodium hydride In tetrahydrofuran; mineral oil at 20℃; for 2h; Inert atmosphere;
Stage #2: tetraethylene glycol di(p-toluenesulfonate) In tetrahydrofuran; mineral oil at 20℃; for 20h; Inert atmosphere;
6.6. 23-Azido-3.6.9.12.15.18.21-heptaoxatricosyl 4-methylbenzenesulfonate (s4b)13
To a solution of s5 (10.00 g; 45.61 mmol; 1 eq.) dissolved in anhydrous THF (70 mL), cooled to 0 °C, NaH (60% inmineral oil; 2190 mg; 54.74 mmol; 1.2 eq.) was added and the suspension was stirred at room temperature for2 h. Then, a solution of s614(45.85 g; 91.23 mmol; 2 eq.) dissolved in anhydrous THF (90 mL) was addeddropwise and the resulting mixture was stirred at room temperature for 20 h. After adding water (100 mL), theresulting mixture was extracted with ethyl acetate (4 x 70 mL). The combined organic layers were washed withbrine (100 mL), dried over MgSO4, filtered and evaporated under reduced pressure. The crude product waspurified on silica gel eluted with ethyl acetate/cyclohexane (60/40; v/v) to eliminate excess of s6. A secondelution with ethyl acetate/dichloromethane/ethanol (65/25/10; v/v/v) afforded s4b (10.99 g; 19.99 mmol;44%). Revelator: Dragendorff and UV. 1H NMR (400 MHz, CDCl3) 2.45 (s, 3H, H7’); 3.39 (t, J = 5.0 Hz, 2H, H23);3.75-3.52 (m, 28H, CH2-O); 4.16 (t, 2H, J = 4.9 Hz, H1); 7.34 (t, 2H, J = 8.3 Hz, H3’, 5’); 7.80 (d, 2H, J = 8.3 Hz, H2’, 6’).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping