There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
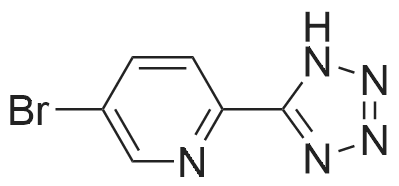
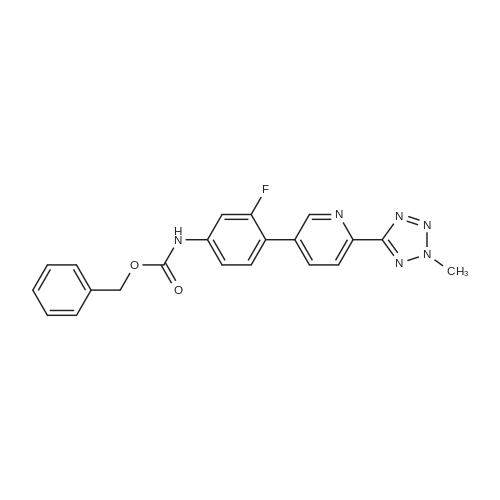
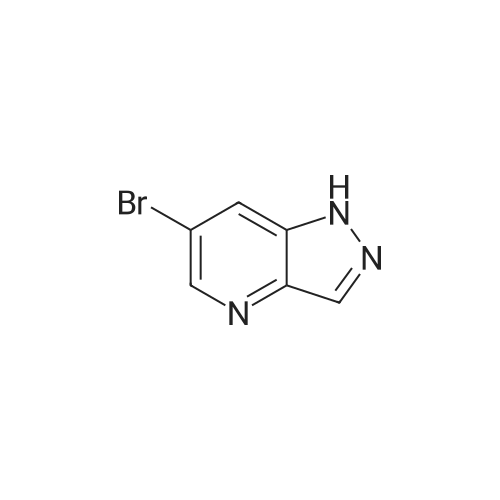
Structure of 380380-64-3 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With 10% palladium on activated carbon; Degussa type; hydrogen In methanol at 20℃; for 3 h;
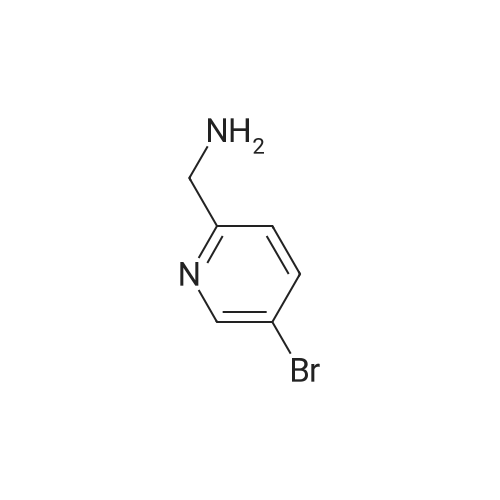
1.5 L of methanol,5-bromo-2- (2H-tetrazol-5-yl) pyridine (226 g)Formaldehyde (90g),10percent palladium on carbon (20 g) was added to the reactor,Under a hydrogen atmosphere (0.2 MPa)Stirred at room temperature for 3 hours, the reaction was completed,Organic phase evaporated,Recrystallization from acetonitrile,To give the product 2-methyl-5- (5-bromo-2-yl) tetrazolium,217 g, yield: 90.4percent.
Reference:
[1] Patent: CN106632240, 2017, A, . Location in patent: Paragraph 0018-0019
2
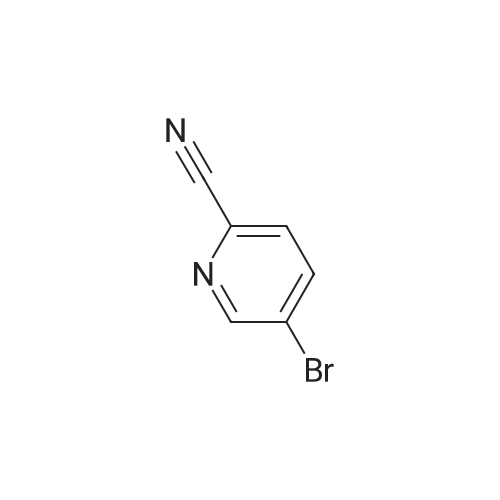
[ 97483-77-7 ]
[ 77-78-1 ]
[ 380380-64-3 ]
Yield
Reaction Conditions
Operation in experiment
75%
Stage #1: With sodium azide; ammonium chloride In dimethyl sulfoxide at 95℃; for 4 h; Stage #2: at 50℃; for 24 h; Stage #3: for 3 h;
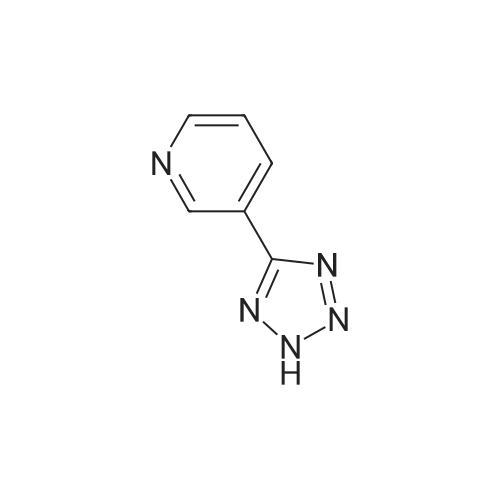
The 5 - bromo -2 - cyano pyridine (800g, 4.4 µM), dimethyl sulfoxide (4L) adding 20L in the bottle, addition of sodium azide (425.7g, 6 . 55 µM), ammonium chloride (350.3g, 6 . 55 µM), stirring, heated to 95 degrees, stirring 4 hours, cooling to room temperature.By adding sodium hydroxide (470g, 11.8 µM), dropping the dimethyl sulfate (665g, 5.3 µM), heating up to 50 degrees, stirring 24 hours, the reaction is finished, the addition of water (11L), centrifugal drying to obtain the crude product.The crude product by adding dilute hydrochloric acid, stirring 3 hours, filtration, the aqueous phase is 40percent of NaOH to adjust the PH=9 - 10, separating out 2 - methyl -5 - (5 - bromo pyridine -2 - yl) tetrazole, (792g, 3.3 µM), yield: 75percent.
Reference:
[1] Patent: CN106699730, 2017, A, . Location in patent: Paragraph 0016; 0017; 0018; 0019; 0020; 0021
3
[ 97483-77-7 ]
[ 380380-64-3 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 2004, vol. 12, # 22, p. 5909 - 5915
[2] Patent: CN106432182, 2017, A,
[3] Patent: CN106432182, 2017, A,
[4] Patent: CN106432182, 2017, A,
[5] Patent: CN106432182, 2017, A,
[6] Patent: CN106432182, 2017, A,
[7] Patent: WO2008/108988, 2008, A1,
[8] Patent: WO2005/116023, 2005, A1, . Location in patent: Page/Page column 67
4
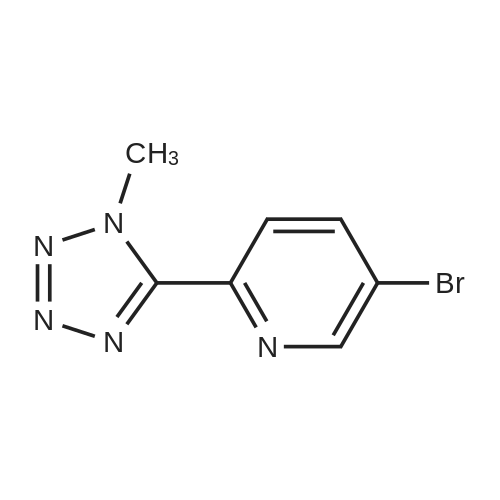
[ 380380-60-9 ]
[ 74-88-4 ]
[ 380380-64-3 ]
Yield
Reaction Conditions
Operation in experiment
70.5%
With calcium hydroxide In dichloromethane; N,N-dimethyl-formamide at 0 - 40℃; for 24 h;
The 5-bromo-2-(2H-tetrazol-5-yl)pyridine (20.0 g, 88.4 mmol) prepared in Example 1 was added with 20.0 mL of N,N-dimethyl formamide and 180.0 mL of methylene chloride and then further added with calcium hydroxide (3.94 g, 53.0 mmol), after which iodomethane (33.0 mL, 530.4 mmol) was slowly added dropwise thereto at 0°C. Thereafter, the reaction solution was warmed to 40°C and stuffed for 24 hr. After the termination of the reaction, the reaction solution was added with water, thus extracting an organic layer. The extracted organic layer was washed with saline and further extracted. The resulting organic layer was added with 300.0 mL of a 6 N hydrochloric acid aqueous solution to thus extract an aqueous layer, after which the separated organic layer was added with 60.0 mL of a 6 N hydrochloric acid aqueous solution, so that the aqueous layer was further extracted. Extraction was performed using HPLC until the amount of Ni was less than 5percent. The separated aqueous layer was collected, and the pH thereof was adjusted to 10.6 at 40°C or less using a 50percent sodium hydroxide aqueous solution. The produced solid was filtered, washed with water, and concentrated under reduced pressure, thus obtaining a desired compound. Yield (16.2 g, 70.5percent), N2/N] ratio percent (98/2)
Reference:
[1] European Journal of Organic Chemistry, 2016, vol. 2016, # 7, p. 1305 - 1313
[2] Patent: WO2004/48350, 2004, A2, . Location in patent: Page 60-61
[3] Patent: WO2004/56819, 2004, A1, . Location in patent: Page 52-53
[4] Patent: WO2004/56818, 2004, A1, . Location in patent: Page 52-53
[5] Patent: WO2004/56816, 2004, A1, . Location in patent: Page 52-53
[6] Patent: WO2008/108988, 2008, A1, . Location in patent: Page/Page column 52
6
[ 1374651-37-2 ]
[ 380380-64-3 ]
Yield
Reaction Conditions
Operation in experiment
33%
With sodium hydroxide; methyl iodide In tetrahydrofuran; N,N-dimethyl-formamide at 48℃; Cooling with ice/brine
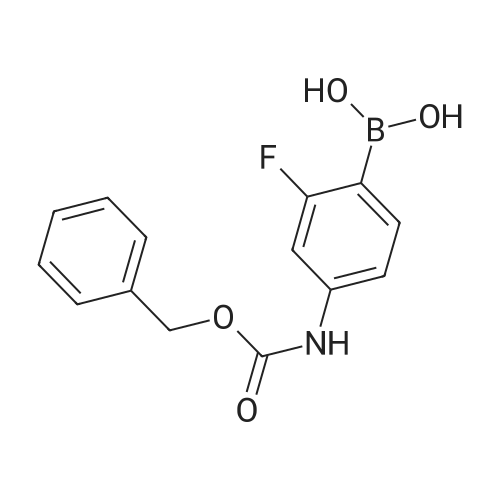
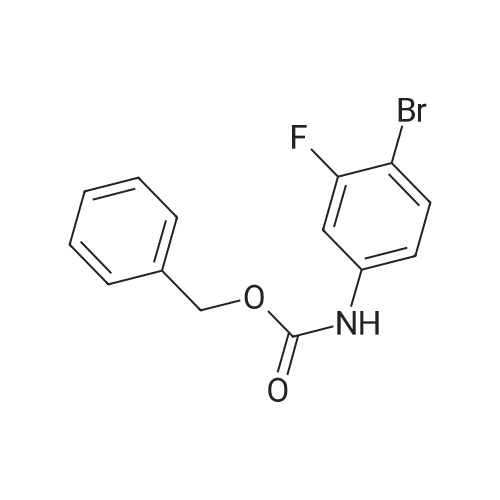
To a 22-L, four-neck, round-bottom flask equipped with an overhead stirrer, nitrogen inlet/outlet, and thermocouple placed in an ice/brine bath was charged the tetrazole ammonium salt (835.0 g, 3.44 mol, 1 weight), tetrahydrofuran (7.5 L, 9 volumes), ΛζjV-dimethylformamide (2.5 L, 3 volumes) and sodium hydroxide powder (343.5 g, 8.59 mol, 2.5 equivalents) while stirring. The internal reactor temperature was allowed to reach 12°C, whereupon iodomethane (1.22 kg, 8.59 mol, 2.5 equivalents) was added dropwise over 50 minutes, maintaining the reaction temperature below 200C. After 20 minutes addition time, due to a rapid increase in temperature, the addition was paused and the reaction continued to self-heat from 15-200C over ten minutes. The remainder of the addition was completed at constant temperature (18°C ). Upon completion of the addition, the ice/brine bath was removed and the reactor was equipped with a water condenser and a heating mantle. The internal reactor temperature was adjusted to 400C, however the reaction continued to self- heat to 48°C. The reaction was judged to be complete after 6 hours by HPLC analysis by complete consumption of the starting material. The reaction mixture was cooled to room temperature overnight for convenience. The THF was removed by distillation, and water (8.35 L, 10 volumes) was charged to the reactor. The slurry was stirred for 30 minutes and filtered by vacuum filtration and the reactor and filter cake were washed with water (4.2 L, 5 volumes) to afford crude 4/Nl isomer mixture as a peach colored solid (500.7 g, 61percent yield, 3.85: 1 4: Nl).[0047] The solids (500.7 g) were dissolved in CH2Cl2 (2.5 L, 5 volumes) to which 6 N aqueous HCl (7.5 L, 15 volumes) was added. The biphasic mixture was stirred and the layers were separated. At this point, the desired product is in the aqueous HCl layer. The CH2Cl2 layer was washed with 6 N aqueous HCl (4.5 L, 3 x 3 volumes) until <5percent AUC 4 was present by HPLC analysis. The combined 6 N HCl extracts were transferred to a reactor and the pH was adjusted to 10.6 with 50percent aqueous NaOH (-3.2 L) while maintaining the internal temperature below 400C. The solids were isolated by vacuum filtration and the reactor and filter cake were rinsed with water (1 L, 2 volumes) to afford crude 4 as a yellow/orange solid (322.4 g, 64percent recovery, 39percent yield, 93.5percent AUC 4, 4.1percent AUC N-I isomer) as confirmed by HPLC and 1H NMR analyses.[0048] The crude 4 was further purified by an isopropyl acetate (IPAc) reslurry (1.61 L, 5 volumes) at 500C for 1 hour. Upon cooling to room temperature, the solids were filtered and the reactor and filter cake were washed with additional IPAc (500 mL, 1.6 volumes) to afford purified 4 as a off-white/yellow solid (275.5 g, 85percent recovery, 33percent yield, 98.2percent AUC) as confirmed by HPLC and 1H NMR analyses. DSC analysis of 4 showed a decomposition exotherm at approximately 245°C. Example 3: Preparation of benzyl (4-bromo-3-fluorophenyl)carbamate, 5 [0049] To a 12-L, three-neck, round-bottom flask equipped with an overhead stirrer, nitrogen inlet/outlet, addition funnel and thermocouple was charged 4-bromo-3- fluoroaniline (800.0 g, 4.21 mol, Matrix lot No. Q13H), THF (6.4 L, 8 vol), and solid sodium bicarbonate (530.5 g, 6.32 mol, 1.5 eq). The addition funnel was charged with benzyl chloroformate (861.9 g, 5.05 mol, 1.2 eq), which was added dropwise to the reactor over 70 minutes. The temperature of the reaction was maintained below 200C with an ice water bath. The batch was aged 1 hour at room temperature at which point HPLC analysis indicated that the reaction was complete. The reaction mixture was transferred to a 22-L flask and the mixture was diluted with water (6.4 L, 8 vol). The two-phase mixture was warmed to 50°C and held at temperature for 16 hours to quench the excess benzyl chloroformate. The mixture was transferred hot to a separatory funnel to remove the lower aqueous phase. A rag layer was observed which was taken with the aqueous layer. The THF layer was filtered through Whatman No.1 filter paper to remove some particulates, and the mixture was transferred back to a 22-L flask equipped for distillation. Heptane was added in portions and distilled to remove the THF. (Note that it is best to distill some of the THF out first before adding the first amount of heptane.) A total of 26.5 L of heptane was added, and the total distillate collected was 25 L. At this point, the pot temperature had reached 97.7°C and the distillate coming over contained 0.9percent THF by 1H NMR analysis. The mixture was cooled to room temperature and the thick white slurry was filtered. The filter cake was washed with heptane (4 L). The product was dried in a vacuum oven at 400C to give 1257.0 g of intermediate 5 (92percent yield). The HPLC assay was 98.3percent (AUC).
With sodium hydroxide In N,N-dimethyl-formamide at 0 - 20℃; Inert atmosphere
To a solution of 2-(tetrazol-5-yl)-5-bromopyridine 8 (10.5 g, 46.5 mmol) in DMF (100 mL) at 0 °C was slowly added NaOH (6.50 g, 162 mmol) and iodomethane (4.08 mL, 65.5 mmol). After being stirred at room temperature for 6 h, the reaction mixture was poured into water and extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography to obtain the title compound (5 g, 45percent). 1H NMR (CDCl3): δ 8.80 (d, J = 2.0 Hz, 1H), 8.13 (d, J = 8.4 Hz, 1H), 7.98 (dd, J = 8.4 Hz, 2.4 Hz, 1H), 4.42 (s, 3H).
4 g
With sodium hydroxide In N,N-dimethyl-formamide at 0 - 20℃; for 6 h;
Will be 10.5 grams2- (tetrazol-5-yl) -5-bromopyridine was dissolved100 ml of dimethylformamide,Then 6.5 g of sodium hydroxide was added to the solution,And 9.3 g of methyl iodide was slowly added to the solution at 0 ° C. The solution was stirred at room temperature for 6 hours,Water was then added and extracted with ethyl acetate.The resulting organic layer was washed with brine, dehydrated, filtered, concentrated in vacuo and purified by column chromatography to give 4 g of 2- (1-methyltetrazol-5-yl) -5-bromopyridine and 5 g of 2- (2 -methyltetrazol-5-yl) -5-bromopyridine.
Reference:
[1] European Journal of Medicinal Chemistry, 2011, vol. 46, # 4, p. 1027 - 1039
[2] Bioorganic and Medicinal Chemistry, 2004, vol. 12, # 22, p. 5909 - 5915
[3] Patent: WO2005/58886, 2005, A1, . Location in patent: Page/Page column 26; 27
[4] Patent: WO2006/38100, 2006, A1, . Location in patent: Page/Page column 67-68
[5] Patent: CN106146559, 2016, A, . Location in patent: Paragraph 0050
[6] Patent: US2003/166620, 2003, A1,
[7] Patent: WO2005/116022, 2005, A1, . Location in patent: Page/Page column 50
[8] Patent: WO2005/116023, 2005, A1, . Location in patent: Page/Page column 67-68
Reference:
[1] European Journal of Medicinal Chemistry, 2011, vol. 46, # 4, p. 1027 - 1039
[2] European Journal of Organic Chemistry, 2016, vol. 2016, # 7, p. 1305 - 1313
[3] Patent: CN106146559, 2016, A,
[4] Patent: WO2005/116022, 2005, A1,
10
[ 624-28-2 ]
[ 380380-64-3 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 2004, vol. 12, # 22, p. 5909 - 5915
11
[ 624-28-2 ]
[ 380380-64-3 ]
[ 380380-63-2 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2011, vol. 46, # 4, p. 1027 - 1039
[2] Patent: CN106146559, 2016, A,
12
[ 380380-64-3 ]
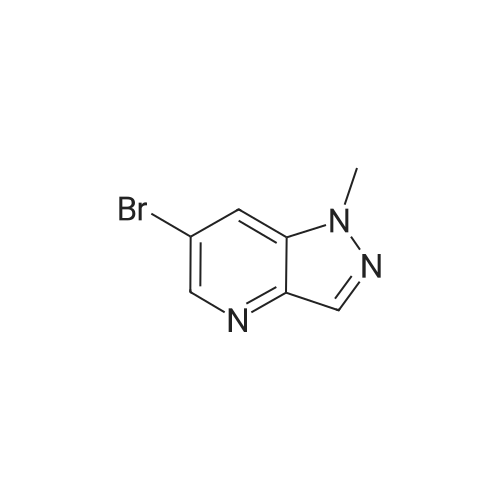
[ 856866-72-3 ]
Yield
Reaction Conditions
Operation in experiment
48%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; lithium chloride In N,N-dimethyl-formamide at 90℃; for 4 h;
In a 100 ml reaction flask,Compound 7 (5.0 g, 10 mmol) and 50 ml of DMF were added,2- (1-methyl-tetrazol-5-yl) -5-bromopyridine (2.65, 11 mmol)Lithium chloride (1.7 g),1,1'-bis (diphenylphosphino) ferrocene] palladium chloride (1.0 g),The mixture was stirred at 90 ° C for 4 hours,After completion of the reaction,To room temperature,And extracted three times with water / ethyl acetate (V / V = 1: 1)The organic layers were combined,Dried over anhydrous sodium sulfate,And concentrated to give 1.7 g of the compound (1)The yield was 48percent
26%
With lithium chloride In 1-methyl-pyrrolidin-2-one at 20 - 120℃; for 4 h;
In 150ml of 1-methyl-2-pyrrolidone was dissolved 37g of (R)-3- (4- tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol. The solution was added with 19.7g of 2- (2-methyltetrazol-5-yl)-5-bromopyridine, 10.44g of lithium chloride and 2.9g of dichlorobistriphenylphospine palladium (I I) at room temperature and then stirred at the temperature of 120 C for 4 hours. The reaction mixture was added with water and then extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to provide 8g of the title compound. Yield 26percent. 1H NMR (DMSO-d6) 5 8.90 (s, lH), 8. 18 (m, 2H), 7.70 (m, 2H), 7.49 (dd, 1H), 5.25 (t, 1H), 4.74 (m, 1H), 4.46 (s, 3H), 4.14 (t, lH), 3. 88 (dd, lH), 3.68 (m, lH), 3. 58 (m, lH)
26%
With bis-triphenylphosphine-palladium(II) chloride; lithium chloride In 1-methyl-pyrrolidin-2-one at 120℃; for 4 h; Inert atmosphere
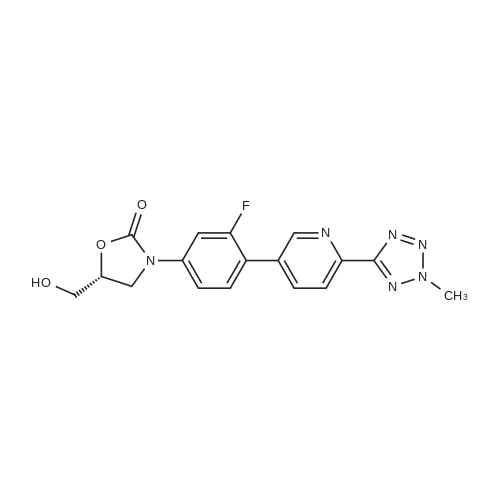
To a solution of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol 5 (37.0 g, 74.0 mmol) in NMP (150 mL) was added 2-(2-methyltetrazol-5-yl)-5-bromopyridine 9 (19.7 g, 81.9 mmol), LiCl (10.4 g, 245 mmol) and Pd(PPh3)2Cl2 (2.90 g, 4.13 mmol). The reaction mixture was stirred for 4 h at 120 °C. After being cooled to room temperature, the reaction mixture was poured into water and extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was further purified by column chromatography to obtain the title compound (8 g, 26percent). Mp: 201 °C. 1H NMR (DMSO-d6): δ 8.90 (s, 1H), 8.18 (m, 2H), 7.70 (m, 2H), 7.49 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 5.25 (t, J = 5.6 Hz, 1H), 4.74 (m, 1H), 4.46 (s, 3H), 4.14 (t, J = 8.8 Hz, 1H), 3.88 (m, 1H), 3.68 (m, 1H), 3.58 (m, 1H). 13C NMR (DMSO-d6): δ 163.56, 159.03, 154.06, 149.18, 144.78, 140.30, 136.96, 131.42, 130.73, 121.89, 118.36, 113.81, 105.18, 73.39, 61.52, 45.96, 39.63. IR (neat, cm-1): 3251.40, 1746.23, 1619.91, 1473.35, 1406.82. [M + H]+: 371.02. HRMS [EI-MS]: m/z, calculated for [C17H15FN6O3] 370.1190, found, 370.1100 [C17H15FN6O3].
Reference:
[1] Patent: CN105859780, 2016, A, . Location in patent: Paragraph 0038
[2] Patent: WO2005/58886, 2005, A1, . Location in patent: Page/Page column 28; 29
[3] European Journal of Medicinal Chemistry, 2011, vol. 46, # 4, p. 1027 - 1039
[4] Patent: CN107400126, 2017, A, . Location in patent: Paragraph 0036
13
[ 444335-16-4 ]
[ 380380-64-3 ]
[ 856866-72-3 ]
Yield
Reaction Conditions
Operation in experiment
83%
Stage #1: With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; potassium acetate; bis(pinacol)diborane In dimethyl sulfoxide at 80℃; for 14 h; Inert atmosphere Stage #2: With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; caesium carbonate In 1,4-dioxane; water at 70℃; for 3 h; Inert atmosphere
DMSO (100 ml) was added to a 250 ml reaction flask,(5R) -3- (4-bromo-3-fluorophenyl) -5-hydroxymethyloxazolidin-2-one(10 g, 34.5 mmol), pinacolate (17.52 g, 69 mmol),[1,1'-bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (1.41 g, 1.73 mmol)And potassium acetate (13.5 g, 138 mmol), and the temperature was raised to 80 ° C under a nitrogen atmosphere,For 14 hours. The heating was stopped, the solution was cooled to room temperature, and extracted with water / ethyl acetate 3 times. The organic layers were combined and the organic layer was washed with saturated waterBrine, dried over anhydrous sodium sulfate and concentrated by suction filtration. The concentrated product of the above step was added to a 250 ml reaction flask,1,4-dioxane (100 ml) was added,5-bromo-2- (2-methyl-2H-tetrazol-5-yl) pyridine(8.28 g, 34.5 mmol),[1,1'-bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (0.56 g, 0.69 mmol)And cesium carbonate aqueous solution (50 ml, containing 33.72 g of cesium carbonate, 103.5 mmol),Under nitrogen protection,The temperature was raised to 70 ° C,The reaction was carried out for 3 hours,Dichloromethane was added for extraction.The separated organic phase was washed with saturated brine,Anhydrous sodium sulfate dehydration,filter,Concentrated in vacuo and purified by column chromatography,10.6 g of a solid was obtained,The yield was 83.0percentHPLC purity was 98.34percent (area normalization method).
Reference:
[1] Patent: CN105418678, 2016, A, . Location in patent: Paragraph 0070; 0071; 0072; 0073; 0074; 0075
14
[ 380380-64-3 ]
[ 856866-72-3 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 2015, vol. 63, # 2, p. 143 - 146
15
[ 73183-34-3 ]
[ 380380-64-3 ]
[ 1056039-83-8 ]
Yield
Reaction Conditions
Operation in experiment
86.3%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 80℃; for 3 h; Sealed tube; Inert atmosphere
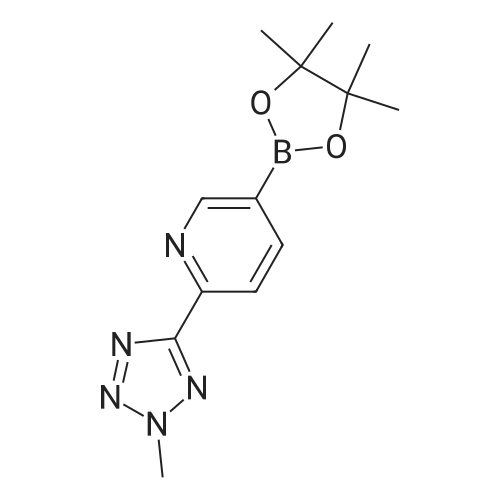
To a solution of 10 (1.00 g, 4.17 mmol) in dry degassed l,4-dioxane (15 mL) was added bis(pinacolato)diboron (1.27 g, 5.00 mmol), potassium acetate (1.35 g, 13.75 mmol), and Pd(dppf)Cl2 (0.26 g, 0.35 mmol). The reaction mixture was heated at 80 °C for 3 h in a sealed tube under argon. The reaction mixture was filtered through silica gel flash column with CH2Cl2 and the organic layer was evaporated under reduced pressure, and the residue was washed with n-hexane to obtain the title compound (1.03 g, 86.3 percent). 1H-NMR (400 MHz, CDCl3) δ: 9.14 (s, 1H), 8.30 (m, 2H), 4.48 (s, 3H), 1.38 (s, 12H).
66%
With potassium acetate In dimethyl sulfoxide at 80℃; for 2 h;
To the solution of 5-bromo-2-(2-methyl-2H-tetrazol-5-yl)pyridine (480 mg, 2 mmol) in DMSO (5 mL) was added pinacol diborane (1.02 g, 4 mmol), KOAc (588 mg, 6 mmol) and PdCl2(dppf)DCM (160 mg, 0.2 mmol), degassed with N2. The mixture was stirred at 80 0C for 2 h. The reaction mixture was diluted with DCM (100 mL) and washed with brine (2 x 100 mL), dried (Na2SO4) and evaporated under vacuum, then purified by prep-TLC (hexanes - EtOAc) to give 2-(2-methyl-2H-tetrazol-5-yl)-5-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)pyridine as white solid (380 mg, 66percent yield).
Reference:
[1] Chemical and Pharmaceutical Bulletin, 2015, vol. 63, # 2, p. 143 - 146
[2] Patent: WO2008/108988, 2008, A1, . Location in patent: Page/Page column 53
[3] European Journal of Organic Chemistry, 2016, vol. 2016, # 7, p. 1305 - 1313
[4] Patent: WO2009/74812, 2009, A1, . Location in patent: Page/Page column 41
[5] Patent: US2010/69441, 2010, A1, . Location in patent: Page/Page column 21-22
[6] Patent: US2010/204477, 2010, A1, . Location in patent: Page/Page column 31
[7] Patent: CN105367547, 2016, A, . Location in patent: Paragraph 0034; 0035; 0036
[8] Patent: CN107382995, 2017, A, . Location in patent: Paragraph 0007; 0014; 0015; 0016; 0117; 0018; 0019-0022
16
[ 874290-59-2 ]
[ 380380-64-3 ]
[ 1220910-89-3 ]
Yield
Reaction Conditions
Operation in experiment
87%
With potassium carbonate In 1,4-dioxane; water at 70℃; for 1 h; Inert atmosphere
To a 5 -L, three-neck, round-bottom flask was charged 4 (200.0 g, 0.833 mol) followed by 1,4-dioxane (3 L, 15 vol). Crude compound 6 (361.2 g, 1.249 mol, 1.5 equiv.), Pd2(dba)3 (11.44 g, 0.0125 g, 0.015 equiv.), and PCy3 (7.0 g, 0.025 mol, 0.03 equiv.) was charged and degassed with nitrogen for 30 minutes. A solution Of K2CO3 (195.7 g, 1.7 equiv.) in water (800 mL, 4 vol) was charged, and the reaction was heated to 70 0C. The reaction was complete after 1 hour with 0.5 area percent of 4 remaining. The reaction was cooled to 50 0C, and Darco G-60 (40 g, 0.2 wt) was added and stirred for 30 minutes. Celite 545 (40 g, 0.2 wt) was charged and then the reaction was filtered through Celite 545 (100 g, 0.5 wt) wetted with water (300 mL). The hot filtration into the water from the Celite caused precipitation of the product. Tetrahydrofuran (1.2 L, 6 vol) and brine (600 mL, 3 vol) were added, and the product re-dissolved at room temperature. The phase split was accomplished cleanly (Vmax = 28 volumes). The dioxane was concentrated and ethanol (1 L, 5 vol) was added and concentrated. Then the product was reslurried in ethanol: water (4: 1, 2 L, 10 vol) at 700C, cooled to room temperature over 3 hours, filtered and washed with ethanol (2 x 400 mL). Compound 7 was isolated in 87percent yield (292.6 g) with a purity of 97.7 percent (AUC) by HPLC analysis. The 1H NMR and 19F NMR indicated the presence of one compound. Pd analysis showed 135 ppm Pd was in the product.
With sodium hydroxide; In DMF (N,N-dimethyl-formamide); at 0 - 20℃; for 2h;
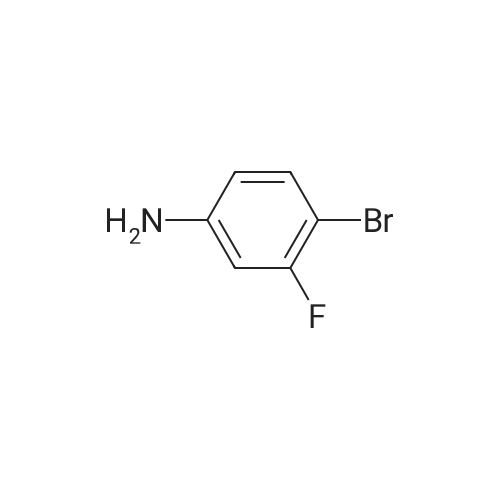
5-Bromo-2-(2-methyl-2H-tetrazol-5-yl) pyridine and 5-bromo-2- (1-methyl-1H-tetrazol-5- yl) pyridine. 5-Bromo-2-(2-methyl-2H-tetrazol-5-yl)pyridine and 5-bromo-2-(1-methyl-1H-tetrazol-5- yl) pyridine were prepared according to the procedure described by Dong A Pharmaceuticals (WO 01/94342). A mixture of 6.5 g unpurified 5-bromo-2-tetrazol-5-ylpyridine [Dong A Pharmaceuticals (WO 01/94342) ] (~28 mmol) and sodium hydroxide (9 g, 125 mmol) in dry dmf was evaporated to dryness under reduced pressure. A stirred solution of the involatile residue in dry DMF (50 mL) was treated dropwise at ice-bath temperature with iodomethane (3.0 ML, 48 mmol). The stirred reaction mixture was allowed to warm and then maintained at room temperature for 2 hours. The reaction mixture was partitioned between iced water and ethyl acetate. The organic phase was washed with water, dried over magnesium sulfate, and tehn evaporated under reduced pressure to give a residue that was purified by chromatography on silica gel [elution with dichloromethane : ethyl acetate (60: 1) ] to give: 1. 5-bromo-2- (1-methyl-1H-tetrazol-5-yl)pyridine (1.397 g), a colorless solid, (TLC: silica-gel, hexanes: ethyl acetate (4: 1), Rf: 0. 3), 1H-NMR (DMSO-d6) (300 MHz) 8 : 4.38 (s, 3H); 8.17 (d, 1H); 8.35 (dd, 1H); 8.96 (d, 1H). 2. 5-bromo-2-(2-methyl-2H-tetrazol-5-yl)pyridine (1.07 g), a colorless solid, (TLC: silica-gel, hexanes: ethyl acetate (4: 1), Rf : 0. 1). 1H-NMR (DMSO-d6) (300 MHz) No. : 4.46 (s, 3H); 8.09 (d, 1H) ; 8.28 (dd, 1H) ; 8.88 (d, 1H).
With sodium hydroxide; In DMF (N,N-dimethyl-formamide); at 0 - 20℃; for 2h;
5-BROMO-2- (2-METHYL-2H-TETRAZOL-5-YL) PYRIDINE AND 5-BROMO-2- (L-METHYL-LH-TETRAZOL-5- YL) pyridine 5-Bromo-2-(2-methyl-2H-tetrazol-5-yl) pyridine and 5-BROMO-2- (1-METHYL-LH-TETRAZOL-5- YL) pyridine were prepared according to the procedure described by Dong A Pharmaceuticals (WO 01/94342). A mixture of 6.5 g unpurified <strong>[380380-60-9]5-BROMO-2-TETRAZOL-5-YLPYRIDINE</strong> [Dong A Pharmaceuticals (WO 01/94342)] (-28 mmol) and sodium hydroxide (9 g, 125 mmol) in dry DMF was evaporated to dryness under reduced pressure. A stirred solution of the involatile residue in dry DMF (50 ML) was treated dropwise at ice-bath temperature with iodomethane (3.0 mL, 48 mmol). The stirred reaction mixture was allowed to warm and then maintained at room temperature for 2 hours. The reaction mixture was partitioned between iced water and ethyl acetate. The organic phase was washed with water, dried over magnesium sulfate, and tehn evaporated under reduced pressure to give a residue that was purified by chromatography on silica gel [elution with dichloromethane : ethyl acetate (60: 1) ] to give: 1. 5-bromo-2-(1-methyl-1H-tetrazol-5-yl) pyridine (1.397 g), a colorless solid, (TLC: silica-gel, hexanes: ethyl acetate (4: 1), Rf: 0.3),'H-NMR (DMSO-D) (300 MHz) 8 : 4.38 (s, 3H); 8.17 (d, 1H); 8.35 (dd, 1H); 8.96 (d, 1H). 2. 5-bromo-2-(2-methyl-2H-tetrazol-5-yl) pyridine (1.07 g), a colorless solid, (TLC: silica-gel, hexanes: ethyl acetate (4: 1), Rf: 0. 1). LH-NMR (DMSO-DSLT300 MHz) 8 4.46 (s, 3H); 8.09 (d, 1H); 8.28 (dd, 1H) ; 8.88 (d, 1H). Structure assignment based on nmr HMBC experiments, in which long range coupling of the protons of CH3 to the C5 of the tetrazole ring is observed in the 1-methyl-1H-isomer of Rf 0.3, but not in the 2-methyl-2H-isomer of Rf 0.1). The compound referred to as 5-bromo-2- (1-METHYL-LH-TETRAZOL-5-YL) pyridine is thus the isomer of Rf 0.3 and the compound referred to as 5-BROMO-2-(2-METHYL-2H-TETRAZOL-5-YL) PYRIDINE is thus the ISOMER OF RF 0. 1
With sodium hydroxide; In DMF (N,N-dimethyl-formamide); at 0 - 20℃; for 2h;
5-BROMO-2-(2-METHYL-2H-TETRAZOL-5-YL) PENDINE AND 5-BROMO-2-(1-METHEL-LH-TETRAZOL-5- 1 pyridine 5-BROMO-2- (2-METHYL-2H-TETRAZOL-5-YL) PYRIDINE AND 5-BROMO-2- (L-METHYL-LH-TETRAZOL-5- yl) pyridine were prepared according to the procedure described by Dong A Pharmaceuticals (WO 01/94342). A mixture of 6.5 g unpurified <strong>[380380-60-9]5-BROMO-2-TETRAZOL-5-YLPYRIDINE</strong> [Dong A Pharmaceuticals (WO 01/94342)] (-28 mmol) and sodium hydroxide (9 g, 125 mmol) in dry DMF was evaporated to dryness under reduced pressure. A stirred solution of the involatile residue in dry DMF (50 ML) was treated dropwise at ice-bath temperature with iodomethane (3.0 mL, 48 mmol). The stirred reaction mixture was allowed to warm and then maintained at room temperature for 2 hours. The reaction mixture was partitioned between iced water and ethyl acetate. The organic phase was washed with water, dried over magnesium sulfate, and tehn evaporated under reduced pressure to give a residue that was purified by chromatography on silica gel [ELUTION WITH DICHLOROMETHANE: ETHYL ACETATE (60: 1) ] TO GIVE: 1. 5-BROMO-2-(1-METHYL-LH-TETRAZOL-5-YL) pyridine (1.397 g), a colorless solid, (TLC: silica-gel, hexanes: ethyl acetate (4: 1), Rf: 0. 3), 1H-NMR (DMSO-D6) (300 MHZ) 8 : 4.38 (s, 3H); 8.17 (d, 1H); 8.35 (dd, 1H); 8.96 (d, 1H). 2. 5-BROMO-2- (2-METHYL-2H-TETRAZOL-5-YL) pyridine (1.07 g), a colorless solid, (TLC: silica-gel, hexanes: ethyl acetate (4: 1), Rf: 0. 1). LH-NMR (DMSO-D) (300 MHZ) 8 4.46 (s, 3H); 8.09 (d, 1H) ; 8.28 (dd, 1H); 8.88 (d, 1H). Structure assignment based on HMBC experiments, in which long range coupling of the protons of CH3 to the C5 of the tetrazole ring is observed in the 1-METHYL-LH-ISOMER of Rf 0.3, but not in the 2-methyl-2H-isomer of Rf 0.1). The compound referred to as 5-bromo-2- (1-METHYL-1H-TETRAZOL-5-YL) PYRIDINE is thus the isomer of Rf 0.3 and the compound referred to as 5-BROMO-2-(2-METHYL-2H-TETRAZOL-5-YL) PYRIDINE is thus the isomer OF RF 0. 1
A mixture of 6.5 g unpurified 5-bromo-2-tetrazol-5-ylpyridine [Dong A Pharmaceuticals (WO 01/94342)] (-28 mmol) and sodium hydroxide (9 g, 125 mmol) in dry DMF was evaporated to dryness under reduced pressure. A stirred solution of the involatile residue in dry DMF (50 ML) was treated dropwise at ice-bath temperature with iodomethane (3.0 mL, 48 mmol). The stirred reaction mixture was allowed to warm and then maintained at room temperature for 2 hours. The reaction mixture was partitioned between iced water and ethyl acetate. The organic phase was washed with water, dried over magnesium sulfate, and tehn evaporated under reduced pressure to give a residue that was purified by chromatography on silica gel [elution with dichloromethane: ethyl acetate (60: 1) ] to give: 1. 5-BROMO-2- (L-METHYL-LH-TETRAZOL-5-YL) pyridine (1.397 g), a colorless solid, (TLC: silica-gel, hexanes: ethyl acetate (4: 1), Rf: 0. 3), (DMSO-D6) (300 MHZ) 8 : 4.38 (s, 3H); 8.17 (d, 1H) ; 8.35 (dd, 1H); 8.96 (d, 1H). 2. 5-BROMO-2- (2-METHYL-2H-TETRAZOL-5-YL) pyridine (1.07 g), a colorless solid, (TLC: silica-gel, hexanes: ethyl acetate (4: 1), Rf: 0. 1). 1H-NMR (DMSO-D6) (300 MHZ) 8 4.46 (s, 3H); 8.09 (d, 1H); 8.28 (dd, 1H); 8.88 (d, 1H). Structure assignment based on HMBC experiments, in which long range coupling of the protons of CH3 to the C5 of the tetrazole ring is observed in the 1-METHYL-LH-ISOMER of Rf 0.3, but not in the 2-methyl-2H-isomer of Rf 0.1). The compound referred to as 5-bromo-2- (1-METHYL-LH-TETRAZOL-5-YL) pyridine is thus the isomer of Rf 0.3 and the compound referred to as 5-BROMO-2- (2-METHYL-2H-TETRAZOL-5-YL) pyridine is thus the isomer OF RF 0. 1
With potassium hydroxide; In N,N-dimethyl-formamide; at 20℃; for 23h;
To the solution of crude <strong>[380380-60-9]5-bromo-2-(2H-tetrazol-5-yl)pyridine</strong>(3.15 g,.13.9 mmol) in DMF (32 mL) was added MeI (7.92 g, 55.8 mmol) and KOH (1.95 g, 34.8 mmol) at room temperature. The mixture was stirred for 23 h at r.t.. The reaction mixture was poured into ice water (100 mL) and extracted with EtOAc. The organic layer was washed with brine, dried (Na2SO4), and evaporated under vacuum to give a residue, further purified by flash column chromatography (hexanes - EtOAc from 50: 1 to 10:1) to give 5-bromo-2- (2-methyl-2H-tetrazol-5-yl)pyridine as yellow solid (1.32 g, 43% yield over 2 steps) and 5- bromo-2-(l-methyl-lH-tetrazol-5-yl)pyridine as a white solid (0.97 g, 32% yield, 2 steps).
(5R)-3-[3-fluoro-4-(tributylstannyl)phenyl]-5-(hydroxymethyl)-1,3-oxazolidin-2-one[ No CAS ]
[ 380380-64-3 ]
[ 856866-72-3 ]
Yield
Reaction Conditions
Operation in experiment
48%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; lithium chloride; In N,N-dimethyl-formamide; at 90℃; for 4h;
In a 100 ml reaction flask,Compound 7 (5.0 g, 10 mmol) and 50 ml of DMF were added,2- (1-methyl-tetrazol-5-yl) -5-bromopyridine (2.65, 11 mmol)Lithium chloride (1.7 g),1,1'-bis (diphenylphosphino) ferrocene] palladium chloride (1.0 g),The mixture was stirred at 90 C for 4 hours,After completion of the reaction,To room temperature,And extracted three times with water / ethyl acetate (V / V = 1: 1)The organic layers were combined,Dried over anhydrous sodium sulfate,And concentrated to give 1.7 g of the compound (1)The yield was 48%
26%
With lithium chloride;bis-triphenylphosphine-palladium(II) chloride; In 1-methyl-pyrrolidin-2-one; at 20 - 120℃; for 4h;
In 150ml of 1-methyl-2-pyrrolidone was dissolved 37g of (R)-3- (4- tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol. The solution was added with 19.7g of 2- (2-methyltetrazol-5-yl)-5-bromopyridine, 10.44g of lithium chloride and 2.9g of dichlorobistriphenylphospine palladium (I I) at room temperature and then stirred at the temperature of 120 C for 4 hours. The reaction mixture was added with water and then extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to provide 8g of the title compound. Yield 26%. 1H NMR (DMSO-d6) 5 8.90 (s, lH), 8. 18 (m, 2H), 7.70 (m, 2H), 7.49 (dd, 1H), 5.25 (t, 1H), 4.74 (m, 1H), 4.46 (s, 3H), 4.14 (t, lH), 3. 88 (dd, lH), 3.68 (m, lH), 3. 58 (m, lH)
26%
With bis-triphenylphosphine-palladium(II) chloride; lithium chloride; In 1-methyl-pyrrolidin-2-one; at 120℃; for 4h;Inert atmosphere;
To a solution of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol 5 (37.0 g, 74.0 mmol) in NMP (150 mL) was added 2-(2-methyltetrazol-5-yl)-5-bromopyridine 9 (19.7 g, 81.9 mmol), LiCl (10.4 g, 245 mmol) and Pd(PPh3)2Cl2 (2.90 g, 4.13 mmol). The reaction mixture was stirred for 4 h at 120 C. After being cooled to room temperature, the reaction mixture was poured into water and extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was further purified by column chromatography to obtain the title compound (8 g, 26%). Mp: 201 C. 1H NMR (DMSO-d6): delta 8.90 (s, 1H), 8.18 (m, 2H), 7.70 (m, 2H), 7.49 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 5.25 (t, J = 5.6 Hz, 1H), 4.74 (m, 1H), 4.46 (s, 3H), 4.14 (t, J = 8.8 Hz, 1H), 3.88 (m, 1H), 3.68 (m, 1H), 3.58 (m, 1H). 13C NMR (DMSO-d6): delta 163.56, 159.03, 154.06, 149.18, 144.78, 140.30, 136.96, 131.42, 130.73, 121.89, 118.36, 113.81, 105.18, 73.39, 61.52, 45.96, 39.63. IR (neat, cm-1): 3251.40, 1746.23, 1619.91, 1473.35, 1406.82. [M + H]+: 371.02. HRMS [EI-MS]: m/z, calculated for [C17H15FN6O3] 370.1190, found, 370.1100 [C17H15FN6O3].
With bis-triphenylphosphine-palladium(II) chloride; lithium chloride; at 120℃; for 4h;
37 g of (R) -3- (4-tributyltin-3-fluorophenyl) -2-oxo-5-oxazolidinylmethanol was dissolved in 150 ml of 2- (1- -yl) -5-bromopyridine.19.7 g of 2- (2-methyltetrazol-5-yl) -5-bromopyridine,10.44 g of lithium chloride and 2.9 g of dichlorobis (triphenylphosphine) palladium (II) were added to the solution,Then stirred at 120 C for 4 hours.Water is added to the reaction mixture,Then extracted with ethyl acetate.The organic layer separated is washed with brine, dried, filtered, concentrated in vacuo and purified by column chromatography to give the title compound.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate; In 1,4-dioxane; at 80℃; for 3h;Sealed tube; Inert atmosphere;
To a solution of 10 (1.00 g, 4.17 mmol) in dry degassed l,4-dioxane (15 mL) was added bis(pinacolato)diboron (1.27 g, 5.00 mmol), potassium acetate (1.35 g, 13.75 mmol), and Pd(dppf)Cl2 (0.26 g, 0.35 mmol). The reaction mixture was heated at 80 C for 3 h in a sealed tube under argon. The reaction mixture was filtered through silica gel flash column with CH2Cl2 and the organic layer was evaporated under reduced pressure, and the residue was washed with n-hexane to obtain the title compound (1.03 g, 86.3 %). 1H-NMR (400 MHz, CDCl3) delta: 9.14 (s, 1H), 8.30 (m, 2H), 4.48 (s, 3H), 1.38 (s, 12H).
66%
With potassium acetate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In dimethyl sulfoxide; at 80℃; for 2h;
To the solution of 5-bromo-2-(2-methyl-2H-tetrazol-5-yl)pyridine (480 mg, 2 mmol) in DMSO (5 mL) was added pinacol diborane (1.02 g, 4 mmol), KOAc (588 mg, 6 mmol) and PdCl2(dppf)DCM (160 mg, 0.2 mmol), degassed with N2. The mixture was stirred at 80 0C for 2 h. The reaction mixture was diluted with DCM (100 mL) and washed with brine (2 x 100 mL), dried (Na2SO4) and evaporated under vacuum, then purified by prep-TLC (hexanes - EtOAc) to give 2-(2-methyl-2H-tetrazol-5-yl)-5-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)pyridine as white solid (380 mg, 66% yield).
A solution of 5-bromo-2-(2-methyl-2H-tetrazol-5-yl)pyridine (0.15 g, 0.63 mmol), bispinacolatodiboron (0.176 g, 0.69 mmol) and KOAc (0.076 g, 0.93 mmol) in 1,4- dioxane (5.0 ml.) was degassed by flushing with nitrogen for 15 min. Tricyclohexylphosphine (0.02 g, 0.07 mmol) and tris(dibenzyledineacetone) dipalladium (0) (0.03 g, 0.03 mmol) was then added to the reaction mixture that was again degassed by nitrogen for 15 min. The resulting reaction mixture was heated to 1000C for 3 h. After the completion of the reaction (TLC monitoring), the reaction mixture was filtered through celite bed and the filtrate was concentrated to get the crude residue that was carried forward to the next step without further purification. MS: 288.10 (M+H)+.
With potassium acetate;dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; In 1,4-dioxane; at 80℃;Inert atmosphere;
To the solution of Intermediate 9a (200 mg, 0.83 mmol, prepared as described in PCT WO 2005/058886) in dioxane (2 mL) was added pinacol diborane (270 mg, 1.06 mmol), KOAc (270 mg, 2.75 mmol) and PdCl2(dppf)DCM (60 mg, 0.07 mmol), degassed and protected with N2. The mixture was stirred at 80 C. for 2-3 h. The reaction mixture was diluted with DCM (100 mL) and washed with brine (2×100 mL), dried (Na2SO4) and evaporated under vacuum, then purified by preparation TLC to give 2-(1-methyl-1H-tetrazol-5-yl)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine as white solid (160 mg). 1H NMR (300 MHz, CDCl3, ppm): 9.07 (s, 1H), 8.22 (m, 2H), 4.58 (s, 3H), 1.39 (s, 12H).
With potassium acetate;dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; In dimethyl sulfoxide; at 80℃;Inert atmosphere;
Compound of Example 225-Bromo-2-(1-methyl-1H-tetrazol-5-yl)pyridine (2.44 g, 10 mmol) was dissolved in 30 mL of anhydrous DMSO. To this solution was added bis-(pinocalato)diboron (5.08 g, 20 mmol), followed by KOAc (4.00 g, 40 mmol) and PdCl2(dppf)DCM (0.75 g, 1 mmol). The reaction mixture was degassed, and then stirred at 80 C. o.n. Resulted solution was filtered through Celite, and the precipitate was washed with EtOAc (100 mL). The filtrate was concentrated and washed with 10% NH4Cl, brine, and dried (Na2SO4). Solvent was removed under vacuum, and the residue was dissolved in ether and filtered through a short silica gel pad. The filtrate was concentrated and the formed solid was washed with methanol. Thus isolated [2-(1-methyl-1H-tetrazol-5-yl)pyridyl-5-yl)(pinacolato)boron was obtained as a white solid [1H NMR (400 MHz): 9.10 (s, 1H); 8.25 (s, 2H); 4.48 (s, 3H); 1.48 (s, 12H)].
With 1,1'-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane complex; potassium acetate; In 1,4-dioxane; at 80℃; for 3h;Inert atmosphere;
Compound 2 (120g, 0.5mol), potassium acetate (145g, 0.5mol) and frequencymellow joint boron is (150g, 0.6mol) 10L in four-mouth bottle, by adding 1,4-dioxane (3L) and Pd (dppf)2Cl2·CH2Cl2(20g, 25mmol). After the protection of nitrogen, heating to 80 C, reaction 3 hours. Combined liquid detection for determining compound 2 is fully converted to compound 3 the rear, the compound is added 4 (146g, 0.45mol), potassium carbonate (173g, 1.2mol) and water (1L), re-protection of nitrogen, heating to 80 C reaction 12 hours. LCMS determining the reaction is complete. Filtering, filtering the solid material, the filtrate obtained by reducing pressure and evaporating the evaporimeter 1,4-dioxane, is added to the aqueous phase 500 ml ethanol, beating sleepovers. Filtering, the filter cake is washed with cold-b activity, reduced-pressure drying 4 hours. The crude product in ethyl acetate, heating reflow, holding 5 minutes, the heat filters. Then reducing the temperature to room temperature, the beating sleepovers, filtering, washing the filter cake by ethyl acetate, to obtain compound 1 (177g, 88%), yellow solid.
With tetrakis(triphenylphosphine) palladium(0); potassium acetate; In 1,4-dioxane;Inert atmosphere; Reflux;
Under stirring, 1,4-dioxane (400 ml), 5-bromo-2-(2-methyl-2H-tetrazol-5-yl)pyridine (20.0 g, 83.3 mmol, 1.0 equiv), bis(pinacolato)diboron (25.3 g, 100 mmol, 1.2 equivalents), potassium acetate (20.4 g, 208 mmol, 2.5 eq) and bis(triphenylphosphine)palladium dichloride (1.17 g, 1.4 mmol, 0.015 eq) was added in sequence to the reaction vessel. The solution was warmed to reflux under nitrogen and TLC followed 5-bromo-2-(2-methyl-2H-tetrazol-5-yl)pyridine for complete reaction. After the reaction is completed, it is cooled to below 20 C. (5R)-3-(4-bromo-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one (21.7 g, 75.0 mmol, 0.9 eq.), potassium carbonate (34.5 g, 250 mmol, 3.0 equiv), palladium acetate (0.14 g, 0.62 mmol, 0.0075 equiv) and triphenylphosphine (0.65 g, 2.5 mmol, 0.03 equiv) was added in sequence to the reaction vessel and nitrogen protected. Warming to reflux, TLC tracks 2-(2-methyl-2H-tetrazol-5-yl)-5-(4,4,5,5-tetramethyl-1,3-dioxoboran-2-yl)pyridine until reaction is complete. After the reaction was completed, it was cooled to room temperature and stirred for 16 hours. It was filtered under reduced pressure and the cake was rinsed with water (200 ml) and methanol (200 ml). The filter cake was collected and air dried at 80 C. to give 25.8 g (compound I) as an off-white powder in a yield of 84%.
With tetrakis(triphenylphosphine) palladium(0); potassium acetate; In 1,4-dioxane; at 70℃; for 4h;Inert atmosphere; Large scale;
Add to the reactor4.3kg2-methyl-5-(5-bromopyridin-2-yl)tetrazolium, 5.4kg bis(pinacolato)diboron, 3.5kg potassium acetate, 35.0kg 1,4-dioxane, start stirring, nitrogen Replace twice, under the protection of nitrogen, add 0.29kg of tetrakis(triphenylphosphine)palladium, heat to 70, react for 4h, stop heating after the reaction is completed, add 22.0kg of n-heptane, and cool down to 5, Keep warm and stir for 1h, remove the nitrogen protection, drain the material and filter with suction. The filter cake is rinsed once with 4.0kg n-heptane, suction filtered until no filter droplets are collected, and the filter cake is collected and dried in a vacuum drying oven at 50C. Weighing to obtain pinacol borate;
With potassium carbonate;tris-(dibenzylideneacetone)dipalladium(0); tricyclohexylphosphine; In 1,4-dioxane; water; at 70℃; for 1h;Inert atmosphere;
To a 5 -L, three-neck, round-bottom flask was charged 4 (200.0 g, 0.833 mol) followed by 1,4-dioxane (3 L, 15 vol). Crude compound 6 (361.2 g, 1.249 mol, 1.5 equiv.), Pd2(dba)3 (11.44 g, 0.0125 g, 0.015 equiv.), and PCy3 (7.0 g, 0.025 mol, 0.03 equiv.) was charged and degassed with nitrogen for 30 minutes. A solution Of K2CO3 (195.7 g, 1.7 equiv.) in water (800 mL, 4 vol) was charged, and the reaction was heated to 70 0C. The reaction was complete after 1 hour with 0.5 area % of 4 remaining. The reaction was cooled to 50 0C, and Darco G-60 (40 g, 0.2 wt) was added and stirred for 30 minutes. Celite 545 (40 g, 0.2 wt) was charged and then the reaction was filtered through Celite 545 (100 g, 0.5 wt) wetted with water (300 mL). The hot filtration into the water from the Celite caused precipitation of the product. Tetrahydrofuran (1.2 L, 6 vol) and brine (600 mL, 3 vol) were added, and the product re-dissolved at room temperature. The phase split was accomplished cleanly (Vmax = 28 volumes). The dioxane was concentrated and ethanol (1 L, 5 vol) was added and concentrated. Then the product was reslurried in ethanol: water (4: 1, 2 L, 10 vol) at 700C, cooled to room temperature over 3 hours, filtered and washed with ethanol (2 x 400 mL). Compound 7 was isolated in 87% yield (292.6 g) with a purity of 97.7 % (AUC) by HPLC analysis. The 1H NMR and 19F NMR indicated the presence of one compound. Pd analysis showed 135 ppm Pd was in the product.
55.1%
With tris-(dibenzylideneacetone)dipalladium(0); potassium carbonate; tricyclohexylphosphine; In 1,4-dioxane; water; at 70℃; for 1h;Inert atmosphere;
Under the protection of nitrogen, add 17.7g of 2-methyl-5- (5-bromopyridin-2-yl) tetrazole, 27.7g of borate intermediate, 0.663g of tricyclohexylphosphine and 0.23L of dioxane Stir to dissolve; add a solution of potassium carbonate 175g in water 70mL at one time; add 0.88g of tris (dibenzylideneacetone) dipalladium, vacuum-nitrogen replacement three times, then heat the reaction to 70C, and keep the reaction for 1 hour, filter the reaction Liquid, rinse, add 15% saline solution to the mother liquor (the mass concentration refers to the mass of sodium chloride as a percentage of the total mass of saline solution) 100mL, separate layers, separate organic phase, and concentrate in vacuum (temperature 45 65 , pressure -0.08MPa -0.1MPa) to dry, the crude product is stirred in ethyl acetate 120mL) for 12 hours to 16 hours and filtered, and the wet product is dried in a vacuum (pressure -0.01MPa -0.1MPa) oven Dry at 45 C to 55 C for 8 hours to 12 hours to obtain 26.0 g of off-white solid as terdazolamide phosphate II, with a total yield of 55.1% (based on benzyl 3-fluoro-4-bromophenylcarbamate) ). HPLC purity 96.23%.
DMSO (100 ml) was added to a 250 ml reaction flask,(5R) -3- (4-bromo-3-fluorophenyl) -5-hydroxymethyloxazolidin-2-one(10 g, 34.5 mmol), pinacolate (17.52 g, 69 mmol),[1,1'-bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (1.41 g, 1.73 mmol)And potassium acetate (13.5 g, 138 mmol), and the temperature was raised to 80 C under a nitrogen atmosphere,For 14 hours. The heating was stopped, the solution was cooled to room temperature, and extracted with water / ethyl acetate 3 times. The organic layers were combined and the organic layer was washed with saturated waterBrine, dried over anhydrous sodium sulfate and concentrated by suction filtration. The concentrated product of the above step was added to a 250 ml reaction flask,1,4-dioxane (100 ml) was added,5-bromo-2- (2-methyl-2H-tetrazol-5-yl) pyridine(8.28 g, 34.5 mmol),[1,1'-bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (0.56 g, 0.69 mmol)And cesium carbonate aqueous solution (50 ml, containing 33.72 g of cesium carbonate, 103.5 mmol),Under nitrogen protection,The temperature was raised to 70 C,The reaction was carried out for 3 hours,Dichloromethane was added for extraction.The separated organic phase was washed with saturated brine,Anhydrous sodium sulfate dehydration,filter,Concentrated in vacuo and purified by column chromatography,10.6 g of a solid was obtained,The yield was 83.0%HPLC purity was 98.34% (area normalization method).
With palladium 10% on activated carbon; hydrogen; In methanol; at 20℃; under 1500.15 Torr; for 3h;
1.5 L of methanol,5-bromo-2- (2H-tetrazol-5-yl) pyridine (226 g)Formaldehyde (90g),10% palladium on carbon (20 g) was added to the reactor,Under a hydrogen atmosphere (0.2 MPa)Stirred at room temperature for 3 hours, the reaction was completed,Organic phase evaporated,Recrystallization from acetonitrile,To give the product 2-methyl-5- (5-bromo-2-yl) tetrazolium,217 g, yield: 90.4%.
With calcium hydroxide; In dichloromethane; N,N-dimethyl-formamide; at 0 - 40℃; for 24h;
The <strong>[380380-60-9]5-bromo-2-(2H-tetrazol-5-yl)pyridine</strong> (20.0 g, 88.4 mmol) prepared in Example 1 was added with 20.0 mL of N,N-dimethyl formamide and 180.0 mL of methylene chloride and then further added with calcium hydroxide (3.94 g, 53.0 mmol), after which iodomethane (33.0 mL, 530.4 mmol) was slowly added dropwise thereto at 0C. Thereafter, the reaction solution was warmed to 40C and stuffed for 24 hr. After the termination of the reaction, the reaction solution was added with water, thus extracting an organic layer. The extracted organic layer was washed with saline and further extracted. The resulting organic layer was added with 300.0 mL of a 6 N hydrochloric acid aqueous solution to thus extract an aqueous layer, after which the separated organic layer was added with 60.0 mL of a 6 N hydrochloric acid aqueous solution, so that the aqueous layer was further extracted. Extraction was performed using HPLC until the amount of Ni was less than 5%. The separated aqueous layer was collected, and the pH thereof was adjusted to 10.6 at 40C or less using a 50% sodium hydroxide aqueous solution. The produced solid was filtered, washed with water, and concentrated under reduced pressure, thus obtaining a desired compound. Yield (16.2 g, 70.5%), N2/N] ratio % (98/2)
To a stirred mixture of 5-bromo-2-(2//-tctrazol-5-yl)pyridinc (452 mg, 2 mmol) in THF (5 mL) was added (diazomethyl)trimethylsilane in hexane (2M, 5 mL, 10 mmol) at 0 C. The reaction mixture was stirred at room temperature for 3 h. TLC showed the reaction completed. The reaction mixture was quenched with water (30 mL) and extracted with ethyl acetate (10 mL><3). The combined organic layers were washed with water and brine, dried over sodium sulfate and concentrated in vacuo. The crude product was purified by column chromatography to afford the title compound as white solid (l80mg,28.5 % yield). NMR (500 MHz, DMSO-r/d) S: 9.00 - 8.89 (m, 1H), 8.32 (dd, J= 8.5, 2.4 Hz, 1H), 8.14 (d, J= 8.4 Hz, 1H), 4.34 (s, 3H).
2-fluoro-4-benzyloxycarbonylamino-1-tributylstannylbenzene[ No CAS ]
[ 380380-64-3 ]
[ 1220910-89-3 ]
Yield
Reaction Conditions
Operation in experiment
2.91 g
With potassium phosphate; tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; at 35 - 85℃;Inert atmosphere;
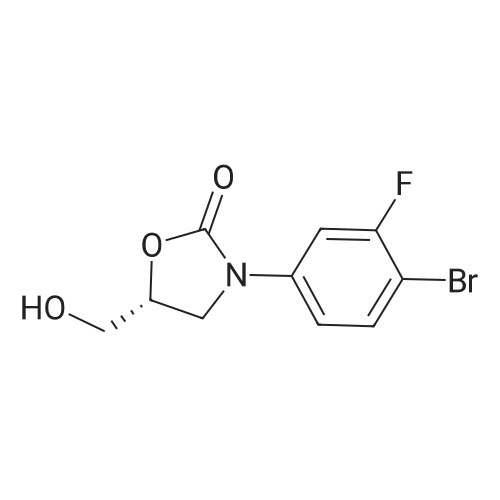
Under nitrogen protection, 5.44 g of 3-fluoro-4-bromophenylcarbamic acid benzyl ester, 67 mL of dioxane and 5.76 g of hexabutylditin are added,Dichlorobistriphenylphosphine palladium 0.93g. Stir at 10 C to 15 C for 2 hours to 3 hours, filter and wash with dioxane to obtain a tributyltin intermediate in dioxane solution.The dioxane solution of the tributyltin intermediate is stirred at 35 C to 45 C, and 2-methyl-5- (5-bromopyridin-2-yl) tetrazolium 4.23g, potassium phosphate 2.09g, tetrakis ( Triphenylphosphine) palladium 0.083g, heated to 75 C-85 C and stirred for 3 hours to 4 hours. After cooling, filter 6g of diatomaceous earth, add 40mL of ethyl acetate and 15% saline (the mass concentration refers to the mass of sodium chloride as a percentage of the total mass of saline), and separate the organic phase Dry, concentrate in vacuum (temperature 35 C- 65 C , pressure -0.08MPa -0.1MPa), add ethyl acetate 26mL, heat to 70 C-75 C , stir for 0.5 hours 1 hour, cool to 0 C- 10 C, stir 1 Hours to 2 hours, filtered, rinsed with ethyl acetate 12mL three times, the wet product was dried in a vacuum (pressure -0.01MPa ~ -0.1MPa) drying oven at 45 C to 55 C for 8 hours to 12 hours, to obtain an off-white solid as tedizolid phosphate intermediate II 2.91g, total yield 42.9%, HPLC purity 97.18%.
With tris-(dibenzylideneacetone)dipalladium(0); sodium carbonate; In tetrahydrofuran; at 20 - 70℃;Inert atmosphere; Large scale;
Under nitrogen protection, to 35.3L of tetrahydrofuran was added 3-fluoro-4-bromophenylcarbamic acid benzyl ester 4.7Kg (14.5mol), stirred at 20 ~ 30 and added zinc powder 3.8Kg (58.1mol), 27.4 g (0.145 mol) of cobaltocene, stirred at 20 C. to 30 C. for 4 hours to 5 hours, filtered, and washed with 11.7 L of tetrahydrofuran to obtain a solution stored under nitrogen, which is a solution of terdazolamide intermediate III.The solution of teridazole amine phosphate intermediate III was stirred at 20 C to 30 C, and 2-methyl-5- (5-bromopyridin-2-yl) tetrazole 3.83Kg (15.95mol) and sodium carbonate 3.1Kg were added (29.2mol), tris (dibenzylideneacetone) dipalladium 26.5g (0.029mol), heated to 65 C to 70 C and stirred for 5 hours to 6 hours.After cooling, filter through 2.5Kg of diatomaceous earth, add 10L of tetrahydrofuran and 15% saline in mass concentration to the mother liquor (the mass concentration refers to the mass of sodium chloride as a percentage of the total mass of saline). After 4L, organic Separate the phases to dry, concentrate in vacuum (temperature 35 C- 55 C , pressure -0.08MPa -0.1MPa) to dryness, draw 30L of ethyl acetate, heat to 70 C- 75 C and stir for 0.5 hours to 1 hour, cool to Stir at 0 to 10 C for 1 to 2 hours, centrifuge, rinse three times with 3L of ethyl acetate, vacuum dry (vacuum degree -0.01MPa to -0.1MPa, temperature 45 C to 55 C) for 16 hours to obtain an off-white solid Tedizolid phosphate intermediate II 4.36kg, total yield 74.4%. HPLC purity 98.74%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +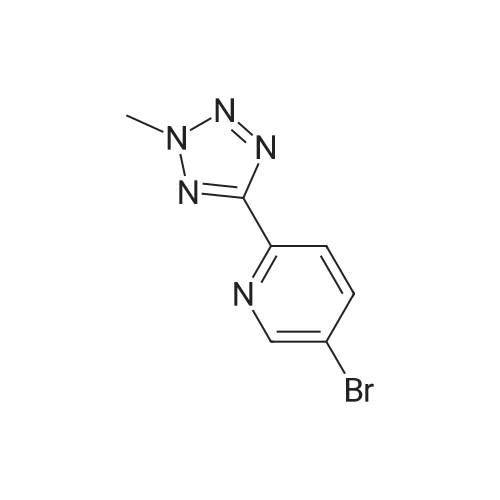

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping