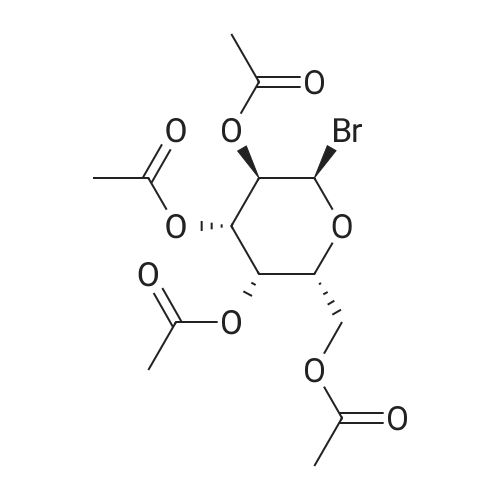
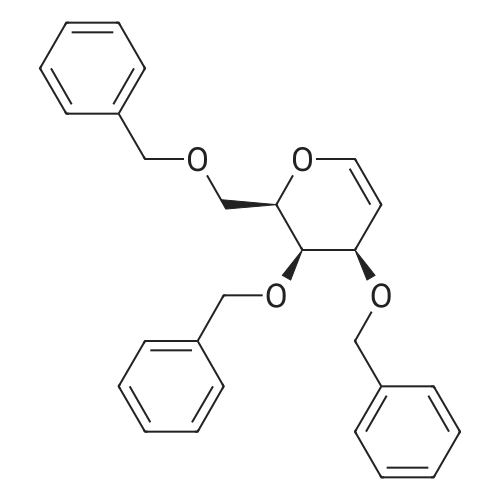
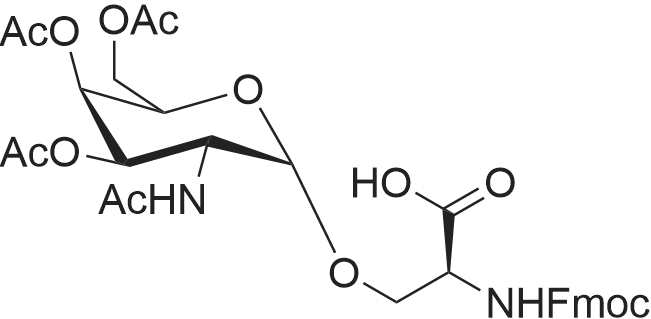
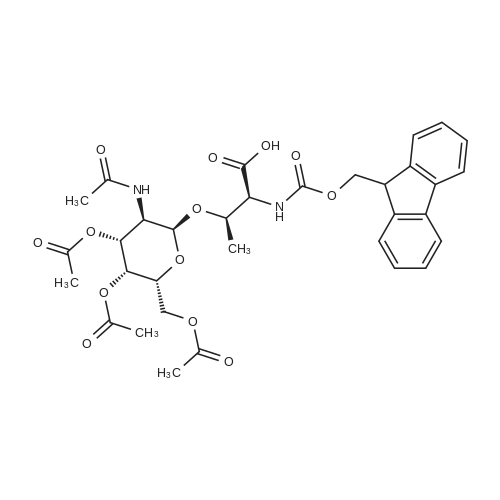
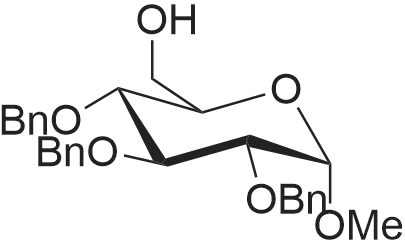
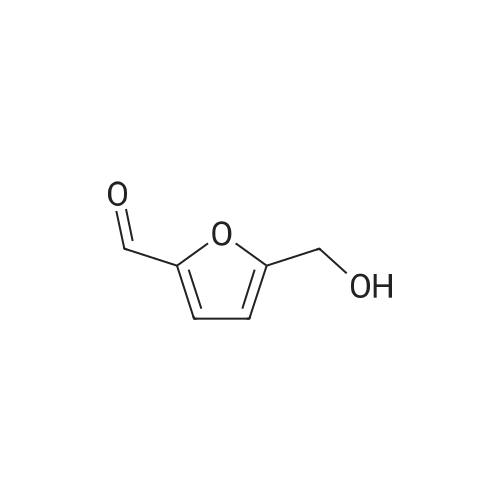
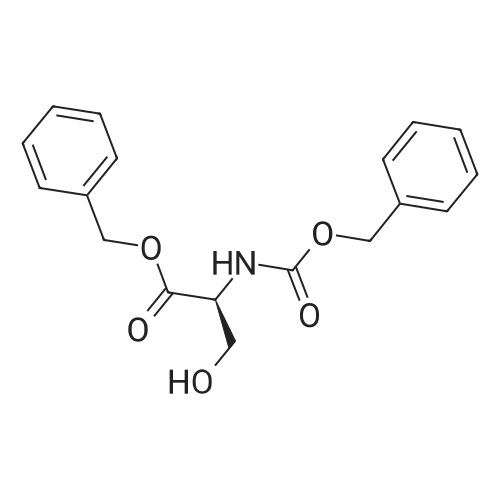
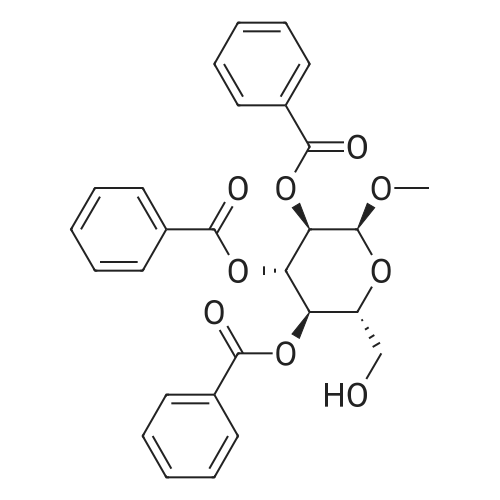
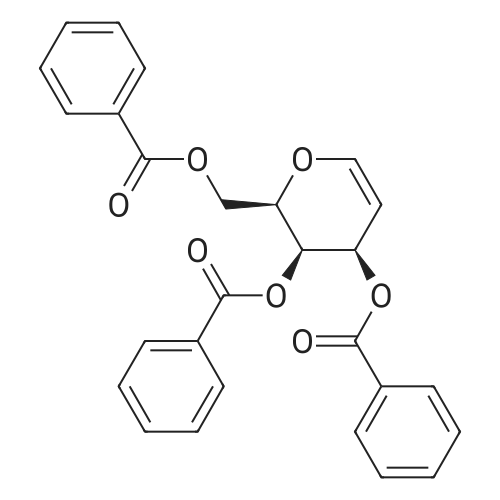
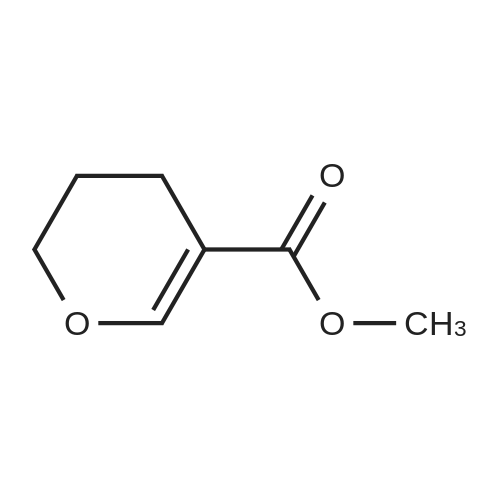
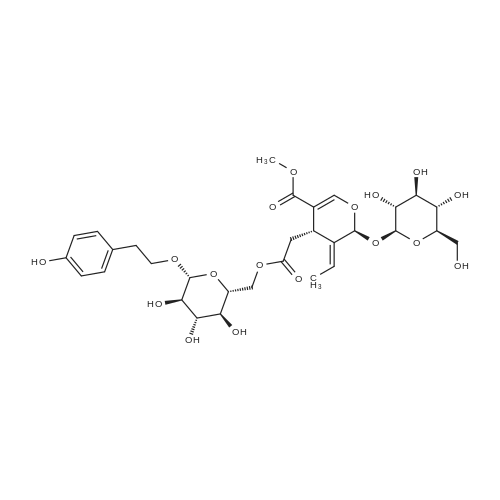
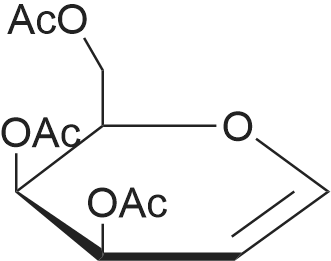
| 93% |
With zinc; sodium acetate; acetic acid In tetrahydrofuran at 20℃; for 1.5h; |
|
| 91% |
With acetic acid; zinc In acetonitrile for 0.833333h; |
|
| 90% |
With ammonium chloride; zinc In acetonitrile at 60℃; for 4h; Alkaline conditions; |
|
| 89% |
With bis(cyclopentadienyl)-titanium(III) chloride In tetrahydrofuran Ambient temperature; |
|
| 89% |
With bis(cyclopentadienyl)-titanium(III) chloride In tetrahydrofuran for 0.25h; Ambient temperature; |
|
| 88.7% |
Stage #1: 1-bromo-1-deoxy-2,3,4,6-tetra-O-acetyl-a-D-galactopyranoside With ammonium chloride In acetonitrile at 20℃;
Stage #2: With zinc In acetonitrile for 1.51667h; |
2.b
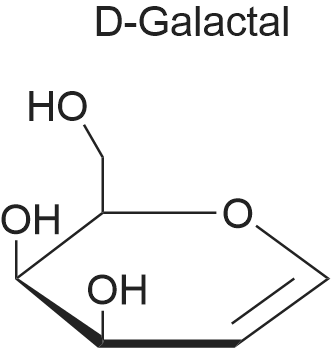
Preparation of 3,4, 6-Tri-O-acetyl-D-galactal; Ammonium chloride (30 g, 0.561 mol) was added to 2,3, 4, 6-tetra-O-acetyl-a-D- galactopyranosyl bromide (97 g, 0.236 mol) in acetonitrile (500 mL) and this reaction mixture was stirred and kept under a blanket of nitrogen at room temperature. Most of the ammonium chloride remained undissolved. Zinc powder (36 g, 0.551 mmol) was added cautiously to the mixture within 1 minute followed by VO (Salen) (0.25 g, mmol) in one batch. After an induction time of about 30 minutes, an exothermic occurred. The solution became lighter and the temperature slowly cools down. Stirring was continued for a further 60 minutes. The progress of the reaction was checked by NMR and after 60 minutes showed that the reaction had finished. Workup of this reaction mixture was carried out in the same manner as for 3,4, 6-tri-O-acetyl-D-glucal. Yield of crude 3,4, 6-tri-O-acetyl-D- galactal (57.0 g, 88. 7%). 1H-NMR (300 MHz, CDC13) : 8 : 6.388 (dd, J=6. 4,1. 4 Hz, 1H), 5.47 (m, lH), 5.349 (dm, J=4. 4 Hz, 1H), 4.672 (ddd, J=6. 3,3. 7,1. 4 Hz, 1H), 4. 268 (dd, J=5. 0,5. 0 Hz, 1H), 4.166 (dd, J=6. 1,4. 3 Hz, 1H), 4. 05 (m, 1H), 2.048, 2.005, 1. 948 (3x CH3). 13C-NMR (75 MHz, CDCl3) : 8 : 170.23, 169.96, 169.85 (3 x C=O), 98.65 (CH- 0), 72.61 (CH-O), 63.69, 63.60 (2x CH), 61.69 (CH2-O), 20.48, 20.42, 20.32 (3x CH3). |
| 84% |
With ammonium chloride; zinc In acetonitrile at 60℃; for 0.583333h; Inert atmosphere; |
2. General procedure for the synthesis of glycals
General procedure: Under nitrogen, the glycopyranosyl bromide (1.0 mmol) was dissolved in CH3CN, and then zinc dust (7.5 mmol) and ammonium chloride (7.5 mmol) were added, followed by stirring at corresponding temperature. Upon completion of the reaction (monitored by TLC), inorganic salts and excessive zinc dust were removed by filtration. The filtrate was concentrated in vacuo and the residue was purified by silica gel flash column chromatography to afford the corresponding glycal in pure form. |
| 81% |
With sodium dihydrogenphosphate; zinc In acetone at 20℃; for 5h; |
|
| 80% |
With zinc In water at 20℃; for 1h; |
|
| 78.7% |
With 1-methyl-1H-imidazole; zinc In ethyl acetate Heating; |
|
| 77% |
In tetrahydrofuran at 20℃; |
|
| 77% |
With manganese; chloro-trimethyl-silane In tetrahydrofuran at 20℃; for 12h; |
|
| 77% |
With 1-methyl-1H-imidazole; zinc In ethyl acetate Heating; |
|
| 72% |
With 1-methyl-1H-imidazole; zinc In ethyl acetate for 3h; Reflux; |
|
| 71% |
With <Cr(II)(EDTA)>(2-) In water; N,N-dimethyl-formamide for 432h; pH 5.0; |
|
| 70% |
With copper(II) sulfate; zinc In water; acetic acid at 20℃; |
|
| 60% |
With aluminium amalgam In tetrahydrofuran; water for 4h; Ambient temperature; |
|
| 60% |
With copper(ll) sulfate pentahydrate; sodium acetate; zinc In water; acetic acid at 0℃; for 3.25h; Inert atmosphere; |
|
| 48% |
With lithium perchlorate In tetrahydrofuran Electrochemical reaction; |
|
|
With acetic acid; zinc |
|
|
With 1-methyl-1H-imidazole; zinc In ethyl acetate for 0.416667h; Heating; Yield given; |
|
|
With 1-methyl-1H-imidazole; zinc In ethyl acetate for 2h; Heating; |
|
| 7.76 g |
With copper(II) sulfate; zinc In water; acetic acid at 20℃; for 18h; |
|
| 2.54 g |
With acetic acid; zinc In water at 0℃; |
|
| 27.7 g |
With acetic acid; zinc In diethyl ether; water at 0℃; for 1h; Inert atmosphere; |
D-Galactal
Using a modification of Kozikowski’s procedure,1 a magnetically stirred solution of D-galactose (S2, 125 mg, 0.7 mmol) in acetic anhydride (75 mL, 790 mmol) was treated dropwise with conc. perchloric acid (0.45 mL, 6.9 mmol). The remaining D-galactose (19.9 g, 110.3 mmol) was slowly added over 30 minutes, at a rate that maintained a temperature of 40-50 °C. Upon complete addition of D-galactose, the solution was allowed to cool to room temperature, then treated with a 33% (w/w) solution of hydrobromic acid in acetic acid (75 mL, 410 mmol). After 90 minutes, the solution was diluted with dichloromethane (180 mL) and washed with ice-cold water (2 x 50 mL), then with cold saturated sodium bicarbonate solution (4 x 100 mL). The organic phase was dried, filtered and concentrated to afford crude tetra-O-acetyl-α-D-galactopyranosyl bromide as a pale-yellow oil.A mechanically stirred dispersion of zinc dust (48.0 g, 734 mmol) in water (150 mL) was cooled to 0 °C, diluted with acetic acid (150 mL), then treated dropwise over one hour with a solution of tetra-O-acetyl-α-D-galactopyranosyl bromide in diethyl ether (150 mL). The reaction was allowed to warm to room temperature and left to proceed overnight. The solution was filtered, then diluted with dichloromethane (200 mL). The solution was then washed successively with water (3 x 60 mL), saturated sodium bicarbonate solution (3 x 50 mL) and brine (60 mL). The organic phase was dried, filtered and concentrated to provide tri-O-acetyl-D-galactal (S5) (27.7 g) as a pale-yellow oil. |
| 24.424 g |
With copper(II) sulfate; zinc In acetic acid at 25℃; for 0.583333h; Inert atmosphere; |
|
| 5.40 g |
With sodium dihydrogenphosphate; zinc In acetone at 20℃; Inert atmosphere; |
4.7 3,4,6-Tri-O-acetyl-D-galactal (11)
To a solution of compound 10 (9.20g, 22.37mmol) in acetone (44.4mL) was added saturated NaH2PO4 solution (89.5mL) and zinc dust (18.29g, 279.63mmol). The reaction mixture was stirred at the room temperature for overnight. The mixture was then diluted with EtOAc and filtered through a pad of celite, then concentrated to dryness in vacuum. The residue was diluted with EtOAc (500mL), washed with saturated NaHCO3(aq) (100mL×2), water (100mL), and brine (100mL). The organic layer was dried over MgSO4 and concentrated to give a crude product, which was purified by flash chromatography with the eluent of EtOAc/hexanes=3/7 to give the desired product 11 (5.40g, 89%) as a colorless oil. 1H NMR (300MHz, CDCl3) δ 6.46 (dd, J=6.3, 1.5Hz, 1H), 5.58-5.53 (m, 1H), 5.42 (dt, J=4.8, 1.7Hz, 1H), 4.73 (ddd, J=6.3, 2.9, 1.7Hz, 1H), 4.35-4.17 (m, 3H), 2.13 (s, 3H), 2.08 (s, 3H), 2.03 (s, 3H); 13C NMR (75MHz, CDCl3) δ 170.6 (C), 170.1 (C), 170.3 (C), 170.1 (C), 145.4 (CH), 98.8 (CH), 72.8 (CH), 63.8 (CH), 63.7 (CH), 61.9 (CH2), 20.8 (CH3), 20.7 (CH3), 20.6 (CH3). |
|
With acetic acid; zinc In water at 0 - 20℃; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping