* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of Materials Chemistry C, 2018, vol. 6, # 32, p. 8733 - 8737
2
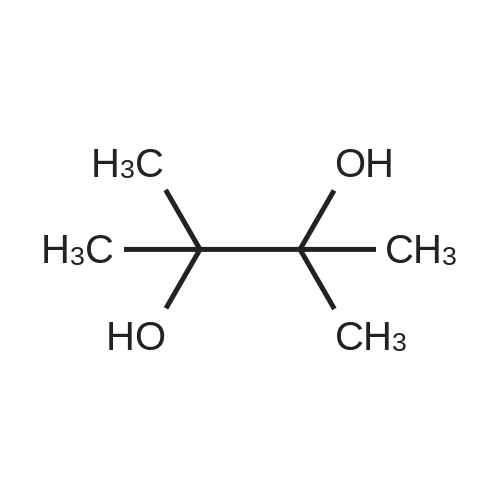
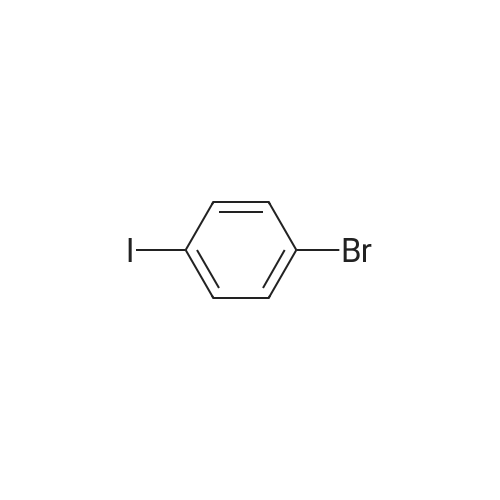
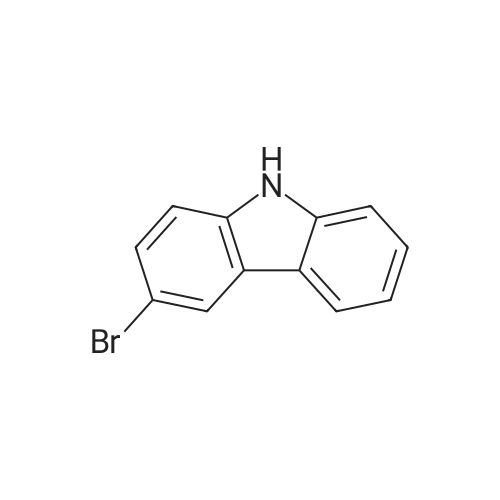
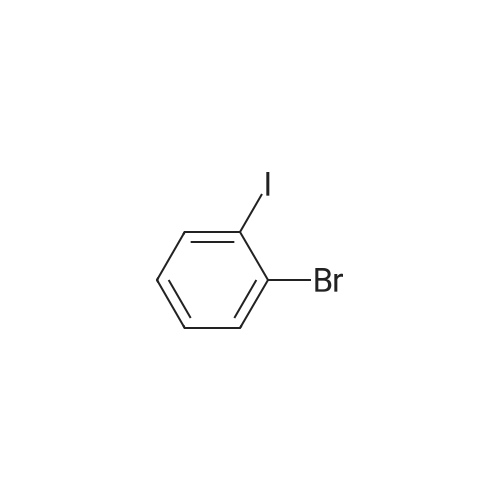
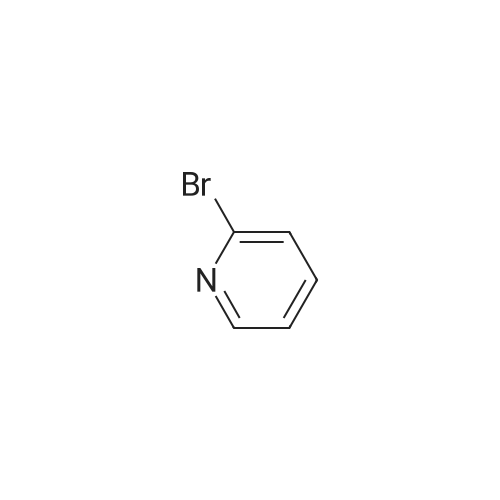
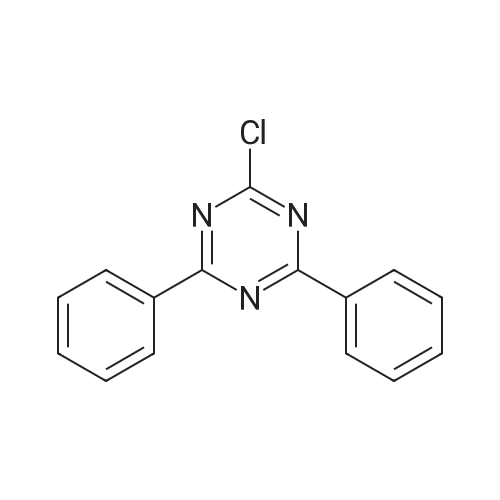
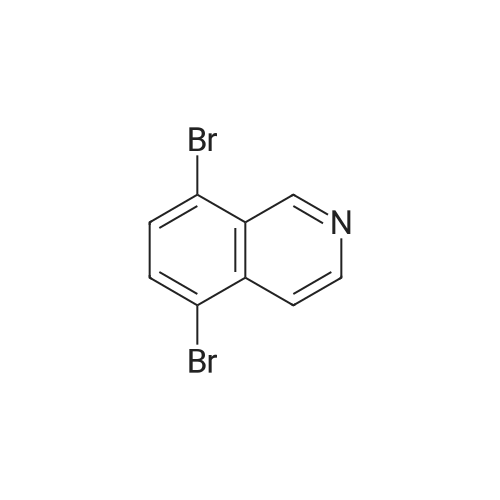
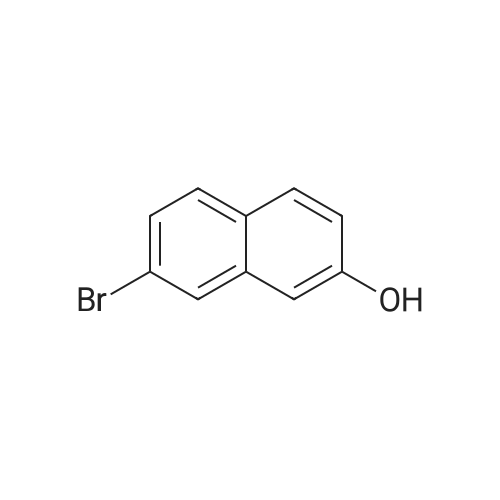
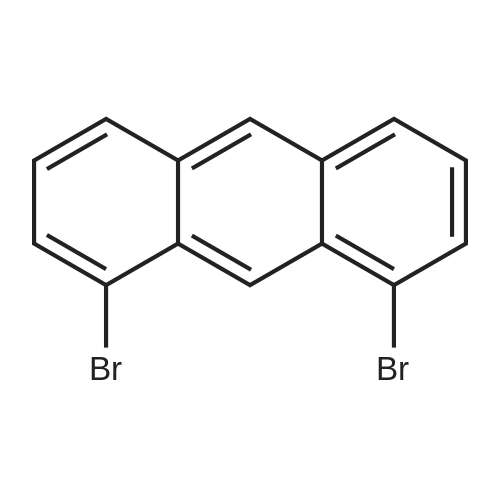
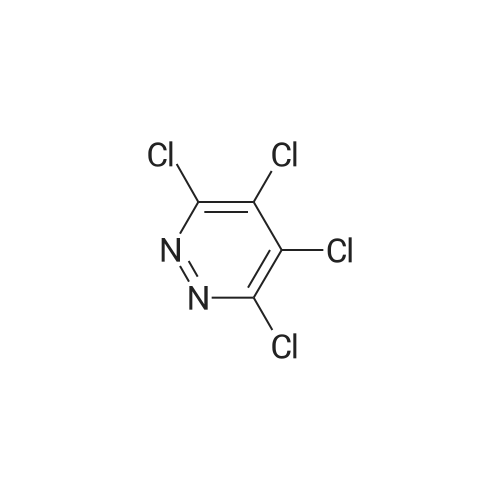
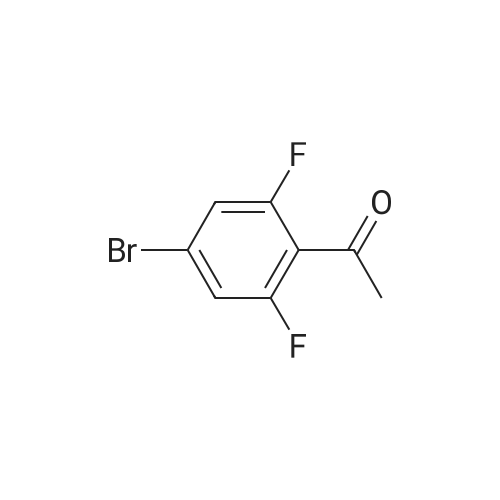
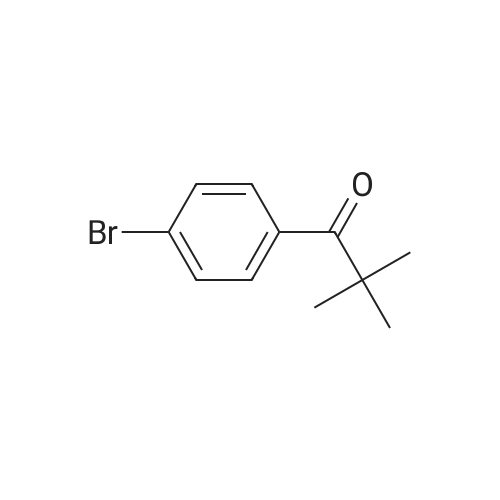
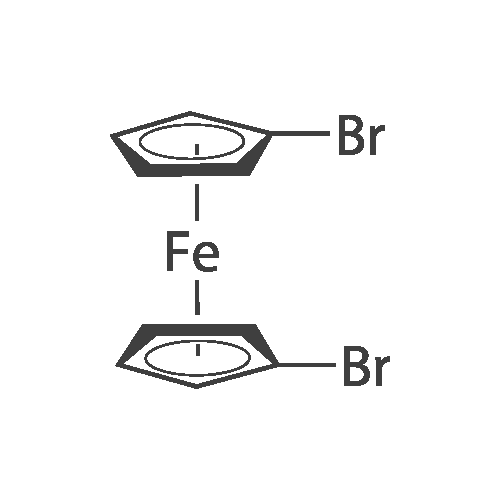
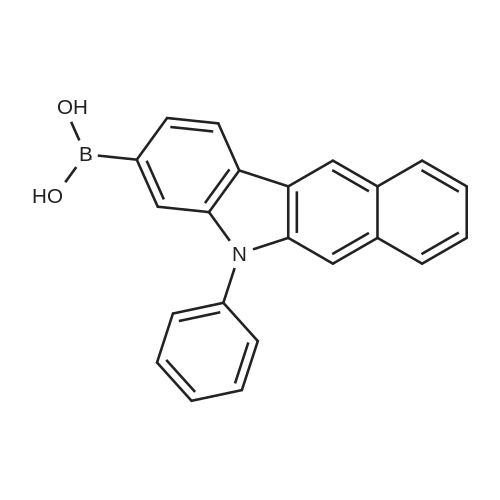
[ 57102-42-8 ]
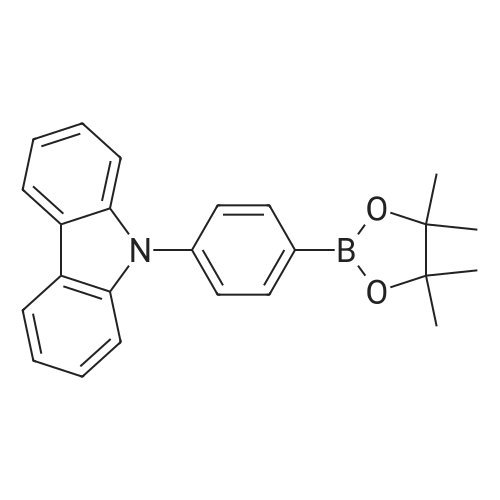
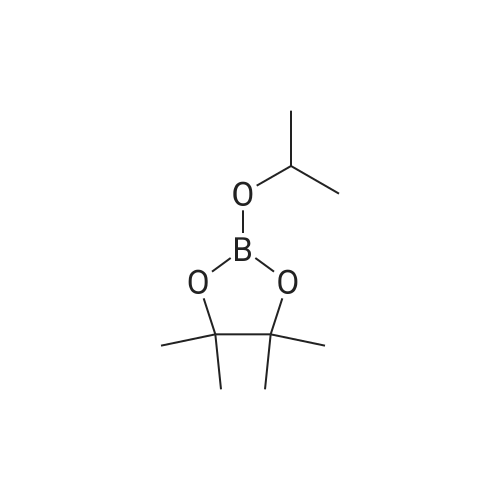
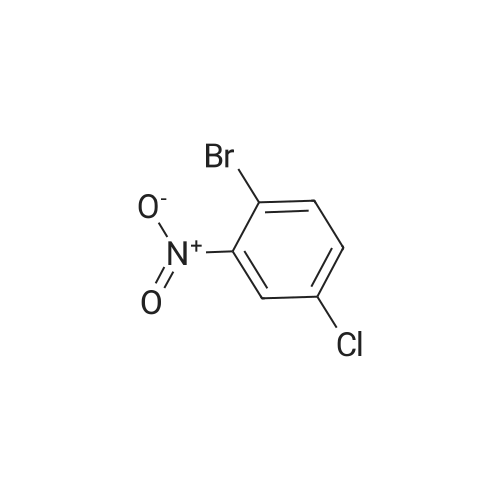
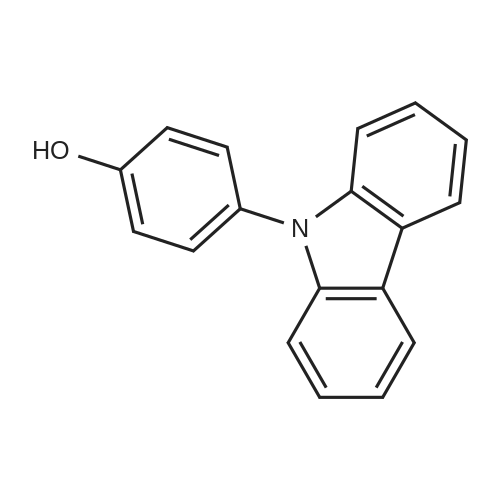
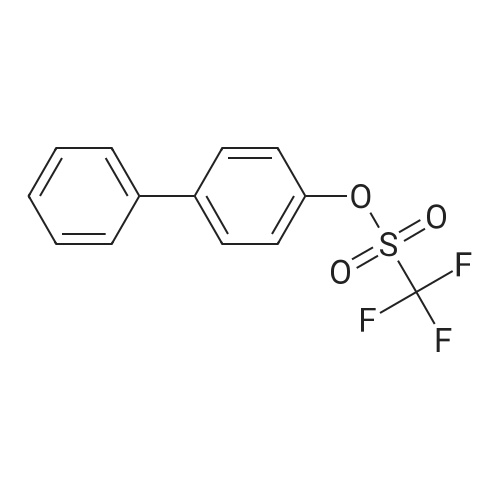
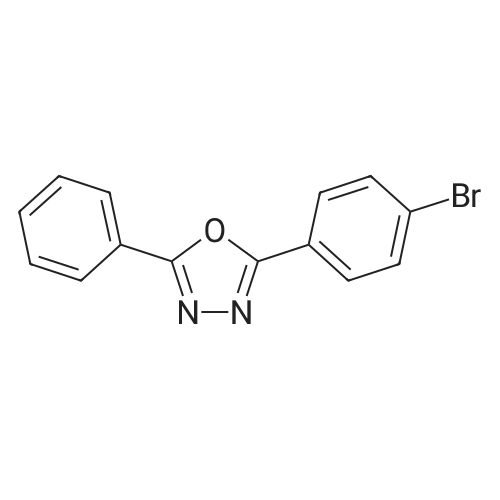
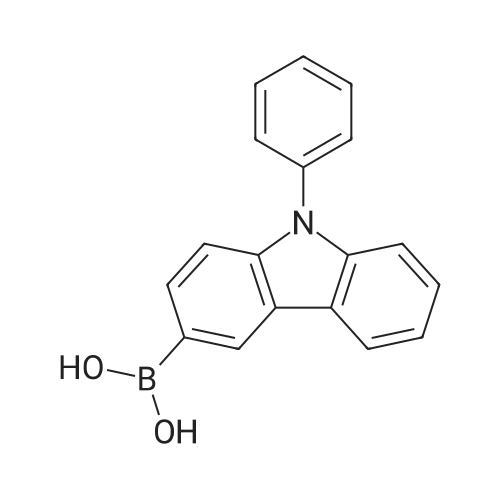
[ 419536-33-7 ]
Yield
Reaction Conditions
Operation in experiment
81%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 1 h; Stage #2: With Triisopropyl borate In tetrahydrofuran; hexane at -78 - 20℃; for 12 h;
After dissolving compound 3-1 (35 g, 108.6 mmol) in THF (600 mL), and adding n-BuLi (52 mL, 130.35 mmol, 2.5 M in hexane) to the reaction mixture at -78°C, the reaction mixture was stirred for 1 hour. The reaction mixture was stirred for 12 hours at room temperature with adding B(Oi-Pr)3 (37 mL, 162.9 mmol) slowly to the reaction mixture, and with slowly increasing the temperature. After extracting with EA, the obtained organic layer was dried with anhydrous MgSO4 to remove the remaining moisture, was distillated under reduced pressure to remove the solvent, and was recrystallized with EA and hexane to obtain compound 3-2 (25 g, 81 percent).
66%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 2 h; Inert atmosphere Stage #2: With Trimethyl borate In tetrahydrofuran; hexane at -78℃; Stage #3: With water In tetrahydrofuran; hexane at 20℃; for 1 h;
(ii) Synthesis of 4-(9H-carbazol-9-yl)phenylboronic acid A synthesis scheme of 4-(9H-carbazol-9-yl)phenylboronic acid is illustrated in (E-2). In a 500 mL three-neck flask was put 21.8 g (67.5 mmol) of 9-(4-bromophenyl)-9H-carbazole. The atmosphere in the flask was replaced with nitrogen. To the mixture was added 200 mL of tetrahydrofuran (THF). The temperature of this solution was set to -78° C., and then 48.9 mL (74.3 mmol) of n-butyllithium (a 1.52 mol/L hexane solution) was dripped to the solution. The mixture was stirred at the same temperature for 2 hours. After that, 17.4 mL (155 mmol) of trimethyl borate was added to the mixture, followed by stirring for 1 hour at the same temperature. Then, the mixture was stirred for 24 hours while the temperature thereof is returned to room temperature. After that, 200 mL of 1.0 mol/L hydrochloric acid was added to this solution, followed by stirring for 1 hour at room temperature. The organic layer of the mixture was washed with water, and the aqueous layer was extracted with ethyl acetate. The extract and the organic layer were combined, washed with saturated brine, and then dried with magnesium sulfate. After that, this mixture was suction filtered. The resulting filtrate was concentrated to give a residue. This residue was recrystallized with a mixed solvent of chloroform and hexane to give 4-(9H-carbazol-9-yl)phenylboronic acid which was the desired substance as 13 g of a white powdered solid in a yield of 66percent.
65.9%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 2 h; Inert atmosphere Stage #2: With Trimethyl borate In tetrahydrofuran at -78 - 20℃; for 25 h; Stage #3: With hydrogenchloride; water In tetrahydrofuran at 20℃; for 1 h;
Then, 21.8 g (67.5 mmol) of 9-(4-bromophenyl)carbazole was put into a 500 mL three-necked flask, and the air in the flask was replaced with nitrogen. Then, 200 mL of tetrahydrofuran (THF) was added thereto. The temperature in the flask was set to be -78° C. and then, 48.9 mL (74.3 mmol) of n-butyllithium (1.52 mol/L, hexane solution) was dropped to the mixture and was stirred for 2 hours at the same temperature. Then, 17.4 mL (155 mmol) of trimethyl borate was added thereto. The mixture was stirred for one hour at the same temperature, then, was stirred for 24 hours while raising the temperature to room temperature. After the reaction, 200 mL of hydrochloric acid (1.0 mol/L) was added to the reaction mixture, and then stirred for one hour at room temperature. After that, the organic layer in the reaction mixture was washed with water and the product was extracted with ethyl acetate from the water which was used for the washing. The obtained extraction solution and the organic layer, which was washed with water, were mixed. The mixture was further washed with a saturated aqueous solution of sodium chloride, and then dried with magnesium sulfate. After the drying, the mixture was suction filtrated, and the filtrate was concentrated. The obtained residue was recrystallized with a mixed solvent of chloroform and hexane to give 12.8 g of a white powdered solid of 4-(carbazol-9-yl)phenylboronic acid (yield: 65.9percent). A synthesis scheme (f-3) of 4-(carbazol-9-yl)phenylboronic acid is shown below.
57%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 1 h; Stage #2: With Trimethyl borate In tetrahydrofuran; hexane for 24 h; Stage #3: With hydrogenchloride; water In tetrahydrofuran; hexane
After Compound B-2 (4 g, 12.4 mmol) was dissolved in THF (40 mL), the mixture was cooled to -78 °C . 10 monutes later, n-butyl lithium(2.5M in hexane) (5.95 mL, 14.88 mmol) was slowly added into a flask and the mixture was stirred for 1 hour. Trimethyl borate (2.35 mL, 18.6 mmol) was slowly added into a flask and the mixture was stirred for 24 hours. When the reaction was completed, IM HCl was added thereto. After extracting with ethyl acetate and removing remaining moisture by using magnesium sulfate, drying followed by column separation yielded Compound B-3 (2 g, 6.96 mmol, 57 percent) .
Reference:
[1] Organic and Biomolecular Chemistry, 2012, vol. 10, # 33, p. 6693 - 6704
[2] Australian Journal of Chemistry, 2007, vol. 60, # 8, p. 603 - 607
[3] Physical Chemistry Chemical Physics, 2011, vol. 13, # 39, p. 17825 - 17830
[4] Organic and Biomolecular Chemistry, 2012, vol. 10, # 47, p. 9481 - 9490
[5] Patent: WO2013/12297, 2013, A1, . Location in patent: Paragraph 111; 114; 115
[6] Inorganica Chimica Acta, 2011, vol. 370, # 1, p. 340 - 345
[7] Patent: US8093399, 2012, B2, . Location in patent: Page/Page column 107
[8] Patent: US7758972, 2010, B2, . Location in patent: Page/Page column 78; 79
[9] Patent: WO2011/14039, 2011, A1, . Location in patent: Page/Page column 20; 21
[10] Inorganica Chimica Acta, 2012, vol. 391, p. 50 - 57
[11] Chemistry Letters, 2008, vol. 37, # 3, p. 262 - 263
[12] Advanced Functional Materials, 2010, vol. 20, # 15, p. 2448 - 2458
[13] Journal of Materials Chemistry, 2012, vol. 22, # 37, p. 19700 - 19708,9
[14] Journal of Materials Chemistry, 2012, vol. 22, # 37, p. 19700 - 19708
3
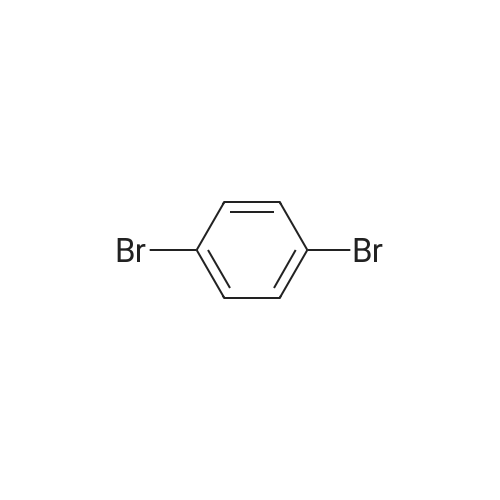
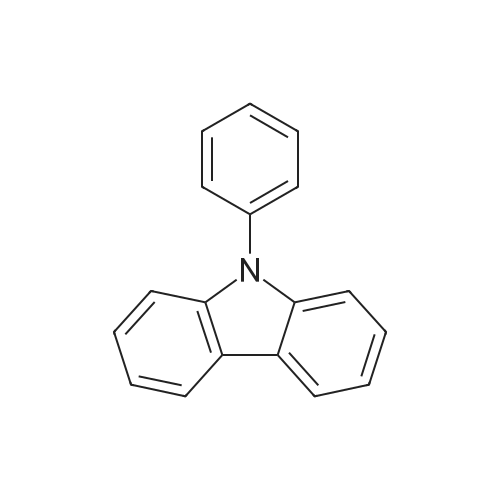
[ 121-43-7 ]
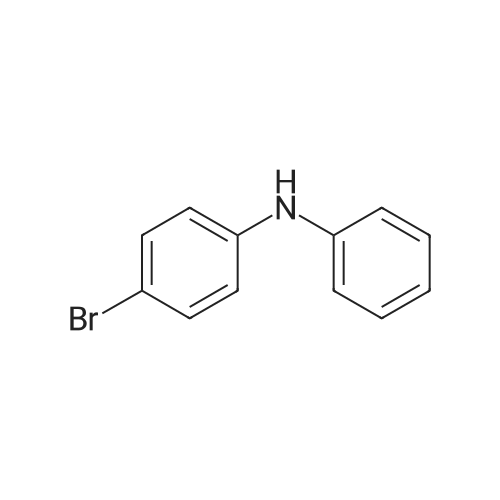
[ 57102-42-8 ]
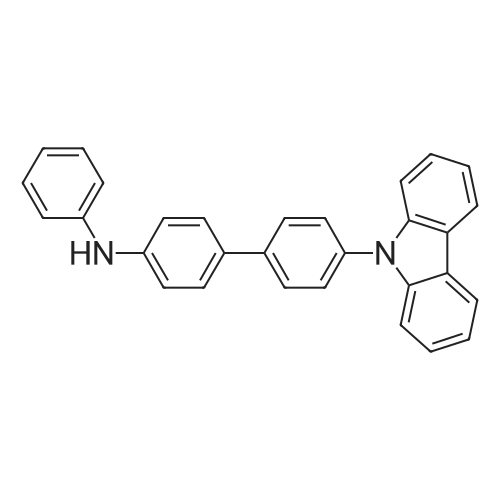
[ 419536-33-7 ]
Yield
Reaction Conditions
Operation in experiment
85%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78 - -70℃; for 1.5 h; Inert atmosphere Stage #2: at -70 - 20℃; Inert atmosphere
To a 200 mL two-necked round bottom flask containing 2-2 (11.13 g, 34.67 mmol) in 150 mL dry THF, 2.4M n-BuLi in hexane (10 mL, 24 mmol) was added dropwise at -78 °C under N2 protection. After stirring for 1.5 hr with the temperature below -70 °C, B(OMe)3 (8 mL, 71.8 mmol) was added dropwise into the reaction mixture. The solution mixture was allowed to stir overnight and slowly warmed to ambient temperature. There after, the solution was quenched with water and extracted using ethyl acetate. The combined organic layer was washed with saturated NaCl (aq) and dried over Na2SO4. Thus, it was concentrated and purified using column chromatography to obtain a white solid 8.46 g in 85percent yield which was directly used in the next step.
80%
Stage #1: With n-butyllithium In tetrahydrofuran at -78℃; for 1 h; Stage #2: at 20℃;
(2) 9-(4-bromophenyl)-9H-carbazole 4.00g of THF was added 50mL of dry, cooling to -78 deg.] C, n-butyllithium was slowly added 6mL (the 2.4M), for 1 hour, followed by after addition of trimethyl borate 1.8mL, reaction was continued for 2 hours. after the reaction warmed to room temperature overnight, cooled to 0 deg.] C, was added HCl (2.0 M) 40ml hydrolysis, and extracted with methylene chloride, the solvent was evaporated, to give 2.84g (4-(9H-carbazol-9-yl)phenyl)boronic acid in 80percent yield.
66%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 2 h; Stage #2: at -78 - 20℃; for 13 h; Stage #3: With hydrogenchloride; water In tetrahydrofuran; hexane at 20℃; for 1 h;
(ii) Synthesis of 4-(carbazol-9-yl)phenylboronic acid A synthesis scheme of 4-(carbazol-9-yl)phenylboronic acid is shown in (B-4).; 21.8 g (67.5 mmol) of N-(4-bromophenyl)carbazole was put into a 500-mL three-neck flask and nitrogen substitution was carried out. After that, 200 mL of tetrahydrofuran (THF) was added to keep the reaction system at -78 0C inside. 48.9 mL (74.3 mmol) of n-butyllithium (1.52 mol/L hexane solution) was dropped to this reaction solution, and the solution was stirred at the same temperature for 2 hours. 17.4 mL (155 mmol) of trimethyl borate was added and the solution was stirred at -78 <n="78"/>°C for 1 hour, and thereafter, the solution was stirred for 12 hours while the reaction temperature was allowed to gradually increase to the room temperature. After reaction, 200 mL of hydrochloric acid (1 mol/L) was added to the reaction solution, and the solution was stirred at the room temperature for 1 hour. The reaction mixture was washed with water to extract a water layer with ethyl acetate. The extracted solution and an organic layer were washed with saturated saline together and dried with magnesium sulfate. After drying, the mixture was subjected to suction filtration, and the filtrate was condensed to obtain a solid. The solid was recrystallized with a mixed solution of chloroform and hexane, whereby 12.8 g of a white powdery solid that was a target matter was obtained with the yield of 66 percent.
66.5%
Stage #1: With n-butyllithium In tetrahydrofuran at -78℃; for 1 h; Inert atmosphere Stage #2: Inert atmosphere
3.44 g (10.72 mmol) of intermediate 1 was weighed250 mL of dry three-necked flask,80 mL of dry tetrahydrofuran was added as the reaction solvent.Under nitrogen protection,This was cooled to -78 & lt; 0 & gt; C with liquid nitrogen acetone,The mixture was stirred at this temperature for 5 min,10.0 mL (1.6 mol / L, 16.08 mmol) of n-butyllithium was added dropwise to the reaction system using a syringe.After 1 hour of reaction,A syringe was added dropwise to the reaction system2.4 mL (42.89 mmol) of trimethyl borate,And allowed to stir overnight with natural warming.After the reaction, the solution was quenched by adding dilute hydrochloric acid solution.After tetrahydrofuran was spin-dried, the reaction solution was extracted with methylene chloride (100 mL x 3)The extracts were combined and dried over anhydrous magnesium sulfate,The solvent was removed and dried.The crude product was purified by column chromatography using dichloromethane and ethyl acetate as the eluent gradient,To give 2.05 g of a white crystalline solid,Yield 66.5percent.
65.9%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 2 h; Stage #2: at -78 - 20℃; for 25 h; Stage #3: With hydrogenchloride; water In tetrahydrofuran; hexane at 20℃; for 1 h;
[Step 2: Synthesis of 4-(9H-carbazol-9-yl)phenylboronic acid] 21.8 g (67.5 mmol) of 9-(4-bromophenyl)-9H-carbazole was put into a 500 mL three-neck flask. The air in the flask was replaced with nitrogen. To the mixture were added 200 mL of tetrahydrofuran (THF), and then the solution was cooled to -78 °C. Into this solution, 48.9 mL (74.3 mmol) of n-butyllithium (a 1.52 mol/L hexane solution) was dropped, and the solution was stirred at the same temperature for 2 hours. After the stirring, 17.4 mL (155 mmol) of trimethyl borate was added to the solution, and the solution was stirred for about 1 hour at the same temperature. Then, the mixture was stirred for 24 hours while the temperature of the solution was being increased to room temperature. Thereafter, to the solution was added about 200 mL increased to room temperature. Thereafter, to the solution was added about 200 mL (1.0 mol/L) of hydrochloric acid, and then the solution was stirred at room temperature for 1 hour. The organic layer of the mixture was washed with water, and then the aqueous layer was extracted with acetate ether. The extract was combined with the organic layer and then washed with a saturated saline solution. The organic layer was dried with magnesium sulfate. After the drying, the mixture was subjected to suction filtration, and then the obtained filtrate was condensed. The obtained residue was recrystallized with chloroform/hexane to give 12.8 g of a white powdered solid, which was the object of the synthesis, at a yield of 65.9 percent. A synthesis scheme of Step 2 is shown in (f-2) given below.
65.9%
With hydrogenchloride; n-butyllithium; sodium chloride In tetrahydrofuran
Then, 21.8 g (67.5 mmol) of 9-(4-bromophenyl)carbazole was put into a 500 mL three-necked flask, and the air in the flask was replaced with nitrogen. Then, 200 mL of tetrahydrofuran (THF) was added thereto. The temperature in the flask was set to be -78° C. and then, 48.9 mL (74.3 mmol) of n-butyllithium (1.52 mol/L, hexane solution) was dropped to the mixture and was stirred for 2 hours at the same temperature. Then, 17.4 mL (155 mmol) of trimethyl borate was added thereto. The mixture was stirred for one hour at the same temperature, then, was stirred for 24 hours while raising the temperature to room temperature. After the reaction, 200 mL of hydrochloric acid (1.0 mol/L) was added to the reaction mixture, and then stirred for one hour at room temperature. After that, the organic layer in the reaction mixture was washed with water and the product was extracted with ethyl acetate from the water which was used for the washing. The obtained extraction solution and the organic layer, which was washed with water, were mixed. The mixture was further washed with a saturated aqueous solution of sodium chloride, and then dried with magnesium sulfate. After the drying, the mixture was suction filtrated, and the filtrate was concentrated. The obtained residue was recrystallized with a mixed solvent of chloroform and hexane to give 12.8 g of a white powdered solid of 4-(carbazol-9-yl)phenylboronic acid (yield: 65.9percent).
Reference:
[1] Chemical Communications, 2013, vol. 49, # 34, p. 3597 - 3599
[2] Journal of the Chilean Chemical Society, 2015, vol. 60, # 2, p. 2971 - 2974
[3] Inorganica Chimica Acta, 2011, vol. 370, # 1, p. 340 - 345
[4] Patent: CN103588770, 2016, B, . Location in patent: Paragraph 0052
[5] Journal of Materials Chemistry C, 2015, vol. 3, # 22, p. 5835 - 5843
[6] Patent: WO2008/26614, 2008, A1, . Location in patent: Page/Page column 76-77
[7] Patent: CN106243086, 2016, A, . Location in patent: Paragraph 0012
[8] Patent: EP1972619, 2008, A1, . Location in patent: Page/Page column 282
[9] Patent: US2008/145700, 2008, A1,
[10] Chemistry Letters, 2008, vol. 37, # 3, p. 262 - 263
[11] Organic and Biomolecular Chemistry, 2012, vol. 10, # 33, p. 6693 - 6704
[12] Dyes and Pigments, 2013, vol. 96, # 3, p. 705 - 713
[13] Patent: EP2857395, 2015, A1, . Location in patent: Paragraph 0105
[14] Turkish Journal of Chemistry, 2015, vol. 39, # 5, p. 917 - 929
[15] Chemistry of Heterocyclic Compounds, 2016, vol. 52, # 6, p. 379 - 387[16] Khim. Geterotsikl. Soedin., 2016, vol. 52, # 6, p. 379 - 387,9
4
[ 5419-55-6 ]
[ 57102-42-8 ]
[ 419536-33-7 ]
Yield
Reaction Conditions
Operation in experiment
86%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane for 1 h; Inert atmosphere Stage #2: Inert atmosphere
Cz-PhBr (15 mmol) was solved into THF in flask with gasreplacement by Ar for three times. After cooling down to 78 °C for10 min. N-butyl lithium (t-BuLi, 11 mL, 1.6 mol L1 in hexane solution)was dropped into and stirred for 1 h at 78 °C. The Triisopropylborate (23 mmol) was dropped into flask and stirred foranother 1 h at 78 °C the slowly warmed to room temperature andstirred overnight. Hydrochloric acid (HCl, 30 mL, 2 mol L1) wasadded into the flask and stirred for another 30 min. The productswas extracted by Dichloromethane (CH2Cl2). After evaporation, thecrude product was washed with pure petroleum ether. A whitepowder achieved with yield of 86percent.
85.91%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 1 h; Inert atmosphere Stage #2: at -78 - 20℃; Inert atmosphere
Compound 1 (4.83g, 15.00mmol) was put into the flask. THF (60mL) was added with the syring. The air in the flask was replaced with Ar three times. Then the reaction solution was cooled down to−78°C for 10min. N-butyl lithium (11.25mL, 1.6mol/L in hexane solution) was dropped into above flask dropwise with the syring, then stirred for 1hat−78°C. Triisopropyl borate ([(CH3)2CHO]3B, 5.20mL, 22.50mmol) was added into above solution and then stirred for 1h at−78°C. After that, the reaction solution was warmed slowly to room temperature and stirred overnight. Hydrochloric acid (HCl, 30mL, 2mol/L) was added into the flask and stirred for another 30min. Dichloromethane was used to extract the product and the organic layer was dried over MgSO4. After filtration, the solution was concentrated using rotary evaporator. Then the crude product was washed with pure petroleum ether to afford white powder, compound 2, 3.70g (85.91percent). 1H NMR (400MHz, DMSO-d6): δ 8.25 (d, J=7.2Hz, 2H), 8.08 (dd, J=23.0, 7.2Hz, 2H), 7.61 (d, J=7.4Hz, 2H), 7.43 (s, 4H), 7.30 (s, 2H). 13C NMR (100MHz, DMSO-d6): δ 140.43, 138.92, 136.39, 126.72, 125.84, 123.28, 120.97, 120.57, 110.17.
67%
Stage #1: With n-butyllithium In tetrahydrofuran at -78℃; for 1 h; Inert atmosphere Stage #2: at 20℃; for 12 h; Inert atmosphere Stage #3: With hydrogenchloride In tetrahydrofuran; water for 0.166667 h;
Under a nitrogen atmosphere, 9.8 g (0.030 mol) of the intermediate E compound and 152 mL (0.2 M) of tetrahydrofuran were placed in a 500 mL two-neck round bottom flask and the temperature was lowered to -78 ° C. To this, a n-butyllithium dilution solution (1.6M, 38.02 mL) was slowly added, and the mixture was stirred for 1 hour while maintaining the temperature at -78 ° C. 17.2 g (0.091 mol) of triisopropylborate was slowly added to the above mixed solution, the temperature of the mixed solution was slowly raised to room temperature, and the mixture was stirred for 12 hours. After the reaction was completed, 1N hydrochloric acid was added, and after 10 minutes, the reaction mixture was washed with water and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate and concentrated under reduced pressure to remove the solvent. The resulting material was separated by column separation using a mixed solvent of hexane and ethyl acetate to obtain 5.9 g (67percent) of intermediate F.
67%
Stage #1: With n-butyllithium In tetrahydrofuran at -78℃; for 1 h; Inert atmosphere Stage #2: at 20℃; for 12 h; Inert atmosphere
Under nitrogen, 9.8 g (0.030 mol) of Intermediate-E compound and 152 mL (0.2 M) of tetrahydrofuran were added to a 500 mL 2-neck round bottom flask and the temperature was lowered to -78 ° C. To this, a n-butyllithium dilution solution (1.6M, 38.02 mL) was added slowly, and the mixture was stirred for 1 hour while maintaining the temperature at -78 ° C. 17.2 g (0.091 mol) of triisopropylborate was slowly added to the above mixture, the temperature of the mixture was slowly raised to room temperature, and the mixture was stirred for 12 hours. After the reaction was completed, 1N hydrochloric acid was added, and after 10 minutes, the reaction mixture was washed with water and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate and concentrated under reduced pressure to remove the solvent. The resulting material was separated by column separation using a mixed solvent of hexane and ethyl acetate to obtain 5.9 g (67percent) of intermediate F.
Reference:
[1] Journal of Materials Chemistry C, 2014, vol. 2, # 46, p. 9858 - 9865
[2] Angewandte Chemie - International Edition, 2014, vol. 53, # 19, p. 4850 - 4855[3] Angew. Chem., 2014, vol. 126, # 19, p. 4950 - 4955,6
[4] Angewandte Chemie - International Edition, 2016, vol. 55, # 9, p. 3049 - 3053[5] Angew. Chem., 2016, vol. 128, # 9, p. 3101 - 3105,5
[6] Dyes and Pigments, 2017, vol. 142, p. 175 - 182
[7] Dyes and Pigments, 2016, vol. 129, p. 34 - 42
[8] Patent: KR2015/29332, 2015, A, . Location in patent: Paragraph 0052; 0066; 0067
[9] Patent: KR2015/29331, 2015, A, . Location in patent: Paragraph 0055; 0066; 0067
5
[ 150-46-9 ]
[ 57102-42-8 ]
[ 419536-33-7 ]
Yield
Reaction Conditions
Operation in experiment
87%
With n-butyllithium In tetrahydrofuran at -78 - 20℃; for 12 h; Inert atmosphere
In N2in the gas purification system, the compound dissolved in tetrahydrofuran "g" and stirring. At -78 ° C a temperature of n-BuLi (26.9mmol) is slowly added into the solution, and the mixing solution 1 hour. By keeping the low-temperature conditions, adding boric acid triethyl ester (21.6mmol), and stirring the mixed solution at room temperature. In stirring the mixed solution at room temperature 12 hours, and complete reaction. Slowly adding distilled water, and adding distilled water/salt acid (8:2) to the mixed solution of pH2. The extraction solution with distilled water and ethyl acetate. From the extraction of using magnesium sulphate remove the moisture in the organic layer, and removing the solvent. The material through the use of hexane and ethyl acetate to carry out wet refining column chromatography, thereby obtaining compound "h". (Yield: 87percent)
Reference:
[1] Patent: CN105585577, 2016, A, . Location in patent: Paragraph 0225; 0026; 0227; 0228
6
[ 121-43-7 ]
[ 57102-42-8 ]
[ 7732-18-5 ]
[ 419536-33-7 ]
Yield
Reaction Conditions
Operation in experiment
66.5%
Stage #1: With n-butyllithium In tetrahydrofuran at -78℃; for 1 h; Inert atmosphere Stage #2: at 20℃; Inert atmosphere Stage #3: With hydrogenchloride In tetrahydrofuranInert atmosphere
Weigh 1.72 g (5.36 mmol) of intermediate 1 in 100 mL of dry two-necked flask, 50 mL of dry tetrahydrofuran was added as the reaction solvent. Under nitrogen protection, It was cooled to -78 °C with liquid nitrogen acetone and stirred at this temperature for 5 min, Under nitrogen, 5.0 mL (1.6 mol / L, 8.04 mmol) of n-butyllithium was added dropwise to the reaction system. After reacting at -78 °C for 1 hour, 1.2 mL (10.72 mmol) of trimethyl borate was added dropwise to the reaction system, and the mixture was stirred at room temperature overnight. After the completion of the reaction, dilute hydrochloric acid solution was added and the solution was made acidic. The reaction solution was extracted with dichloromethane (50 mL x 3) The extracts were combined and dried over anhydrous magnesium sulfate, and the solvent was taken out and dried. The crude product was purified by column chromatography using dichloromethane and ethyl acetate as the eluent gradient to give 1.02 g of a white crystalline solid in 66.5percent yield.
60%
Stage #1: With n-butyllithium In tetrahydrofuran at -78℃; for 1 h; Inert atmosphere Stage #2: for 2 h; Inert atmosphere Stage #3: With hydrogenchloride In tetrahydrofuran at 0℃; Inert atmosphere
Under nitrogen 9- (4-bromophenyl) -9H- carbazole THF was cooled to 150 ml of the mixture (10g, 1.0eq) in -78 . After dropwise addition of n-BuLi (18.6 ml, 1.5eq) it was slowly stirred for 1 hour. After adding the trimethyl borate (5.2ml, 1.5eq) it was stirred for 2 hours. Cooled to 0 back extracted with MC was added to 6M HCl. The concentrate EA: hexane = 1: 1 was obtained by column separation of white solid compound 70-5. (5.4g, 60percent)
Reference:
[1] Patent: CN106243091, 2016, A, . Location in patent: Paragraph 0020
[2] Patent: KR2015/75169, 2015, A, . Location in patent: Paragraph 0192-0193; 0202-0203
7
[ 7647-01-0 ]
[ 121-43-7 ]
[ 57102-42-8 ]
[ 419536-33-7 ]
Yield
Reaction Conditions
Operation in experiment
58%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 3 h; Inert atmosphere Stage #2: at -78 - 20℃; for 24 h;
19.6 g (60.7 mmol) of N-(4-bromophenyl)carbazole synthesized in Step 1 of Example 3 was put into a 500 mL three-neck flask, and the atmosphere in the flask was substituted by nitrogen. 100 mL of tetrahydrofuran (THF) was added to the flask and cooled to -78° C. Into this solution, 66.8 mL (42.3 mmol) of n-butyllithium hexane solution (1.58 mol/L) wad dropped under a nitrogen gas flow, and the solution was stirred at the same temperature for three hours. Then, 13.5 mL (140 mmol) of trimethyl borate was added into this solution at the same temperature, and while the temperature of the solution was being increased to room temperature, the solution was stirred for about 24 hours. About 200 mL of 2.0 mol/L hydrochloric acid was added into this solution and stirred at room temperature for one hour. This solution was extracted with ethyl acetate and the extracted solution and an organic layer were washed together with a saturated brine and dried with magnesium sulfate. The mixture was filtrated, and the filtrate was concentrate to obtain a solid substance. The obtained solid substance was recrystallized with a mixed solvent of chloroform and hexane, so that 10.2 g of the white powder of 4(9H-carbazol-9-yl)phenylboronic acid was obtained in yield 58percent.
With potassium carbonate;palladium diacetate; tris-(o-tolyl)phosphine; In ethanol; water; toluene; at 100℃; for 6.5h;
[Step 2] Synthesis of 4-[4(9H-carbazol-9-yl)phenyl]diphenylamine (abbreviation: YGBA).; A synthetic scheme of 4-[4(9eta-carbazol-9-yl)phenyl]diphenylamine (abbreviation: YGBA) is shown in (C-36). [0505] . . ;(C-36)[0506]2.2 g (8.8 mmol) of 4-bromodiphenylamine, 2.5 g (8.8 mmol) of triphenylamine-4-boronic acid, and 398 mg (1.3 mmol) of tri(otolyl)phosphine were put into a 200 mL three-neck flask, and the inside of the flask was substituted with nitrogen.30 mL of toluene, 20 mL of ethanol, and 14 mL of potassium carbonate aqueous solution(0.2 mol/L) were added to this mixture. This mixture was degassed under reduced pressure while being stirred, and 59 mg (0.26 mmol) of palladium acetate (II) was added.This mixture was refluxed for 6.5 hours at 100 0C. After this mixture was left to cool for about 15 hours, a light blackish-brown solid was precipitated. This solid was collected by suction filtration, and 2.5 g of the target compound was obtained as light <n="201"/>blackish-brown solid in 70% yield.
70%
With tri-tert-butyl phosphine;palladium diacetate; In ethanol; water; toluene; at 100℃; for 6.5h;Inert atmosphere;
2.2 g (8.8 mmol) of 4-bromodiphenylamine, 2.5 g (8.8 mmol) of 4-(9H-carbazol-9-yl)phenylboronic acid, 398 mg (1.3 mmol) of tri(o-tolyl)phosphine were put in a 200 mL three-neck flask, and the atmosphere in the flask was substituted by nitrogen. 30 mL of toluene, 20 mL of ethanol and 14 mL of potassium carbonate aqueous solution (0.2 mol/L) were added into this mixture. This mixture was deaerated under reduced pressure while being stirred, and 59 mg (0.26 mmol) of palladium(II) acetate was added thereto. The mixture was refluxed at 100 C. for 6.5 hours. This mixture was left to be cooled for about 15 hours, so that a gray solid substance was precipitated. The gray solid substance was collected by suction filtration, so that the gray solid substance was obtained in 70% yield.
7.9 g
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; for 24h;Reflux;
4-bromodiphenylamine and 6 g (20.24 mmol) of 9-carbazole phenyl-4-boronic acid (20.24 mmol) in 70 ml of tetrahydrofuran and a solution of 8.4 g (60.7 mmol) of potassium carbonate in 70 ml of water And the mixture was stirred at an elevated temperature. When refluxing0.47 g (0.41 mmol) of tetrakis (triphenylphosphine) palladium was added thereto and reacted for 24 hours.After cooling, the water and the organic layer were separated, and the organic layer was dried. The organic layer was dried over anhydrous magnesium sulfate, filtered through celite, concentrated under reduced pressure to obtain a column, and 7.9 g of 1-A was obtained after drying.
With sodium carbonate;palladium diacetate; tris-(o-tolyl)phosphine; In 1,2-dimethoxyethane; water; at 90℃; for 4h;
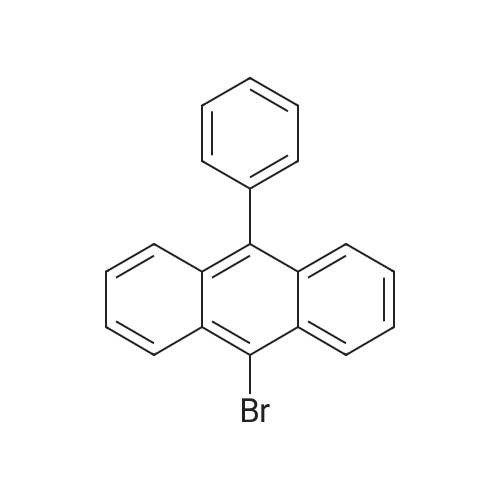
[Step 3] Synthesis of 9-[4-(JV-carbazolyl)]phenyl-10-phenylanthracene (abbreviation: CzPA); A synthesis scheme of CzPA is shown in (B-5).; 1.44 g (4.32 mmol) of 9-bromo-lO-phenylanthracene, 1.49 g (5.19 mmol) of <strong>[419536-33-7]4-(carbazol-9-yl)phenylboronic acid</strong>, 38.0 mg (0.17 mmol) of palladium acetate(II), and 0.36 g (1.21 mmol) of tris(o-tolyl)phosphine were put into a 100-mL three-neck flask and nitrogen substitution was carried out. Then, 10 mL of ethylene glycol dimethyl <n="79"/>ether (DME) and 6.5 mL (13.0 mmol) of a sodium carbonate aqueous solution (2.0 mol/L) were added and the solution was stirred at 90 0C for 4 hours. After that, the reaction mixture was subjected to suction filtration to collect a precipitated solid. The obtained solid was dissolved into chloroform, and the solution was subjected to suction filtration through Florisil, celite, and then alumina. The filtrate was condensed to obtain a solid, and the solid was recrystallized with a mixture solvent of chloroform and hexane, whereby 1.81 g of a light yellow powdery solid that was a target matter was obtained with the yield of 85 %. By a nuclear magnetic resonance measurement (NMR), it was confirmed that this compound was 9-[4-(JV-carbazolyl)]phenyl-10-phenylanthracene (abbreviation: CzPA). [0229]1H NMR data of CzPA is shown below. 1H NMR (300MHz, CDCl3) ; delta = 8.22(d, J = 7.8Hz, 2H), 7.86-7.82 (m, 3H), 7.61-7.36 (m, 20H). The 1K NMR chart is shown in FIGS. 1OA and 1OB. It is to be noted that the range of 6.5 ppm to 8.5 ppm in FIG 10A, which is expanded, is shown in FIG 1OB.
9-(biphenyl-4-yl)-10-[4-(carbazol-9-yl)phenyl]anthracene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
72%
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; at 80℃; for 12h;
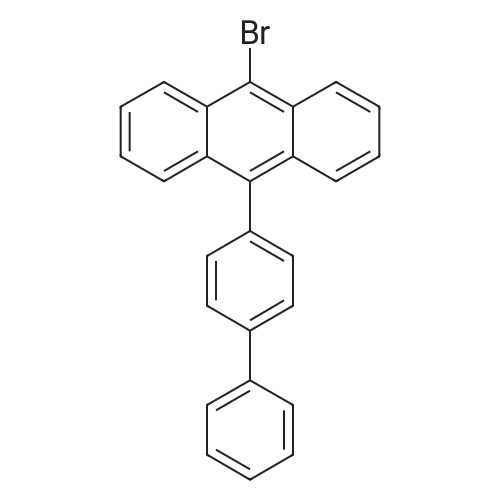
[Step 2] Synthesis of 9-(biphenyl-4-yl)-10-[4-(carbazol-9-yl)phenyl]anthracene (abbreviation: PPCzPA); A synthesis scheme of PPCzPA is shown in (C-3).; 3.0 g (7.3 mmol) of 9-(biphenyl-4-yl)-10-bromoanthracene and 2.1 g (7.3 mmol) of <strong>[419536-33-7]4-(carbazol-9-yl)phenylboronic acid</strong> were put into a 100-mL three-neck flask, and nitrogen substitution in the system was carried out. 25 mL of ethylene glycol dimethyl ether (DME) and 10 mL (2.0 mol/L) of a potassium carbonate solution were added to this mixture, and the mixture was stirred under reduced pressure and degassed. After that, 85 mg (0.017 mmol) of tetrakis(triphenylphosphine)palladium(0) was added. <n="85"/>This reaction mixture was stirred at 80 C for 12 hours under nitrogen gas stream. Then, the reaction mixture was cooled to the room temperature, and a solid that was precipitated was collected by suction filtration. The collected solid was dissolved in toluene, and the solution was subjected to suction filtration through Florisil, celite, and then alumina. The filtrate was condensed to obtain a solid, and the solid was recrystallized with a mixture solvent of chloroform and hexane, whereby 2.9 g of a light yellow powdery solid, which was a target matter, was obtained with the yield of 72 %. By a nuclear magnetic resonance measurement (NMR), it was confirmed that this compound was 9-(biphenyl-4-yl)-10-[4-(carbazol-9-yl)phenyl]anthracene (abbreviation: PPCzPA). [0248]1H NMR data of PPCzPA is shown below. 1H NMR (300MHz, CDCl3) ; delta = 7.33-7.61(m,13H), 7.68-7.88(m,14H), 8.21(d,J = 7.8Hz, 2H). The 1H NMR chart is shown in FIGS. 15A and 15B. It is to be noted that the range of 6.5 ppm to 8.5 ppm in FIG 15A, which is expanded, is shown in FIG 15B.
9-bromo-10-(4-trifluoromethylphenyl)anthracene[ No CAS ]
9-[4-(carbazol-9-yl)phenyl]-10-(4-trifluoromethylphenyl)anthracene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
38%
With potassium carbonate;palladium diacetate; tris-(o-tolyl)phosphine; In water; toluene; at 80℃; for 12h;
[Step 2] Synthesis of9-[4-(carbazol-9-yl)phenyl]-10-(4-trifluoromethylphenyl)anthracene (abbreviation:CF3CzPA); A synthesis scheme of CF3CzPA is shown in (E-4). [0291]Pd(PPh3)4, 2M Na2CO3 aq., toluene (E-4)[0292] 3.0 g (7.5 mmol) of 9-bromo-10-(4-trifluoromethylphenyl)anthracene, 2.2 g(7.5 mmol) of <strong>[419536-33-7]4-(carbazol-9-yl)phenylboronic acid</strong>, and 200 mg (0.66 mmol) of tri(o-tolyl)phosphine were put into a 100-mL three-neck flask, and nitrogen substitution in the system was carried out. 25 mL of toluene and 10 mL (2.0 mol/L) of a potassium carbonate solution were added to this mixture, and the mixture was stirred under reduced pressure and degassed. After that, 60 mg (0.27 mmol) of palladium acetate(II) was added. This reaction mixture was stirred at 80 C for 12 hours under nitrogen gas stream. Then, the reaction mixture was washed with water three times. A water layer of the mixture was extracted three times using ethyl acetate, the extracted solution and an organic layer were washed together using saturated saline, and the organic layer was dried with magnesium sulfate. The mixture was filtrated naturally to remove magnesium sulfate, and the filtrate was condensed to obtain a solid, and the obtain was <n="99"/>purified by silica gel column chromatography (hexane: toluene = 65:35). The resulting solid was recrystallized with hexane, whereby 1.6 g of a light yellow powdery solid, which was a target matter, was obtained with the yield of 38 %. By a nuclear magnetic resonance measurement (NMR), it was confirmed that this compound was 9-[4-(carbazol-9-yl)phenyl]-10-(4-trifluoromethylphenyl)anthracene (abbreviation: CF3CzPA). [0293]1H NMR data of CF3CzPA is shown below. 1H NMR (300MHz, CDCl3) ; delta = 7.33-7.54(m,8H), 7.60-7.74(m,8H), 7.83-7.92(m,6H), 8.22(d,J = 7.8Hz, 2H). The 1R NMR chart is shown in FIGS. 25A and 25B. It is to be noted that the range of 6.5 ppm to 8.5 ppm in FIG 25A, which is expanded, is shown in FIG. 25B.
9-(4-(10-(naphthalen-2-yl)anthracen-9-yl)phenyl)-9H-carbazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
57%
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; at 80℃; for 12h;
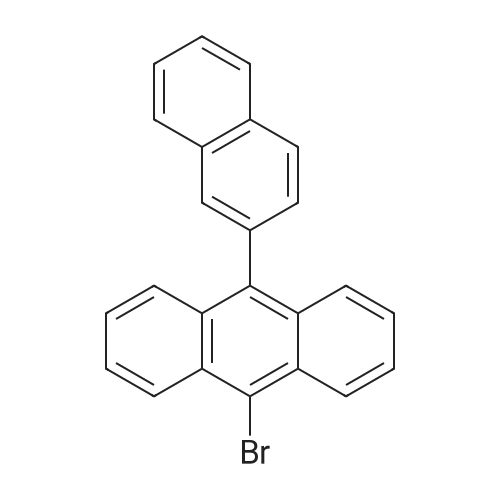
[Step 2] Synthesis of 9-[4-(carbazol-9-yl)phenyl]-10-(2-naphthyl)anthracene (abbreviation: betaNCzPA); A synthesis scheme of betaNCzPA is shown in (F-3).; 3.0 g (7.8 mmol) of 9-bromo-10-(2-napthyl)anthracene and 2.3 g (7.8 mmol) of<strong>[419536-33-7]4-(carbazol-9-yl)phenylboronic acid</strong> were put into a 100-mL three-neck flask, and nitrogen substitution in the system was carried out. 25 mL of ethylene glycol dimethyl ether (DME) and 10 mL (2.0 mol/L) of a potassium carbonate solution were added to this mixture, and the mixture was stirred under reduced pressure and degassed. After <n="105"/>that, 90 mg (0.017 mmol) of tetrakis(triphenylphosphine)palladium(0) was added. This reaction mixture was stirred at 80 C for 12 hours under nitrogen gas stream. Then, the reaction mixture was washed with water. A water layer of the mixture was extracted using ethyl acetate, the extracted solution and an organic layer were washed together using saturated saline, and the organic layer was dried with magnesium sulfate. The mixture was filtrated naturally to remove magnesium sulfate. The filtrate was condensed to obtain a solid, and the solid was purified by silica gel column chromatography (hexanertoluene = 7:3). The resulting solid was recrystallized with hexane, whereby 2.4 g of a light yellow solid, which was a target matter, was obtained with the yield of 57 %. By a nuclear magnetic resonance measurement (NMR), it was confirmed that this compound was9-[4-(carbazol-9-yl)phenyl]-10-(2-naphthyl)anthracene (abbreviation: betaNCzPA). [0314]1H NMR data of betaNCzPA is shown below. 1H NMR (300MHz, CDCl3) ; delta = 7.33-7.56(m,9H), 7.59-7.78(m,9H), 7.83-7.89(m,4H), 7.92-7.95(m,lH), 8.01-8.06(m, 2H), 8.10(d,J = 8.7Hz, IH), 8.22(d,J = 7.2Hz, 2H). The 1H NMR chart is shown in FIGS. 3OA and 3OB. It is to be noted that the range of 6.5 ppm to 8.5 ppm in FIG 30A, which is expanded, is shown in FIG 3OB.
After dissolving compound 3-1 (35 g, 108.6 mmol) in THF (600 mL), and adding n-BuLi (52 mL, 130.35 mmol, 2.5 M in hexane) to the reaction mixture at -78C, the reaction mixture was stirred for 1 hour. The reaction mixture was stirred for 12 hours at room temperature with adding B(Oi-Pr)3 (37 mL, 162.9 mmol) slowly to the reaction mixture, and with slowly increasing the temperature. After extracting with EA, the obtained organic layer was dried with anhydrous MgSO4 to remove the remaining moisture, was distillated under reduced pressure to remove the solvent, and was recrystallized with EA and hexane to obtain compound 3-2 (25 g, 81 %).
66%
(ii) Synthesis of 4-(9H-carbazol-9-yl)phenylboronic acid A synthesis scheme of 4-(9H-carbazol-9-yl)phenylboronic acid is illustrated in (E-2). In a 500 mL three-neck flask was put 21.8 g (67.5 mmol) of 9-(4-bromophenyl)-9H-carbazole. The atmosphere in the flask was replaced with nitrogen. To the mixture was added 200 mL of tetrahydrofuran (THF). The temperature of this solution was set to -78 C., and then 48.9 mL (74.3 mmol) of n-butyllithium (a 1.52 mol/L hexane solution) was dripped to the solution. The mixture was stirred at the same temperature for 2 hours. After that, 17.4 mL (155 mmol) of trimethyl borate was added to the mixture, followed by stirring for 1 hour at the same temperature. Then, the mixture was stirred for 24 hours while the temperature thereof is returned to room temperature. After that, 200 mL of 1.0 mol/L hydrochloric acid was added to this solution, followed by stirring for 1 hour at room temperature. The organic layer of the mixture was washed with water, and the aqueous layer was extracted with ethyl acetate. The extract and the organic layer were combined, washed with saturated brine, and then dried with magnesium sulfate. After that, this mixture was suction filtered. The resulting filtrate was concentrated to give a residue. This residue was recrystallized with a mixed solvent of chloroform and hexane to give 4-(9H-carbazol-9-yl)phenylboronic acid which was the desired substance as 13 g of a white powdered solid in a yield of 66%.
65.9%
Then, 21.8 g (67.5 mmol) of 9-(4-bromophenyl)carbazole was put into a 500 mL three-necked flask, and the air in the flask was replaced with nitrogen. Then, 200 mL of tetrahydrofuran (THF) was added thereto. The temperature in the flask was set to be -78 C. and then, 48.9 mL (74.3 mmol) of n-butyllithium (1.52 mol/L, hexane solution) was dropped to the mixture and was stirred for 2 hours at the same temperature. Then, 17.4 mL (155 mmol) of trimethyl borate was added thereto. The mixture was stirred for one hour at the same temperature, then, was stirred for 24 hours while raising the temperature to room temperature. After the reaction, 200 mL of hydrochloric acid (1.0 mol/L) was added to the reaction mixture, and then stirred for one hour at room temperature. After that, the organic layer in the reaction mixture was washed with water and the product was extracted with ethyl acetate from the water which was used for the washing. The obtained extraction solution and the organic layer, which was washed with water, were mixed. The mixture was further washed with a saturated aqueous solution of sodium chloride, and then dried with magnesium sulfate. After the drying, the mixture was suction filtrated, and the filtrate was concentrated. The obtained residue was recrystallized with a mixed solvent of chloroform and hexane to give 12.8 g of a white powdered solid of 4-(carbazol-9-yl)phenylboronic acid (yield: 65.9%). A synthesis scheme (f-3) of 4-(carbazol-9-yl)phenylboronic acid is shown below.
57%
After Compound B-2 (4 g, 12.4 mmol) was dissolved in THF (40 mL), the mixture was cooled to -78 C . 10 monutes later, n-butyl lithium(2.5M in hexane) (5.95 mL, 14.88 mmol) was slowly added into a flask and the mixture was stirred for 1 hour. Trimethyl borate (2.35 mL, 18.6 mmol) was slowly added into a flask and the mixture was stirred for 24 hours. When the reaction was completed, IM HCl was added thereto. After extracting with ethyl acetate and removing remaining moisture by using magnesium sulfate, drying followed by column separation yielded Compound B-3 (2 g, 6.96 mmol, 57 %) .
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In water; toluene; at 80℃; for 6h;
Under an argon atmosphere, Intermediate 4-1 (0.70 g, 2.0 mmol), 4-(N-carbazolyl)phenylboronic acid (1.26 g, 4.4 mmol), tetrakis(triphenylphosphine)palladium (0.14 g, 0.12 mmol), toluene (10 mL), and a 2-M aqueous solution of sodium carbonate (3 mL, 6 mmol) were added to a 200-mL three-necked flask, and the whole was heated at 80C for 8 hours. Water (100 mL) was added to the reaction liquid in such a manner that a solid would be precipitated. Then, the solidwas filtered. The resultant was purified by means of silica gel column chromatography (amount 1.0 g, yield 74 %) . The purified product was identified as Compound 6 on the basis of 1H-NMR and FD-MS. The measured value of the FD-MS was 676.
To a 200 mL two-necked round bottom flask containing 2-2 (11.13 g, 34.67 mmol) in 150 mL dry THF, 2.4M n-BuLi in hexane (10 mL, 24 mmol) was added dropwise at -78 C under N2 protection. After stirring for 1.5 hr with the temperature below -70 C, B(OMe)3 (8 mL, 71.8 mmol) was added dropwise into the reaction mixture. The solution mixture was allowed to stir overnight and slowly warmed to ambient temperature. There after, the solution was quenched with water and extracted using ethyl acetate. The combined organic layer was washed with saturated NaCl (aq) and dried over Na2SO4. Thus, it was concentrated and purified using column chromatography to obtain a white solid 8.46 g in 85% yield which was directly used in the next step.
80%
(2) 9-(4-bromophenyl)-9H-carbazole 4.00g of THF was added 50mL of dry, cooling to -78 deg.] C, n-butyllithium was slowly added 6mL (the 2.4M), for 1 hour, followed by after addition of trimethyl borate 1.8mL, reaction was continued for 2 hours. after the reaction warmed to room temperature overnight, cooled to 0 deg.] C, was added HCl (2.0 M) 40ml hydrolysis, and extracted with methylene chloride, the solvent was evaporated, to give 2.84g (4-(9H-carbazol-9-yl)phenyl)boronic acid in 80% yield.
75.3%
In a 1 L round bottom flask, 72.0 g (0.223 mol) of synthesized in Scheme 11 under nitrogen atmosphere was dissolved in 576.0 ml of tetrahydrofuran and cooled to minus 70 degrees. After cooling, 167.60 ml (0.268 mol) of n-butyllithium (1.6 M hexane solution) is slowly added dropwise.After stirring for 1 hour while maintaining a low temperature, 75.7 g (0.402 mol) of trimethylborate was added dropwise, followed by stirring at room temperature for 12 hours. After completion of the reaction, 200 ml of 2 N HCl solution was added dropwise, followed by extraction with ethyl acetate and water. The organic layer is anhydrous and then depressurized to remove the organic solvent. After concentration under reduced pressure, 500 ml of hexane was added and recrystallized. The resulting solid was filtered to give 48.3 g (yield 75.3%) of .
66%
(ii) Synthesis of 4-(carbazol-9-yl)phenylboronic acid A synthesis scheme of 4-(carbazol-9-yl)phenylboronic acid is shown in (B-4).; 21.8 g (67.5 mmol) of N-(4-bromophenyl)carbazole was put into a 500-mL three-neck flask and nitrogen substitution was carried out. After that, 200 mL of tetrahydrofuran (THF) was added to keep the reaction system at -78 0C inside. 48.9 mL (74.3 mmol) of n-butyllithium (1.52 mol/L hexane solution) was dropped to this reaction solution, and the solution was stirred at the same temperature for 2 hours. 17.4 mL (155 mmol) of trimethyl borate was added and the solution was stirred at -78 <n="78"/>C for 1 hour, and thereafter, the solution was stirred for 12 hours while the reaction temperature was allowed to gradually increase to the room temperature. After reaction, 200 mL of hydrochloric acid (1 mol/L) was added to the reaction solution, and the solution was stirred at the room temperature for 1 hour. The reaction mixture was washed with water to extract a water layer with ethyl acetate. The extracted solution and an organic layer were washed with saturated saline together and dried with magnesium sulfate. After drying, the mixture was subjected to suction filtration, and the filtrate was condensed to obtain a solid. The solid was recrystallized with a mixed solution of chloroform and hexane, whereby 12.8 g of a white powdery solid that was a target matter was obtained with the yield of 66 %.
66.5%
3.44 g (10.72 mmol) of intermediate 1 was weighed250 mL of dry three-necked flask,80 mL of dry tetrahydrofuran was added as the reaction solvent.Under nitrogen protection,This was cooled to -78 & lt; 0 & gt; C with liquid nitrogen acetone,The mixture was stirred at this temperature for 5 min,10.0 mL (1.6 mol / L, 16.08 mmol) of n-butyllithium was added dropwise to the reaction system using a syringe.After 1 hour of reaction,A syringe was added dropwise to the reaction system2.4 mL (42.89 mmol) of trimethyl borate,And allowed to stir overnight with natural warming.After the reaction, the solution was quenched by adding dilute hydrochloric acid solution.After tetrahydrofuran was spin-dried, the reaction solution was extracted with methylene chloride (100 mL x 3)The extracts were combined and dried over anhydrous magnesium sulfate,The solvent was removed and dried.The crude product was purified by column chromatography using dichloromethane and ethyl acetate as the eluent gradient,To give 2.05 g of a white crystalline solid,Yield 66.5%.
65.9%
[Step 2: Synthesis of 4-(9H-carbazol-9-yl)phenylboronic acid] 21.8 g (67.5 mmol) of 9-(4-bromophenyl)-9H-carbazole was put into a 500 mL three-neck flask. The air in the flask was replaced with nitrogen. To the mixture were added 200 mL of tetrahydrofuran (THF), and then the solution was cooled to -78 C. Into this solution, 48.9 mL (74.3 mmol) of n-butyllithium (a 1.52 mol/L hexane solution) was dropped, and the solution was stirred at the same temperature for 2 hours. After the stirring, 17.4 mL (155 mmol) of trimethyl borate was added to the solution, and the solution was stirred for about 1 hour at the same temperature. Then, the mixture was stirred for 24 hours while the temperature of the solution was being increased to room temperature. Thereafter, to the solution was added about 200 mL increased to room temperature. Thereafter, to the solution was added about 200 mL (1.0 mol/L) of hydrochloric acid, and then the solution was stirred at room temperature for 1 hour. The organic layer of the mixture was washed with water, and then the aqueous layer was extracted with acetate ether. The extract was combined with the organic layer and then washed with a saturated saline solution. The organic layer was dried with magnesium sulfate. After the drying, the mixture was subjected to suction filtration, and then the obtained filtrate was condensed. The obtained residue was recrystallized with chloroform/hexane to give 12.8 g of a white powdered solid, which was the object of the synthesis, at a yield of 65.9 %. A synthesis scheme of Step 2 is shown in (f-2) given below.
65.9%
With hydrogenchloride; n-butyllithium; sodium chloride; In tetrahydrofuran;
Then, 21.8 g (67.5 mmol) of 9-(4-bromophenyl)carbazole was put into a 500 mL three-necked flask, and the air in the flask was replaced with nitrogen. Then, 200 mL of tetrahydrofuran (THF) was added thereto. The temperature in the flask was set to be -78 C. and then, 48.9 mL (74.3 mmol) of n-butyllithium (1.52 mol/L, hexane solution) was dropped to the mixture and was stirred for 2 hours at the same temperature. Then, 17.4 mL (155 mmol) of trimethyl borate was added thereto. The mixture was stirred for one hour at the same temperature, then, was stirred for 24 hours while raising the temperature to room temperature. After the reaction, 200 mL of hydrochloric acid (1.0 mol/L) was added to the reaction mixture, and then stirred for one hour at room temperature. After that, the organic layer in the reaction mixture was washed with water and the product was extracted with ethyl acetate from the water which was used for the washing. The obtained extraction solution and the organic layer, which was washed with water, were mixed. The mixture was further washed with a saturated aqueous solution of sodium chloride, and then dried with magnesium sulfate. After the drying, the mixture was suction filtrated, and the filtrate was concentrated. The obtained residue was recrystallized with a mixed solvent of chloroform and hexane to give 12.8 g of a white powdered solid of 4-(carbazol-9-yl)phenylboronic acid (yield: 65.9%).
Carbazole (30g, 0.18mol), 1,4-dibromobenzene (85g, 0.36mol), CuI (34g, 0.18mol), K3PO4 (114g, 0.54mol), and toluene(1200ml) were put into a 500mL round-bottom flask, and the resultant mixture was stirred at 120 for 10 minutes. Then, ethylenediamine (24ml, 0.36mol) was added thereinto, followed by stirring at 120 for 12 hours. After completion of the reaction, extraction with ethyl acetate was performed on the resultant material, followed by column chromatography, thereby obtaining a compound. This compound (13g, 0.04mol) was put into a 1000mL round-bottom flask of anhydrous condition, followed by addition of dried THF (200ml) thereinto, and then n-BuLi (20ml, 2.25M solution in hexane) was slowly added thereinto at -78 while stirring under nitrogen. The resultant mixture was stirred at -78 for 1 hour, followed by slow addition of B(OMe)3(6.7ml, 0.06mol) thereinto at -78C, and then the temperature was raised to room temperature, followed by reaction for 12 hours. After completion of the reaction, extraction with ethyl acetate was performed on the resultant material, and then the organic layer is dried over MgSO4, followed by filtration. The solvent is removed under reduced pressure, followed by column chromatography, thereby obtaining Compound 15-1 (8.5g, 73%) as white solids
With potassium carbonate;palladium diacetate; tris-(o-tolyl)phosphine; In ethanol; water; toluene; at 110℃; for 5h;
[Step 1: Synthesis of 3-6-bis[4-(9H-carbazol-9-yl)phenyl]-9H-carbazole (CP2C)] 1.0 g (3.1 mmol) of 3,6-dibromo-9H-carbazole, 1.8 g (6.2 mmol) of 4-(9H-carbazol-9-yl)phenylboronic acid, and 457 mg (1.5 mmol) of tri(ortho-tolyl)phosphine were put into a 300 mL three-neck flask. To the mixture were added 20 mL of ethanol, 50 mL of toluene, and 20 mL (2.0 mol/L) of an aqueous solution of potassium carbonate. This mixture was stirred to be degassed while the pressure was reduced. To the mixture was added 70 mg (0.30 mmol) of palladium(II) acetate. This mixture was refluxed at 110 C for 5 hours, cooled to room temperature, and then left for 15 hours; accordingly a black solid was precipitated. The precipitated solid was subjected to suction filtration and then collected. The collected solid was dissolved in toluene which was heated, and this solution was subjected to filtration through celite (produced by Wako Pure Chemical Industries, Ltd., Catalog No. 531-16855), alumina, and Florisil (produced by Wako Pure Chemical Industries, Ltd., Catalog No. 540-00135). The obtained filtrate was condensed to give a white solid. The obtained solid was recrystallized with toluene/hexane to give 1.1 g of a white powder, which was the object of the synthesis, at a yield of 58 %.
With potassium carbonate;tris-(o-tolyl)phosphine; bis(dibenzylideneacetone)-palladium(0); In 1,2-dimethoxyethane; water; at 90℃; for 4.5h;
[Step 3: Synthesis of 3-[4-(9H-carbazol-9-yl)phenyl]-9H-carbazole (CPC)] 5.0 g (20 mmol) of 3-bromo-9H-carbazole, 5.8 g (20 mmol) of 4-(9H-carbazol-9-yl)phenylboronic acid, and 308 mg (1.0 mmol) of tri(ortho-tolyl)phosphine were put into a 300 mL three-neck flask. The air in the flask was replaced with nitrogen. To the mixture were added 100 mL of ethyleneglycoldimethylether and 20 mL of an aqueous solution of potassium carbonate (2.0 mol/L). This mixture was stirred to be degassed under reduced pressure. After the degassing, 46 mg (0.20 mmol) of palladium(II) acetate was added the mixture. This mixture was refluxed at 90 C for 4.5 hours. After the reflux, the organic layer of the mixture was washed with water twice, and the aqueous layer was extracted with acetate ether. The extract was combined with the organic layer and then washed with a saturated saline solution. The obtained organic layer was dried with magnesium sulfate. After the drying, the mixture was subjected to gravity filtration. The obtained filtrate was condensed to give an oily light brown substance. The obtained oily substance was purified by silica gel column chromatography (a developing solvent was a mixed solvent of hexane: toluene = 1: 1) to give 5.4 g of a white powdered solid, which was the object of the synthesis, at a yield of 65 %.
9-{4-[9,10-bis(biphenyl-2-yl)-2-anthryl]phenyl}-9H-carbazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
66%
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In water; toluene; at 110℃; for 10h;Inert atmosphere;
Step 1] Synthesis of 9-{4-[9,10-bis(biphenyl-2-yl)-2-anthryl]phenyl}-9H-carbazole(abbreviation: 2CzPBPhA)A synthesis scheme of 2CzPBPhA is illustrated in (E-I). [0464] <n="160"/> [0465]In a 100 mL three neck flask were put 1.7 g (3.0 mmol) of 2-bromo-9,10-bis(2-biphenyl)anthracene, 0.86 g (3.0 mmol) of 4-(9H-carbazol-9-yl)benzeneboronic acid, and 0.13 g (0.12 mmol) of tetrakis(triphenylphosphine)palladium(0), and the atmosphere in the flask was replaced with nitrogen. To this mixture were added 15 mL of toluene and 7 mL of an aqueous potassium carbonate solution (2.0 mol/L). This mixture was deaerated by being stirred under reduced pressure. This mixture was refluxed at 110 0C for 10 hours. Then, after the mixture was cooled to room temperature, about 100 mL of toluene was added thereto. This mixture was suction filtered through alumina, Florisil (a product of Wako Pure Chemical Industries, Ltd., Catalog No. 540-00135), and Celite (a product of Wako Pure Chemical Industries, Ltd., Catalog No. 531-16855). The obtained filtrate was washed with water and a saturated saline solution in that order, and the organic layer was dried with magnesium sulfate. This mixture was gravity filtered. The obtained filtrate was concentrated to give a solid, which was recrystallized with dichloromethane/hexane to give the object of the synthesis as 1.4 g of a light yellow powdered solid in a yield of 66 %. [0466]Then, 490 mg of the obtained light-yellow powdered solid was sublimated and purified by train sublimation. For sublimation purification conditions, the material was heated at 360 C under a pressure of 200 Pa with argon gas at a flow rate of 15.0 mL/min. After the sublimation purification, 430 mg of 2CzPBPhA was recovered in a <n="161"/>yield of 86 %. [0467]By nuclear magnetic resonance (NMR) measurement, it was confirmed that this compound was 9-{4-[9,10-bis(biphenyl-2-yl)-2-anthryl]phenyl}-9H-carbazole (abbreviation: 2CzPBPhA). [0468]The 1H NMR data of 2CzPBPhA are shown as follows: 1H NMR (CDCl3, 300 MHz):delta = 6.89-6.96 (m, 6H), 7.00-7.06 (m, 4H), 7.21-7.32 (m, 4H), 7.37-7.45 (m, 7H), 7.54-7.74 (m, 15H), 7.85 (d, J = 1.5 Hz, IH), 8.15 (d, J = 8.4 Hz, 2H). Further, FIGS. 52A and 52B show 1H NMR charts. Note that FIG. 52B is a chart in which the range of 6.5 to 8.5 ppm in FIG. 52A is enlarged. [0469]
With potassium carbonate;palladium diacetate; tris-(o-tolyl)phosphine; In ethanol; water; toluene; at 100℃; for 20h;Inert atmosphere;
[Step 1] Synthesis of 9-[4-(9,10-Diphenyl-2-anthryl)phenyl]-9H-carbazole(abbreviation: 2CzPPA)A synthesis scheme of 2CzPPA is illustrated in (D-I). [0371] [0372] <n="139"/>In a 100 niL three neck flask were put 2.0 g (4.9 mmol) of 2-bromo-9,10-diphenylanthracene, 1.4 g (4.9 mmol) of4-(9H-carbazol-9-yl)phenylboronic acid, and 0.15 g (0.50 mmol) of tri(ortho-tolyl)phosphine, and the atmosphere in the flask was replaced with nitrogen. To this mixture were added 15 mL of toluene, 15 mL of ethanol, and 10 mL of an aqueous potassium carbonate solution (2.0 mol/L). This mixture was deaerated while being stirred under reduced pressure. Then, the atmosphere in the flask was replaced with nitrogen. To this mixture was added 23 mg (0.10 mmol) of palladium(II) acetate. This mixture was refluxed at 100 0C for 20 hours. Then, after this mixture was cooled to room temperature, about 50 mL of toluene was added thereto, and the mixture was filtered through a filter paper. The obtained mixture was washed with water, and the aqueous layer was extracted with toluene. The extract solution and the organic layer were combined and washed with a saturated saline solution, and the organic layer was dried with magnesium sulfate. This mixture was gravity filtered. The obtained filtrate was concentrated to give a light yellow solid. This solid was washed with toluene to give the object of the synthesis as 1.5 g of a light yellow powdered solid in a yield of 54 %. [0373]Then, 1.5 g of the obtained light-yellow powdered solid was sublimated and purified by train sublimation. For sublimation purification conditions, 2CzPPA was heated at 260 C with argon gas at a flow rate of 3.0 mL/min. After the sublimation purification, 2CzPPA was recovered as 1.4 g of a light yellow solid in a yield of 94 %. [0374]By nuclear magnetic resonance (NMR) measurement, it was confirmed that this compound was 9-[4-(9,10-diphenyl-2-anthryl)phenyl]-9H-carbazole (abbreviation: 2CzPPA). [0375]The 1H NMR data of 2CzPPA are shown as follows: 1H NMR (CDCl3, 300 MHz):delta = 7.30 (d, J = 6.3 Hz, 2H), 7.33-7.75 (m, 21H), 7.77 (d, J = 8.1 Hz, 2H)5 7.85 (d, J = 9.0 Hz, IH), 8.01 (d, J = 2.1 Hz, IH), 8.15 (d, J = 7.8 Hz, 2H). Further, FIGS. 2OA and 2OB show 1H NMR charts. Note that FIG. 2OB is a chart in which the range of 7.0 <n="140"/>to 8.5 ppm in FIG. 2OA is enlarged. [0376]
With potassium carbonate; tris-(o-tolyl)phosphine In ethanol; water; toluene for 4h; Inert atmosphere; Reflux;
4
1.0 g (3.9 mmol) of (E)-4-dibromostilbene, which was obtained in the synthesis scheme (h-1) in Embodiment 3, 1.1 g (3.9 mmol) of 4-(carbazol-9-yl)phenylboronic acid, which was obtained in the synthesis scheme (f-3), in Embodiment 1, and 0.294 g (0.97 mmol) of tris(ortho-tolyl)phosphine were put into a 100 mL three-necked flask, and the air in the flask was replaced with nitrogen. Then, 20 mL of toluene, 10 mL of ethanol, and 7 mL (14 mmol) of potassium carbonate solution (2.0 mol/L) were added thereto. The mixture was degassed while being stirred under reduced pressure in the flask. Then, 0.043 g (0.19 mmol) of palladium acetate (II) was added thereto. The mixture was refluxed for 4 hours. After the reaction, a precipitate in the reaction mixture was collected by suction filtration. The collected solid was dissolved in toluene and suction filtrated with florisil, celite, and alumina. The filtrate was concentrated to obtain a solid. The obtained solid was recrystallized with a mixed solvent of toluene and hexane to give 0.96 g of white needle-like crystals (yield: 59%). The white needle-like crystal was identified as (E)-4-(carbazol-9-yl)stilbene (abbr.: CzPS) by a nuclear magnetic resonance (NMR) method. A synthesis scheme (i-1) of (E)-4-(carbazol-9-yl)stilbene is shown below. Then, 1H NMR of the compound is shown below. A 1H NMR chart is shown in each of FIGS. 28A and 28B. The range of 7.0 ppm to 8.5 ppm in FIG. 28A is expanded and shown in FIG. 28B.1H NMR (CDCl3, 300 MHz): δ=7.20 (s, 2H), 7.30 (t-d, J1=8.1 Hz, J2=1.2 Hz, 3H), 7.38 (d, J=7.8 Hz, 1H), 7.41-7.42 (m, 1H), 7.45 (d-d, J1=6.9 Hz, J2=0.9 Hz, 2H), 7.49 (d, J=8.4H z, 2H), 7.56 (d, J=7.2 Hz, 2H), 7.64-7.73 (m, 6H), 7.86 (d, J=8.4 Hz, 2H), 8.16 (d, J=7.8 Hz, 2H).An absorption spectrum of CzPS is shown in FIG. 29. In FIG. 29, the horizontal axis indicates a wavelength (nm) and the vertical axis indicates absorption intensity (arbitrary unit (a.u.)). The ultraviolet-visible spectrophotometer (manufactured by JASCO Corporation, V-550) was used for the measurement. The absorption spectrum shown in FIG. 29 is the absorption spectrum in a state where CzPS is dissolved in a toluene solution. According to FIG. 29, CzPS has absorption peaks at 292 nm and 341 nm.
With potassium carbonate; tris-(o-tolyl)phosphine;palladium diacetate; In 1,2-dimethoxyethane; water; at 90℃; for 6h;Reflux; Inert atmosphere;
Then, 2.0 g (5.9 mmol) of (E)-4-4'-dibromostilbene and 3.7 g (13 mmol) of 4-(carbazol-9-yl)phenylboronic, which were obtained as described above, and 0.013 g (0.059 mmol) of palladium acetate (II) and 0.12 g (0.41 mmol) of tris(ortho-tolyl)phosphine were put into a 100 mL three-necked flask, and the air in the flask was replaced with nitrogen. Then, 30 mL of ethylene glycol dimethyl ether (DME) and 9 mL (17 mmol) of potassium carbonate solution (2.0 mol/L) were added to the mixture. The mixture was refluxed for 6 hours at 90 C. After the reaction, a precipitate in the reaction mixture was collected by suction filtration. The obtained solid was washed with toluene to give 2.3 g of a light yellow powdered solid (yield: 59%). Note that the light yellow powdered solid was identified as (E)-4,4'-bis[4-(carbazol-9-yl)phenyl]stilbene (abbr.: CzP2S) by a nuclear magnetic resonance (NMR) method. A synthesis scheme (f-4) of (E)-4,4'-bis[4-(carbazol-9-yl)phenyl]stilbene is shown below.1H NMR of the compound is shown below. A 1H NMR chart is shown in each of FIGS. 7A and 7B. The range of 7.0 ppm to 9.0 ppm in FIG. 7A is expanded and shown FIG. 7B.1H NMR (CDCl3, 300 MHz): delta=7.26-7.33 (m, 7H), 7.36-7.48 (m, 9H), 7.59-7.75 (1 m, 10H), 7.80-7.88 (m, 4H), 8.14-8.17 (m, 4H).
With palladium diacetate; potassium carbonate; tris-(o-tolyl)phosphine; In ethanol; water; toluene; at 90℃; for 5h;Inert atmosphere;
4- (9H- carbazol-9-yl) phenyl boronic acid 5.0g (17mmol), 2- bromoiodobenzene 9.9g (35mmol), palladium (II) 0.039g (0.17mmol), tri (ortho - tolyl) phosphine 0.37g of (1.2 mmol) were placed in a 200mL three-necked flask. The toluene 30mL mixture, ethanol 5 mL, of a 2M potassium carbonate aqueous solution 15mL was added and after degassed under reduced pressure and the mixture was the flask was replaced with nitrogen. The mixture was stirred for 5 hours at 90 C.. After the stirring, toluene was added to the mixture, and the organic layer aqueous saturated sodium carbonate, and washed successively with saturated aqueous sodium chloride solution. After washing and dried by adding magnesium sulfate to the organic layer. After drying, the mixture was suction filtered, and the filtrate was obtained. The resulting filtrate was concentrated and purified by silica gel column chromatography. Column chromatography was performed by, first, using hexane as a developing solvent and then hexaneSan: ethyl acetate = 20: was performed using a mixed solvent of 1 as a developing solvent. The obtained fraction was concentrated and dried to obtain a colorless oily substance Yield 6.0 g, 86% yield.
74%
With potassium carbonate;palladium diacetate; tris-(o-tolyl)phosphine; In ethanol; water; toluene; at 90℃; for 5h;Inert atmosphere;
[Step 1] This step is a step of synthesizing 9-(2'-bromobiphenyl-4-yl)-9H-carbazole. The step is illustrated in a synthetic scheme (E1-1) and will be detailed below. Into a 300 mL three-neck flask were put 15 g (52 mmol) of 4-(9H-carbazol-9-yl)phenylboronic acid, 22 g (78 mmol) of 2-bromoiodobenzene, 0.12 g (0.52 mmol) of palladium(II) acetate, and 1.1 g (3.7 mmol) of tri(o-tolyl)phosphine. After 90 mL of toluene, 15 mL of ethanol, 45 mL of a 2M aqueous solution of potassium carbonate were added to the mixture and this mixture was degassed while being stirred under reduced pressure, the atmosphere in the flask was replaced with nitrogen. The mixture was stirred at 90 C. for 5 hours. After the stirring, toluene was added to the mixture and an organic layer was washed with a saturated aqueous solution of sodium carbonate and saturated saline in that order. After washing, magnesium sulfate was added to the organic layer to dry the organic layer. After drying, this mixture was subjected to suction filtration to obtain a filtrate. An oily substance obtained by concentrating the obtained filtrate was purified by silica gel column chromatography. The column chromatography was performed first using hexane as a developing solvent and then using a mixed solvent of hexane and ethyl acetate (hexane:ethyl acetate=20:1) as a developing solvent. The obtained fraction was concentrated and dried to give 15 g of colorless oily substance in a yield of 74%.
74%
With potassium carbonate;palladium diacetate; tris-(o-tolyl)phosphine; In ethanol; water; toluene; at 90℃; for 5h;Inert atmosphere;
[Step 1] This step is a step of synthesizing 9-(2'-bromobiphenyl-4-yl)-9H-carbazole. The step is shown in a synthetic scheme (E1-1) and will be detailed below. Into a 300 mL three-neck flask were put 15 g (52 mmol) of 4-(9H-carbazol-9-yl)phenylboronic acid, 22 g (78 mmol) of 2-bromoiodobenzene, 0.12 g (0.52 mmol) of palladium(II) acetate, and 1.1 g (3.7 mmol) of tri(o-tolyl)phosphine. After 90 mL of toluene, 15 mL of ethanol, 45 mL of a 2M aqueous solution of potassium carbonate were added to the mixture and this mixture was degassed while being stirred under reduced pressure, the atmosphere in the flask was replaced with nitrogen. The mixture was stirred at 90 C. for 5 hours. After the stirring, toluene was added to the mixture and an organic layer was washed with a saturated aqueous solution of sodium carbonate and saturated saline in this order. After the washing, magnesium sulfate was added to the organic layer to dry the organic layer. After the drying, this mixture was subjected to suction filtration to obtain a filtrate. An oily substance obtained by concentrating the obtained filtrate was purified using silica gel column chromatography. The column chromatography was performed using hexane first as a developing solvent and then using a mixed solvent of hexane and ethyl acetate (hexane:ethyl acetate=20:1) as a developing solvent. The obtained fraction was concentrated and dried, so that 15 g of colorless oily substance was obtained in a yield of 74%.
With potassium carbonate; tris-(o-tolyl)phosphine;palladium diacetate; In 1,2-dimethoxyethane; at 90℃; for 5h;Inert atmosphere;
Step 2: Synthesis of 9-[4'-(Benzoxazol-2-yl)biphenyl-4-yl]-9H-carbazole (abbreviation: CzPBOx)A synthesis scheme of 9-[4'-(benzoxazol-2-yl)biphenyl-4-yl]-9H-carbazole (abbreviation: CzPBOx) is illustrated in (E-3). In a 100 mL three-neck flask were put 1.0 g (3.1 mmol) of 2-(4-iodophenyl)benzoxazole, 0.90 g (3.1 mmol) of 4-(9H-carbazol-9-yl)phenylboronic acid, and 0.067 g (0.22 mmol) of tri(ortho-tolyl)phosphine. To this mixture were added 30 mL of 1,2-dimethoxyethane (abbreviation: DME) and 30 mL of a 2M potassium carbonate aqueous solution. This mixture was degassed under reduced pressure, and then the atmosphere in the flask was replaced with nitrogen. To this mixture was added 7.0 mg (0.031 mmol) of palladium(II) acetate, and the resulting mixture was stirred at 90 C. for 5 hours while being heated. After that, toluene was added to this mixture, and the organic layer was washed with a saturated aqueous sodium carbonate solution and saturated brine in that order. After that, magnesium sulfate was added to the organic layer to dry it. Next, this mixture was suction filtered. The resulting filtrate was suction filtered through Celite (produced by Wako Pure Chemical Industries, Ltd., Catalog No. 531-16855). The resulting filtrate was concentrated, followed by purification by silica gel column chromatography. As a developing solvent for the chromatography, toluene was used. The fraction obtained was concentrated to give a solid. This solid was recrystallized with a mixed solvent of chloroform and methanol to give 1.2 g of a white powdered solid in a yield of 88%.Sublimation purification of 1.2 g of the white solid obtained was performed by a train sublimation method. Under a reduced pressure of 7.0 Pa and with an argon flow rate of 4 mL/min, the sublimation purification was performed at 220 C. for 19 hours, whereby 1.1 g of the resulting substance was obtained in a yield of 90%.The compound obtained through the above Step 2 was measured by a nuclear magnetic resonance (NMR) method. The following are the measurement data: 1H NMR (CDCl3, 300 MHz): delta=7.27-7.52 (m, 8H), 7.59-7.64 (m, 1H), 7.69 (d, J=8.3 Hz, 2H), 7.79-7.92 (m, 5H), 8.17 (d, J=7.8 Hz, 2H), 8.40 (d, J=8.3 Hz, 2H)
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In water; toluene;Inert atmosphere;
General procedure: G3-B(OH)2 (1.04 g, 0.6 mmol), 1,3,6,8-tetrabromo-1,3-dihydropyrene (0.05 g, 0.1 mmol), and tetrakis(triphenylphosphine) palladium (0.012 g, 0.01 mmol) were added to an air-free two-phase system composed of toluene (30 mL) and 2 M K2CO3 aqueous solution (10 mL). The resulting mixture was intensively stirred under an argon atmosphere at 80 C for 24 h. The organic layer was separated and the aqueous phase was extracted with diethyl ether. The organic layers were combined and washed with brine and dried over anhydrous MgSO4. The solvent was evaporated and the residue went through silica-gel column.
22%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,4-dioxane; water; at 100℃; for 72h;Inert atmosphere;
In a 250mL three-necked round-bottomed flask, 1,3,6,8-tetrabromo-pyrene (1.0356g, 2mmol) and 4-(9H-carbozol-9yl)phenylboronic acid (2.8712g, 10mmol) were dissolved in dioxane (100mL) and then an aqueous solution of potassium carbonate (3.15g, 23mmol) in water (10mL) and Pd(PPh3)4 catalyst (0.6g, 0.50mmol) were added to the reaction mixture and it was stirred under N2 at 100C for 3days. After cooling to room temperature, the yellow reaction mixture was transferred to dilute hydrochloric acid solution (100mL). The precipitate was collected by filtration and then washed with water (3×40mL). The solid was transferred to a Soxhlet and continuously extracted with CHCl3 for 48h. The CHCl3 extract was evaporated under reduced pressure. The crude product L was further purified by silica-gel column chromatography using CHCl2 as mobile phase to afford a bright yellow solid (0.5g, 22%). 1H NMR (400MHz, CDCl3): delta(ppm) 8.51(4H), 8.32(2H), 8.22(8H), 8.03(8H), 7.84 (8H), 7.61(8H), 7.51(8H), 7.35(8H). FT-IR (ATR4000-400cm-1) 3418, 3047, 2910, 1600, 1514, 1493, 1451, 1359, 1338, 1312, 1223, 1164, 1004, 839, 746, 717, 628, 561, 533. MS(ESI): m/z for C88H54N4 cacld 1166, M+, 1166.02.
With palladium diacetate; potassium carbonate; In ethanol; water; at 80℃;
In the round-bottom flask is added 2-bromo pyridine (1.0mmol), potassium carbonate (2.0mmol), palladium acetate (0.015mmol), 4 - (9-carbazolyl) phenylboronic acid (1.5mmol) and the volume ratio of 3:1 ethanol/water mixed solution of 12 ml. In the air, 80 C conditions, magnetic stirring, to Suzuki cross-coupling reaction, the reaction process by the thin layer chromatography tracking. After the reaction is complete, add saturated salt water 25 ml, with ethyl acetate (3×25 ml) extraction reaction product, combined with the phase, anhydrous Na 2 SO 4 drying, filtering, using rotary evaporimeter concentration to obtain the crude product, to petroleum ether and ethyl acetate as the eluting agent, column chromatography, to obtain the target product of analytical, yield 99%, the product structure through 1 HNMR, 13 CNMR and mass spectrometric identification. (
With palladium diacetate; potassium carbonate; In ethanol; water; at 80℃;
General procedure: A mixture of the N-heteroaryl halide (1.0mmol), 4-(9H-carbazol-9-yl)-phenylboronic acid (1.5mmol), K2CO3 (2.0mmol), Pd(OAc)2 (1.5mol%), ethanol (6mL) and distilled water (2mL) was stirred at 80C in air for indicated time (<30min). The reaction was monitored by TLC. The reaction mixture was added to brine (30mL) and extracted four times with ethyl acetate (4×30mL). The solvent was concentrated under vacuum, and the product was isolated by short-column chromatography on silica gel (200-300 mesh).
With caesium carbonate; In ethanol; water; toluene; at 20 - 95℃; for 48h;Inert atmosphere;
These compounds were obtained following an essentially similar procedure. An illustrative example is provided for S1: compound 13 (2.40 g, 0.005 mol), compound 4 (2.87 g, 0.01 mol) was dissolved in the mixture of toluene (75 mL), ethanol (25 mL) and cesium carbonate aqueous solution (2.0 M, 15 mL) in a three-necked round-bottom flask fitted with a magnetic stirrer, a condenser and a N2 purge. The mixture was stirred at room temperature for 0.5 h under N2 gas followed by adding Pd(PPh3)4 (0.0003 mol, 0.35 g) and then heated slowly to 95 C for 48 h. After cooling to room temperature, the reaction mixture was filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica gel chromatography with petroleum ether/dichloromethane (2:1) as the eluent to afford a white power. The white power was sublimated by train sublimation (Model D-8-25-880, Nanchang Choice Laboratory Instruments Ltd.) [47] to afford a white crystalline power (S1). 2.2.10.1. (S1) White power, yield: 78.6%, Mp: 271-273 C; FT-IR (KBr) nu = 3035 (Ar-H), 1334 (Ar-N), 1H NMR (400 MHz, CDCl3, TMS) delta: 8.19-8.17 (d, J = 7.74 Hz, 4H); 7.99-7.97 (d, J = 8.30 Hz, 4H); 7.94-7.92 (d, J = 8.62 Hz, 3H); 7.73-7.71 (d, J = 8.31 Hz, 4H); 7.67-7.65 (d, J = 8.58 Hz, 2H); 7.53-7.51 (d, J = 8.10 Hz, 4H); 7.47-7.43 (t, J = 7.62 Hz, 4H); 7.34-7.28 (m, 10H); 7.19-7.17 (d, J = 7.73 Hz, 4H); 7.09-7.05 (t, J = 7.31 Hz, 2H). 13C NMR (101 MHz, CDCl3+DMSO) delta: 147.25, 147.13, 141.77, 141.10, 140.32, 139.66, 136.67, 134.07, 129.04, 128.48, 127.70, 126.94, 125.80, 124.46, 124.10, 123.43, 122.94, 122.84, 119.99, 119.82, 109.52; Anal. calcd. for C60H41N3 (%): C, 89.63; H, 5.14; N, 5.23. Found: C, 89.60; H, 5.16; N, 5.24. ESI - MS (m/z): calcd. for C60H41N3 803.33; Found 803.38 ([M]+). 2.2.10.2.
With caesium carbonate; In ethanol; water; toluene; at 20 - 95℃; for 48h;Inert atmosphere;
General procedure: These compounds were obtained following an essentially similar procedure. An illustrative example is provided for S1: compound 13 (2.40 g, 0.005 mol), compound 4 (2.87 g, 0.01 mol) was dissolved in the mixture of toluene (75 mL), ethanol (25 mL) and cesium carbonate aqueous solution (2.0 M, 15 mL) in a three-necked round-bottom flask fitted with a magnetic stirrer, a condenser and a N2 purge. The mixture was stirred at room temperature for 0.5 h under N2 gas followed by adding Pd(PPh3)4 (0.0003 mol, 0.35 g) and then heated slowly to 95 C for 48 h. After cooling to room temperature, the reaction mixture was filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica gel chromatography with petroleum ether/dichloromethane (2:1) as the eluent to afford a white power. The white power was sublimated by train sublimation (Model D-8-25-880, Nanchang Choice Laboratory Instruments Ltd.) [47] to afford a white crystalline power
With caesium carbonate; In ethanol; water; toluene; at 20 - 95℃; for 48h;Inert atmosphere;
General procedure: These compounds were obtained following an essentially similar procedure. An illustrative example is provided for S1: compound 13 (2.40 g, 0.005 mol), compound 4 (2.87 g, 0.01 mol) was dissolved in the mixture of toluene (75 mL), ethanol (25 mL) and cesium carbonate aqueous solution (2.0 M, 15 mL) in a three-necked round-bottom flask fitted with a magnetic stirrer, a condenser and a N2 purge. The mixture was stirred at room temperature for 0.5 h under N2 gas followed by adding Pd(PPh3)4 (0.0003 mol, 0.35 g) and then heated slowly to 95 C for 48 h. After cooling to room temperature, the reaction mixture was filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica gel chromatography with petroleum ether/dichloromethane (2:1) as the eluent to afford a white power. The white power was sublimated by train sublimation (Model D-8-25-880, Nanchang Choice Laboratory Instruments Ltd.) [47] to afford a white crystalline power
With caesium carbonate; In ethanol; water; toluene; at 20 - 95℃; for 48h;Inert atmosphere;
General procedure: These compounds were obtained following an essentially similar procedure. An illustrative example is provided for S1: compound 13 (2.40 g, 0.005 mol), compound 4 (2.87 g, 0.01 mol) was dissolved in the mixture of toluene (75 mL), ethanol (25 mL) and cesium carbonate aqueous solution (2.0 M, 15 mL) in a three-necked round-bottom flask fitted with a magnetic stirrer, a condenser and a N2 purge. The mixture was stirred at room temperature for 0.5 h under N2 gas followed by adding Pd(PPh3)4 (0.0003 mol, 0.35 g) and then heated slowly to 95 C for 48 h. After cooling to room temperature, the reaction mixture was filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica gel chromatography with petroleum ether/dichloromethane (2:1) as the eluent to afford a white power. The white power was sublimated by train sublimation (Model D-8-25-880, Nanchang Choice Laboratory Instruments Ltd.) [47] to afford a white crystalline power
With palladium diacetate; potassium carbonate; triphenylphosphine; In ethanol; toluene; at 45℃; for 24h;Inert atmosphere;
Compound 2-3 (8.46 g, 29.45 mmol), diethyl 2,5-dibromoterephthalate (22.4 g, 58.9mmol), palladium (II) acetate (65 mg), triphenylphosphine (0.12g), 4M potassium carbonate (15 mL), ethanol (50 mL) and toluene (250 mL) were added to a 500 mL two-necked round bottom flask and stirred. The reaction mixture was heated at 45C with nitrogen protection for 24 h. After cooling down to room temperature, the reaction mixture was quenched with water and acidified by 6 M HCl (3mL). Thereafter, it was extracted with 3 portions of ethyl acetate and then dried over sodium sulphate. Thus, purification of residue was carried out via column chromatography to afford a white solid 5.1 g in 32% yield. 1H NMR (400MHz, CDCl3, delta): 8.25-8.14 (m, 3H), 7.97-7.84 (m,1H), 7.53-7.68 (m, 4H), 7.52-7.42 (m, 4H), 7.36-7.28 (m, 2H), 4.47 (q, J = 7.2Hz, 2H), 4.23 (q, J = 7.2 Hz, 2H), 1.46 (t, J = 7.2 Hz, 3H), 1.59 (t, J = 7.2 Hz,3H). MS(MALDI-TOF, m/z): found 542.0964 [M+H]+.
With palladium diacetate; potassium carbonate; In ethanol; water; at 80℃;
General procedure: A mixture of the N-heteroaryl halide (1.0mmol), 4-(9H-carbazol-9-yl)-phenylboronic acid (1.5mmol), K2CO3 (2.0mmol), Pd(OAc)2 (1.5mol%), ethanol (6mL) and distilled water (2mL) was stirred at 80C in air for indicated time (<30min). The reaction was monitored by TLC. The reaction mixture was added to brine (30mL) and extracted four times with ethyl acetate (4×30mL). The solvent was concentrated under vacuum, and the product was isolated by short-column chromatography on silica gel (200-300 mesh).
With palladium diacetate; potassium carbonate; In ethanol; water; at 80℃;
General procedure: A mixture of the N-heteroaryl halide (1.0mmol), 4-(9H-carbazol-9-yl)-phenylboronic acid (1.5mmol), K2CO3 (2.0mmol), Pd(OAc)2 (1.5mol%), ethanol (6mL) and distilled water (2mL) was stirred at 80C in air for indicated time (<30min). The reaction was monitored by TLC. The reaction mixture was added to brine (30mL) and extracted four times with ethyl acetate (4×30mL). The solvent was concentrated under vacuum, and the product was isolated by short-column chromatography on silica gel (200-300 mesh).
Cz-PhBr (15 mmol) was solved into THF in flask with gasreplacement by Ar for three times. After cooling down to 78 C for10 min. N-butyl lithium (t-BuLi, 11 mL, 1.6 mol L1 in hexane solution)was dropped into and stirred for 1 h at 78 C. The Triisopropylborate (23 mmol) was dropped into flask and stirred foranother 1 h at 78 C the slowly warmed to room temperature andstirred overnight. Hydrochloric acid (HCl, 30 mL, 2 mol L1) wasadded into the flask and stirred for another 30 min. The productswas extracted by Dichloromethane (CH2Cl2). After evaporation, thecrude product was washed with pure petroleum ether. A whitepowder achieved with yield of 86%.
85.91%
Compound 1 (4.83g, 15.00mmol) was put into the flask. THF (60mL) was added with the syring. The air in the flask was replaced with Ar three times. Then the reaction solution was cooled down to-78C for 10min. N-butyl lithium (11.25mL, 1.6mol/L in hexane solution) was dropped into above flask dropwise with the syring, then stirred for 1hat-78C. Triisopropyl borate ([(CH3)2CHO]3B, 5.20mL, 22.50mmol) was added into above solution and then stirred for 1h at-78C. After that, the reaction solution was warmed slowly to room temperature and stirred overnight. Hydrochloric acid (HCl, 30mL, 2mol/L) was added into the flask and stirred for another 30min. Dichloromethane was used to extract the product and the organic layer was dried over MgSO4. After filtration, the solution was concentrated using rotary evaporator. Then the crude product was washed with pure petroleum ether to afford white powder, compound 2, 3.70g (85.91%). 1H NMR (400MHz, DMSO-d6): delta 8.25 (d, J=7.2Hz, 2H), 8.08 (dd, J=23.0, 7.2Hz, 2H), 7.61 (d, J=7.4Hz, 2H), 7.43 (s, 4H), 7.30 (s, 2H). 13C NMR (100MHz, DMSO-d6): delta 140.43, 138.92, 136.39, 126.72, 125.84, 123.28, 120.97, 120.57, 110.17.
67%
Under a nitrogen atmosphere, 9.8 g (0.030 mol) of the intermediate E compound and 152 mL (0.2 M) of tetrahydrofuran were placed in a 500 mL two-neck round bottom flask and the temperature was lowered to -78 C. To this, a n-butyllithium dilution solution (1.6M, 38.02 mL) was slowly added, and the mixture was stirred for 1 hour while maintaining the temperature at -78 C. 17.2 g (0.091 mol) of triisopropylborate was slowly added to the above mixed solution, the temperature of the mixed solution was slowly raised to room temperature, and the mixture was stirred for 12 hours. After the reaction was completed, 1N hydrochloric acid was added, and after 10 minutes, the reaction mixture was washed with water and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate and concentrated under reduced pressure to remove the solvent. The resulting material was separated by column separation using a mixed solvent of hexane and ethyl acetate to obtain 5.9 g (67%) of intermediate F.
67%
Under nitrogen, 9.8 g (0.030 mol) of Intermediate-E compound and 152 mL (0.2 M) of tetrahydrofuran were added to a 500 mL 2-neck round bottom flask and the temperature was lowered to -78 C. To this, a n-butyllithium dilution solution (1.6M, 38.02 mL) was added slowly, and the mixture was stirred for 1 hour while maintaining the temperature at -78 C. 17.2 g (0.091 mol) of triisopropylborate was slowly added to the above mixture, the temperature of the mixture was slowly raised to room temperature, and the mixture was stirred for 12 hours. After the reaction was completed, 1N hydrochloric acid was added, and after 10 minutes, the reaction mixture was washed with water and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate and concentrated under reduced pressure to remove the solvent. The resulting material was separated by column separation using a mixed solvent of hexane and ethyl acetate to obtain 5.9 g (67%) of intermediate F.
9-(4-(4,6-diphenyl-1,3,5-triazin-2-yl)phenyl)-9H-carbazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In ethanol; water; toluene for 12h; Reflux;
1 [Comparative Compound 1]
Compound 1-1 5 g (17.4 mmol), 2-chloro-4,6-diphenyl-1,3,5-triazine 4.6 g (17.4 mmol), Pd(PPh3)41 g (0.9 mmol), potassium carbonate 4.8 g (34.8 mmmol) was suspended in 90 ml of toluene, 18 ml of ethyl alcohol, and 18 ml of distilled water, followed by stirring at reflux for 12 hours.Extract with dichloromethane and distilled water, and filter the organic layer with silica gel.The reaction solution was re-crystallized after neutralization under reduced pressure to obtain Comparative Compound 1, 6.6 g (yield: 80%).
80%
With potassium carbonate In ethanol; water; toluene for 12h; Reflux;
1 Synthesis of Comparative Compound 1
Compound 1-1 5 g (17.4 mmol), 2-chloro-4,6-diphenyl-1,3,5-triazine 4.6 g (17.4 mmol), Pd(PPh3)4 1 g (0.9 mmol), potassium carbonate 4.8 g (34.8 mmmol) was suspended in 90 ml of toluene, 18 ml of ethyl alcohol, and 18 ml of distilled water, followed by stirring at reflux for 12 hours. Extract with dichloromethane and distilled water, and filter the organic layer with silica gel. The reaction solution was subjected to crystallization under reduced pressure to obtain Comparative Compound 1, 6.6 g (yield: 80%).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In ethanol; water; toluene for 26h; Inert atmosphere; Reflux;
TCT
General procedure: TCT: Toluene (12 ml), ethanol (6 ml), and 2M aqueous Na2CO3 (9 ml) were added to a mixture of 2,4,6-trichloro-1,3,5-triazine (0.368 g, 2 mmol), 4-(9H-carbozol-9-yl)phenylboronic acid (1.894 g, 6.6 mmol), and tetrakis(triphenylphosphine)platinum (0.234 g, 0.2 mmol). The mixture was refluxed for 26 h under a nitrogen atmosphere. When cooled to room temperature, the mixture was extracted with dichloromethane and dried over Na2SO4. After the solvent had been removed under reduced pressure, the residue was purified by column chromatography on silica gel using dichloromethane-petroleum ether (1:1) as the eluent to give a light yellow solid (0.547 g, 34%).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In ethanol; water; toluene for 26h; Reflux; Inert atmosphere;
PTC: This compound was synthesized according to the synthesis procedure of TCT from the intermediates of 2-chloro-4,6-diphenyl-1,3,5-triazine and 4-(9H-carbozol-9-yl)phenylboronic acid.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In ethanol; water; toluene for 26h; Inert atmosphere; Reflux;
PTC
General procedure: Toluene (12 ml), ethanol (6 ml), and 2 M aqueous Na2CO3 (9 ml) were added to a mixture of 2,4,6-trichloro-1,3,5-triazine (0.368 g, 2 mmol), 4-(9H-carbozol-9-yl)phenylboronic acid (1.894 g, 6.6 mmol), and tetrakis(triphenylphosphine)platinum (0.234 g, 0.2 mmol). The mixture was refluxed for 26 h under a nitrogen atmosphere. When cooled to room temperature, the mixture was extracted with dichloromethane and dried over Na2SO4. After the solvent had been removed under reduced pressure,the residue was purified by column chromatography on silica gel using dichloromethane-petroleum ether (1:1) as the eluent to give a light yellow solid (0.547 g, 34%).
With tetrakis(triphenylphosphine) palladium(0); caesium carbonate; In 1,4-dioxane; water; for 24h;Inert atmosphere; Reflux;
General procedure: Under argon atmosphere, a mixture of <strong>[464192-28-7]2-bromothiazole-5-carbaldehyde</strong> (0.39 g, 2.00 mmol), 4-(diphenylamino)phenylboronic acid (0.87 g, 3.00 mmol), [Pd(PPh3)4] (0.12 g, 0.10 mmol), cesium carbonate (1.95 g, 6.00 mmol), dioxane (40 mL) and deionized water (4 mL) was degassed for 10 min and heated to reflux for 24 h. The mixture was then cooled to the room temperature and extracted with chloroform. The resulting organic layer was washed thoroughly by brine and then dried with anhydrousNa2SO4 and filtered. After purified by column chromatography over silica gel using hexane and dichloromethane (v/v 1:2), compound 4 was obtained in a yield of 0.62 g (87%) as orange solid.
9-(2-bromobenzyl)-9H-carbazole-3,6-diamine[ No CAS ]
9-(4'-(9H-carbazol-9-yl)biphenyl-2-yl)methyl-9H-carbazole-3,6-diamine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
88%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In water; toluene; at 110℃; for 6h;Inert atmosphere;
A solution of (3) (0.60 g, 1.64 mmol), 4-(9H-carbazol-9-yl)phenylboronic acid (0.71 g, 2.46 mmol), Pd(PPh3)4 (0.038 g, 0.033 mmol), Na2CO3 (0.75 g, 6.56 mmol) in toluene (20 mL) and water (10 mL) was heated 110 C under an nitrogen atmosphere for 6 h. When the reaction was completed, the reaction mixture was cooled and filtered. The filtrate was condensed and recrystallized from ethanol and water (Vethanol: Vwater = 9: 1). The solid was washed twice with water and ethanol respectively. The gray solid was collected and dried in vacuum at 50 C to give product 0.76 g (88%). Mp: 283 C. FT-IR (KBr, cm-1): 3354, 3321 (-NH2), 1324 (C-N). 1H NMR (400 MHz, CDCl3, delta, ppm): 8.17 (d, J = 7.8 Hz, 2 H, ArH), 7.65 (d, J = 9.3 Hz, 4 H, ArH), 7.48 (d, J = 10.0 Hz, 3 H, ArH), 7.45-7.38 (m, 3 H, ArH), 7.33 (d, J = 8.0 Hz, 4 H, ArH), 7.16 (d, J = 8.3 Hz, 1 H, ArH), 6.97 (s, 2 H, ArH), 6.80 (d, J = 8.2 Hz, 3 H, ArH), 5.43 (s, 1 H, CH2), 3.55 (s, 2 H, NH2). 13C NMR (101 MHz, CDCl3, delta, ppm): 140.8, 140.1, 139.8, 138.7, 136.9, 135.9, 135.0, 130.5, 130.1, 128.2, 127.2, 126.9, 126.7, 126.0, 123.5, 123.3, 120.3, 120.1, 115.7, 109.8, 109.3, 106.3, 45.2. Anal. calcd. For C37H28N4: C, 84.06; H, 5.34; N, 10.60. Found: C, 84.09; H, 5.42; N, 10.35.
88%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In water; toluene; at 110℃; for 4h;
Under the protection of nitrogen, 100 ml is added to the flask of the three-port 9 - (2'- bromine phenmethyl ) - 3,6-diamino-carbazole (1.0000g, 2 . 7400mmol), 4 - (9H-carbazolyl) phenyl boronic acid( 0 . 9417g, 3.2880mmol), Pd (PPh3)4(0.0316g, 0 . 0274mmol), anhydrous Na2CO3(0.5808g, 5 . 4801mmol), and then adding toluene 20 ml, distilled water 10 ml, in 110 C reflux reaction 4h. There are a large number of bubbles in the reaction process to produce. After the reaction is complete, cooling to room temperature. Filtered, filtrate layered heating toluene layer, reducing the toluene to obtain the solid, the solid will be combined with the filter cake. Using ethanol, water (ethanol: water volume ratio is 9:1) to recrystallize 2-3 time, filtering, in the vacuum drying oven 50 C drying 12h, to obtain gray solid 1.30g, 9-[2-(4-carbazol-phenyl) benzyl]3,6-diamino-carbazole, molecular formula:Yield 88%.
9-(4'-((4-bromophenyl)sulfonyl)-[1,1'-biphenyl]-4-yl)-9H-carbazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
60%
With potassium carbonate; palladium; In tetrahydrofuran; water; at 80℃;Inert atmosphere;
In N2in the gas purification system, the 4-bromobenzyl sulfone soluble in toluene solvent, and "h" compound is added (0.9 equiv). The K2CO3(4 equivalent) dissolved in distilled water in and added to the solution. By adding tetrahydrofuran solvent, and by adding palladium (0.05 equiv). The mixture at 80 C and the temperature of the refluxing under stirring. After the reaction is finished, the ethyl ester of acetic acid and distilled water to the mixture to extraction. From the extraction of using magnesium sulphate remove the moisture in the organic layer, and removing the remaining organic solvent. The material through the use of hexane and ethyl acetate to carry out wet refining column chromatography, thereby obtaining solid compound "j". (Yield: 60%)
9-(4-(4-chloro-6-phenyl-1,3,5-triazin-2-yl)phenyl)-9H-carbazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
With tetrakis-(triphenylphosphine)-palladium; anhydrous sodium carbonate In 1,4-dioxane; water monomer; toluene for 16h; Inert atmosphere;
16-1 [Reaction formula 16-1]
In N2in the gas purification system, the use of a compound "k", "h" compound (0.9 equivalent) and Na2CO3(0.6 equivalent) is added to a toluene/dioxane/distilled water (1:1: 0.7) and stirring in the solvent. Add another Pd (PPh3)4(four (triphenylphosphine) palladium (0), 0.3 equivalent) and stirring 16 hours. After the completion of reaction, the solution is cooled to the room temperature. The silica gel in distilled water in the washing and filtering organic layer. Remove solvent and distilled water, and recrystallization with chloroform methylpyrrolidinone and dried, so as to obtain compound "l". (Yield: 80%)
78.1%
With tetrakis-(triphenylphosphine)-palladium; potassium carbonate In tetrahydrofuran; water monomer; toluene for 24h; Reflux;
3.3-3
12.5 g (0.055 mol) of synthesized in Scheme 8, 15.9 g (0.055 mol) synthesized in Scheme 12, in a 250 ml round bottom flask,30.6 g (0.221 mol) of potassium carbonate (K2CO3),Tetrakistriphenylphosphinepalladium (Pd (PPh3) 4) 3.2 g (0.003 mol),25.0 mL of water, 62.5 ml of toluene and 62.5 mL of tetrahydrofuran were added and refluxed for 24 hours. When the reaction was completed, the resulting product was separated into layers to remove the aqueous layer, the organic layer was separated and concentrated under reduced pressure, and then obtained by column chromatography to give 18.7 g (yield 78.1%).
78.1%
With tetrakis-(triphenylphosphine)-palladium; potassium carbonate In tetrahydrofuran; water monomer; toluene for 24h; Reflux;
4.4-3
250ml round In Scheme 14 in the bottom flask Synthesized of <2-d>12.5 g (0.055 mol),15.9 g (0.055 mol) of<3-b> synthesized in Scheme 12, 30.6 g g (0.221 mol) of potassium carbonate (K2CO3),3.2 g (0.003 mol) of tetrakistriphenylphosphinepalladium (Pd(PPh3)4),25.0 mL of water, toluene 62.5ml And 62.5 mL of tetrahydrofuran charged and refluxed for 24 hours. When the reaction is over,The resulting product was separated into layers to remove the aqueous layer, and the organic layer was separated and concentrated under reduced pressure, and then subjected to column chromatography. <4-c>18.7 g (78.1%) was obtained.
57%
With tetrakis-(triphenylphosphine)-palladium; potassium carbonate In tetrahydrofuran; water monomer at 120℃; for 3h;
1.1 Preparation of Intermediate A
One neck Round Bottom Flask (One neck Round Bottom Flask, One neck r.b.f) 2,4-dichloro-6-phenyl-1,3,5-triazine (2,4-dichloro-6-phenyl-1,3,5-triazine) (30g, 132.7mmol), ( 4-(9H-carbazol-9-yl)phenyl)boronic acid ((4-(9H-carbazol-9-yl)phenyl)boronic acid) (45.7g, 159.2mmol), tetrakis(triphenylphosphine) Palladium (0) (Tetrakis (triphenylphosphine) palladium (0)) (7.62 g, 6.6 mmol), potassium carbonate (55 g, 398.1 mmol), and tetrahydrofuran/water (THF/water (500 ml/100 ml) The mixture was refluxed for 3 hours at 120° C. After completion of the reaction, the temperature was lowered to room temperature, and the resulting solid was washed with distilled water and methanol (MeOH) to obtain an intermediate A (33 g, 57%) The THF was tetrahydrofuran (Tetrahydrofuran) means
With potassium carbonate; palladium In tetrahydrofuran; water at 80℃; Inert atmosphere;
12-3 [Reaction formula 12-3]
In N2in the gas purification system, the compound dissolved in the toluene solvent "f", and "h" compound is added (0.9 equiv). The K2CO3(4 equivalent) dissolved in distilled water in and added to the solution. By adding tetrahydrofuran solvent, and by adding palladium (0.05 equiv). The mixture at 80 °C and the temperature of the refluxing under stirring. After the reaction is finished, the ethyl ester of acetic acid and distilled water to the mixture to extraction. From the extraction of using magnesium sulphate remove the moisture in the organic layer, and removing the remaining organic solvent. The material through the use of hexane and ethyl acetate to carry out wet refining column chromatography, thereby obtaining compound of the solid "i". (Yield: 65%)
With potassium carbonate; palladium In tetrahydrofuran; water; toluene at 80℃; Inert atmosphere;
15-1 Synthesis of Compound K-1
In N2Gas purification system, Compound I-2 was dissolved in toluene, adding the compound J-2 (0.9 equiv).Dissolved in deionized water K2CO3(4.4 equiv.) Was added to the mixture.After the addition of tetrahydrofuran was added Pd (0.05 equiv).The mixture was stirred at reflux at a temperature of 80 .The recrystallization to isolate Compound I-2, J-2 and Compound byproduct mixture.The resultant was passed through the column with dichloromethane and hexane to obtain compound K-1.
With tetrakis-(triphenylphosphine)-palladium; anhydrous sodium carbonate In tetrahydrofuran; water monomer at 80℃; Inert atmosphere; Schlenk technique;
2.2.1 General procedure synthesis of 1-1∼1-8
General procedure: To a mixture of bromide (1.6eq.), Pd(PPh3)4 (0.1eq.), aqueous solution of Na2CO3 (20mmol L-1) and adequateTHF was added boracic acid (1eq.). The reaction was stirred in three-necked flask at 80°C for 16-24h under Ar atmosphere. After cooling to room temperature, the reaction mixture was extracted with copious DCM (3×50mL), dried (Na2SO4), and concentrated in vacuo. The combined organic layers were concentrated and subjected to flash column chromatography on silica-gel column.
86%
With tetrakis-(triphenylphosphine)-palladium; potassium carbonate In ethanol; water monomer; toluene at 40 - 100℃; for 5h; Inert atmosphere;
5 Synthesis of 9-(4'-bromo-[1,1'-biphenyl]-4-yl)-9H-carbazole
Under a stream of nitrogen 50.0 g (187.18 mmol) of (4 - (9H-carbazol-9-yl) phenyl) boronic acid, 47.53 g (187.18mmol) of 1,4-dibromobenzene, 77.61 g (561.55 mmol) of K2CO3, 1000 ml / 250 ml / stirred into the Toluene / H2O / EtOH in 250 ml. At 40°C into the Pd (PPh3) 4 g of 10.81 (5 mol%) it was stirred at 100°C for 5 hours. halfAfter termination response was extracted with methylene chloride and the filter insert MgSO4. The solvent of the filtered organic layer was purified by column chromatography to 64.12 g (yield: 86%) of 9- (4'-bromo- [1,1'-biphenyl] -4-yl) -9H-carbazole was obtained.
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; for 1h;Reflux;
7-bromo-2-naphthol (21g, 94.1mmol) and carbazole boron acid(29.7g, 103.5mmol) and Was dissolved in potassium carbonate (K2CO3) (39g, 282mmol) in tetrahydrofuran (THF) (300mL), water (H2O) (100ml) was heated to 90 .After addition of tetrakis (triphenylphosphine) palladium (Pd (PPh3) 4) (2.17g, 1.88mmol) was refluxed for 1 hour. After cooling to room temperature to remove the water layer. After the insert was the hwangsang magnesium (MgSO4) the organic layer was filtered. After concentration is purified by column chromatography to obtain the compound of Formula 1A (30g, 85% yield).
In 1L round-bottomed flasks under Scheme 11 under nitrogen atmosphere72.0 g (0.223 mol) of the synthesized <3-a> It is dissolved in 576.0 ml of tetrahydrofuran and cooled to minus 70 degrees. After cooling, 167.60 ml (0.268 mol) of n-butyllithium (1.6 M hexane solution) was slowly added dropwise.1 hour at low temperatureAfter stirring, 75.7 g (0.402 mol) of trimethyl borate was added dropwise, followed by stirring at room temperature for 12 hours. After completion of the reaction, 200 ml of 2N HCl solution was added dropwise, followed by extraction with ethyl acetate and water.The organic layer is anhydrous and then depressurized to remove the organic solvent.After concentration under reduced pressure, 500 ml of hexane was added and recrystallized. The resulting solid was filtered to give 48.3 g (75.3%) of <4-b>.
66.5%
Weigh 1.72 g (5.36 mmol) of intermediate 1 in 100 mL of dry two-necked flask, 50 mL of dry tetrahydrofuran was added as the reaction solvent. Under nitrogen protection, It was cooled to -78 C with liquid nitrogen acetone and stirred at this temperature for 5 min, Under nitrogen, 5.0 mL (1.6 mol / L, 8.04 mmol) of n-butyllithium was added dropwise to the reaction system. After reacting at -78 C for 1 hour, 1.2 mL (10.72 mmol) of trimethyl borate was added dropwise to the reaction system, and the mixture was stirred at room temperature overnight. After the completion of the reaction, dilute hydrochloric acid solution was added and the solution was made acidic. The reaction solution was extracted with dichloromethane (50 mL x 3) The extracts were combined and dried over anhydrous magnesium sulfate, and the solvent was taken out and dried. The crude product was purified by column chromatography using dichloromethane and ethyl acetate as the eluent gradient to give 1.02 g of a white crystalline solid in 66.5% yield.
60%
Under nitrogen 9- (4-bromophenyl) -9H- carbazole THF was cooled to 150 ml of the mixture (10g, 1.0eq) in -78 . After dropwise addition of n-BuLi (18.6 ml, 1.5eq) it was slowly stirred for 1 hour. After adding the trimethyl borate (5.2ml, 1.5eq) it was stirred for 2 hours. Cooled to 0 back extracted with MC was added to 6M HCl. The concentrate EA: hexane = 1: 1 was obtained by column separation of white solid compound 70-5. (5.4g, 60%)
Compound 1-3 (4.86 g, 15.1 mmol) was dissolved in 100 mL of THF under nitrogen gas, and stirred at -78 C. for 30 minutes. N-BuLi (2.5 M Solution in hexane, 6.5 mL, 16.3 mmol) was slowly added dropwise at -78 C., and further stirred for 30 minutes. Trimethylborate (2.5 mL, 22.4 mmol) was added to the reaction mixture, which was stirred for 2 hours at -78 C.The temperature of the reaction mixture was raised to room temperature and stirred for 15 hours. aq, HCl (3.0 M) were added and stirred for 30 minutes. The reaction mixture was extracted with DCM (100 mL X 5), the organic layer was removed with MgSO 4, and all organic solvents were removed to give compound 1-4, which was used in the next reaction without further purification.
With palladium diacetate; potassium carbonate; In ethanol; water; at 80℃;
In the round-bottom flask is added 2 - (4-bromobenzyl) pyridine (1.0mmol), potassium carbonate (2.0mmol), palladium acetate (0.015mmol), 4 - (9-carbazolyl) phenylboronic acid (1.5mmol) and the volume ratio of 3:1 ethanol/water mixed solution of 12 ml. In the air, 80 C conditions, magnetic stirring, to Suzuki cross-coupling reaction, the reaction process by the thin layer chromatography tracking. After the reaction is complete, add saturated salt water 25 ml, with ethyl acetate (3×25 ml) extraction reaction product, combined with the phase, anhydrous Na 2 SO 4 drying, filtering, using rotary evaporimeter concentration to obtain the crude product, to petroleum ether and ethyl acetate as the eluting agent, column chromatography, to obtain the target product of analytical, yield 95%, the product structure through 1 HNMR, 13 CNMR and mass spectrometric identification.
With palladium diacetate; potassium carbonate; triphenylphosphine; In 1,2-dimethoxyethane; for 24h;Reflux;
(4.408 mmol) of 4- (9H-carbazol-9-yl) benzeneboronic acid, 200 mg (0.92 mmol) of <strong>[1780-40-1]2,4,5,6-<strong>[1780-40-1]tetrachloropyrimidine</strong></strong>,103 mg (0.46 mmol) of palladium acetate,241 mg (0.92 mmol) of triphenylphosphine as well1.17 g (11.04 mmol) of anhydrous potassium carbonate was added100ml three-mouth flask,Then, 50 mL of ethylene glycol dimethyl ether was added as a reaction solvent,Air was removed and refluxed for 24 h.After completion of the reaction, the reaction solvent was distilled off under reduced pressure,DCM was extracted three times and then spin-dried through the column.0.26 g of a yellow solid was obtained,Yield 30.6%
1,5-bis[4-(9H-carbazole-9-yl)phenyl]anthracene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
86%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In ethanol; water; toluene; at 90℃; for 14h;Inert atmosphere;
EXAMPLE 2 Synthesis Example 1 (0396) In this example, a synthesis example of an organic compound, 1,5-bis[4-(9H-carbazole-9-yl)phenyl]anthracene (abbreviation: 1,5CzP2A) (Structural Formula:100) is described. Note that chemical formula of 1,5CzP2A is shown below. (0397) A mixture of 1.2 g (3.6 mmol) of dibromoanthracene, 2.3 g (7.9 mmol) of 4-(9H-carbazol-9-yl)phenylboronic acid, 2.2 g (16 mmol) of potassium carbonate, 30 mL of toluene, and 10 mL of ethanol, 8 mL of water, and 83 mg (71 mumol of tetrakis(triphenylphosphine)palladium(0) was stirred under a nitrogen stream at 90 C. for 14 hours. (0398) After the stirring, the mixture was filtered, the obtained solid was washed with water and ethanol and then collected. This solid was purified by silica gel column chromatography (developing solvent: toluene). The purified solid was recrystallized, so that 2.0 g of a pale yellow solid was obtained in a yield of 86%. (0399) The 2.0 g of obtained solid was purified by a train sublimation method under a pressure of 2.7 Pa in an argon stream at 343 C. After the purification, 1.8 g of a pale yellow solid was obtained at a collection rate of 90%. Synthesis scheme (a) of the above synthesis method is shown below. (0400) The following shows analysis results by nuclear magnetic resonance (1H-NMR) spectroscopy of the pale yellow solid obtained by the above-described synthesis method. A 1H-NMR chart is shown in FIGS. 30A and 30B. The 1H NMR charts revealed that 1,5CzP2A, the organic compound of one embodiment of the present invention represented by Structural Formula (100), was obtained in Synthesis Example 1. 1H-NMR (CDCl3, 300 MHz) : delta=7.36 (t, J1=7.8 Hz, 4H), 7.51 (t, J1=8.4 Hz, 4H),7.57 (s, 2H), 7.58 (dd, J1=6.9 Hz, J2=11.7 Hz, 2H), 7.65 (d, J1=7.8 Hz, 4H), 7.80(d, J1=8.4 Hz, 4H), 7.88 (d, J1=8.7 Hz, 4H), 8.07 (dd, J1=2.4 Hz, J2=6.6 Hz, 2H), 8.22 (dd, J1=7.5 H, 4H), 8.72 (s=2H).
1.8 g
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In ethanol; water; toluene; at 90℃; for 14h;Inert atmosphere;
A mixture of 1.2 g (3.6 mmol) of 1,5-dibromoanthracene, 2.3 g (7.9 mmol) of 4-(9H-carbazol-9-yl)phenylboronic acid, 2.2 g (16 mmol) of potassium carbonate, 30 mL of toluene, 10 mL of ethanol, 8 mL of water, and 83 mg (71 mumol) of tetrakis(triphenylphosphine)palladium(0) was stirred under a nitrogen stream at 90 C. for 14 hours. (0443) After the stirring, the mixture was filtered and the obtained solid was washed with water and ethanol and then collected. This solid was purified by silica gel column chromatography (developing solvent: toluene) to give a solid. The obtained solid was recrystallized, so that 2.0 g of a pale yellow solid was obtained in a yield of 86%. (0444) By a train sublimation method, 2.0 g of the obtained solid was purified under a pressure of 2.7 Pa in an argon stream at 343 C. After the purification, 1.8 g of a pale yellow solid was obtained at a collection rate of 90%. A synthesis scheme of the above-described synthesis method is shown in (a). The following shows analysis results by nuclear magnetic resonance (1H-NMR) spectroscopy of the pale yellow solid obtained by the above-described synthesis method. The 1H-NMR charts are shown in FIGS. 37A and 37B. The 1H NMR charts revealed that 1.5CzP2A, the organic compound represented by Structure Foimula (100), was obtained in this synthesis example. (0447) 1H-NMR (CDCl3, 300 MHz): delta =7.36 (t, J1=7.8 Hz, 4H), 7.51 (t, J1=8.4 Hz, 4H), 7.57 (s, 2H), 7.58 (dd, J1=6.9 Hz, J2=11.7 Hz, 2H), 7.65 (d, J1=7.8 Hz, 4H), 7.80 (d, J1=8.4 Hz, 4H), 7.88 (d, J1=8.7 Hz, 4H), 8.07 (dd, J1=2.4 Hz, J2=6.6 Hz, 2H), 8.22 (d, J1=7.5 Hz, 4H), 8.72 (s=2H). (0448) Next, ultraviolet-visible absorption spectra (hereinafter simply referred to as ?absorption spectra?) and emission spectra of 1.5CzP2A in a toluene solution and 1.5CzP2A in a solid thin film were measured. The toluene solution and the solid thin film were each measured in a manner similar to that in Example 2. FIG. 38A shows the measurement results of the obtained absorption and emission spectra of 1.5CzP2A in the toluene solution. The horizontal axis represents wavelength, and the vertical axis represents absorption intensity. FIG. 38B shows the obtained absorption and emission spectra of 1.5CzP2A in the solid thin film. The horizontal axis represents wavelength, and the vertical axis represents absorption intensity. The absorption spectrum shown in FIG. 38A was obtained by subtraction of an absorption spectrum of toluene only put in a quartz cell from the measured absorption spectrum of the toluene solution in a quartz cell. The absorption spectrum shown in FIG. 3 8B was obtained by subtraction of an absorption spectrum of the quartz substrate from an absorption spectrum of the quartz on which 1.5CzP2A was deposited. FIG. 38A shows that 1.5CzP2A in the toluene solution has absorption peaks at around 287 nm, 293 nm, 327 nm, 341 nm, 359 nm, 378 nm, and 397 nm and emission wavelength peaks at around 425 nm and 448 nm (the excitation wavelength: 379 nm). FIG. 38B shows that 1.5CzP2A in the solid thin film has absorption peaks at around 265 nm, 286 nm, 296 nm, 314 nm, 331 nm, 345 nm, 369 nm, 387 nm, and 404 nm and an emission wavelength peak at around 462 nm (the excitation wavelength: 345 nm). (0450) These results show that the organic compound 1.5CzP2A can be used as a blue fluorescent material. (0451) Next, LC/MS analysis was performed. The measurement results are shown in FIG. 39. (0452) In the LC/MS analysis, liquid chromatography (LC) separation was carried out with ACQUITY UPLC (registered trademark) (manufactured by Waters Corporation) and mass spectrometric (MS) analysis was carried out with Xevo G2 Tof MS (manufactured by Waters Corporation). (0453) For the LC separation, ACQUITY UPLC BEH C8 (2.1×100 mm, 1.7 mum) was used as a column, and a mixed solution of acetonitrile and a 0.1% formic acid aqueous solution was used for a mobile phase. (0454) In the MS analysis, ionization was carried out by an electrospray ionization (ESI) method, and the analysis was performed in a positive mode. A component that underwent the ionization was collided with an argon gas in a collision cell to dissociate into product ions. Energy (collision energy) for the collision with argon was 50 eV. A mass range for the measurement was m/z=100-1200. (0455) The result shows that a precursor ion of 1.5CzP2A was detected at around m/z=661, and product ions of 1.5CzP2A were detected at around m/z=495 and around m/z=707. This result is characteristically derived from 1.5CzP2A and thus can be regarded as important data in identification of 1.5CzP2A contained in the mixture. Note that the product ion around m/z=495 is presumed to be a hydrogen ion adduct of a radical expressed as C38H25N·+ in the state where one carbazole is dissociated, and the product ion around m/z=707 is presumed to be acetonitrile and a hydrogen ion adduct. These indicate that a terminal of 1.5CzP2A has a carbazole skeleton and that acetonitrile is easily a...
1,8-bis[4-(9R-carbazole-9-yl)phenyl]anthracene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In ethanol; water; toluene; at 90℃; for 14h;Inert atmosphere;
In a 200 mL three-neck flask, a mixture of 1.2 g (3.7 mmol) of 1,8-dibromoanthracene, 2.3 g (8.1 mmol) of 4-(9H-carbazol-9-yl)phenylboronic acid, 2.2 g (16 mmol) of potassium carbonate, 30 mL of toluene, 10 mL of ethanol, 8 mL of water, and 85 mg (74 mumol) of tetrakis(triphenylphosphine)palladium(0) was stirred under a nitrogen stream at 90 C. for 14 hours. (0460) After the stirring, the mixture was filtered and the obtained solid was washed with water and ethanol and then collected. This solid was purified by silica gel column chromatography (developing solvent: toluene). The purified solid was recrystallized by toluene, so that 2.3 g of a pale yellow solid was obtained in a yield of 93%. By a train sublimation method, 2.2 g of the obtained solid was purified under a pressure of 2.9 Pa in an argon stream at 295 C. After the purification, 1.9 g of a pale yellow solid was obtained at a collection rate of 83%. A synthesis scheme of the above-described synthesis method is shown in (b). (0462) The following shows analysis results by nuclear magnetic resonance (1H-NMR) spectroscopy of the pale yellow solid obtained by the above-described synthesis method. 1H-NMR charts are shown in FIGS. 40A and 40B. The 1H-NMR charts revealed that 1.8CzP2A, the organic compound represented by Structure Formula (110), was obtained. (0463) 1H NMR (CDCl3, 300 MHz): delta =6.88 (t, J1=7.2 Hz, 4H), 7.08 (t, J1=7.8 Hz, 4H), 7.31 (d, J1=8.1 Hz, 4H), 7.55 (dd, J1=1.5 Hz, J2=6.9 Hz, 2H), 7.60-7.68 (m, 6H), 7.80 (d, J1=8.1 Hz, 4H), 8.03 (d, J1=7.8 Hz, 4H), 8.14 (d, J1=7.8 Hz, 2H), 8.66 (s, 1H), 8.95 (s, 1H). (0464) Next, ultraviolet-visible absorption spectra (hereinafter simply referred to as ?absorption spectra?) and emission spectra of 1.8CzP2A in a toluene solution and 1.8CzP2A in a solid thin film were measured. The solution and the solid thin film were each measured in a manner similar to that in Example 2. FIG. 41A shows the obtained absorption and emission spectra of 1.8CzP2A in the toluene solution. The horizontal axis represents wavelength, and the vertical axis represents absorption intensity. FIG. 41B shows the obtained absorption and emission spectra of 1.8CzP2A in the solid thin film. The horizontal axis represents wavelength, and the vertical axis represents absorption intensity. (0465) The result in FIG. 41A shows that 1.8CzP2A in the toluene solution has absorption peaks at around 287 nm, 294 nm, 328 nm, 341 nm, 361 nm, 380 nm, and 399 nm and emission wavelength peaks at around 423 nm and 445 nm (the excitation wavelength: 381 nm). The result in FIG. 41B shows that 1.8CzP2A in the solid thin film has absorption peaks at around 265 nm, 286 nm, 296 nm, 315 nm, 331 nm, 344 nm, 370 nm, 388 nm, and 404 nm and an emission wavelength peak at around 468 nm (the excitation wavelength: 345 nm). These results show that the organic compound 1.8CzP2A can be used as a blue fluorescent material. (0467) Next, LC/MS analysis was performed. The measurement method was similar to that used in Example 2. The measurement result is shown in FIG. 42. (0468) The result shows that precursor ions of 1.8CzP2A were detected at around m/z=661, and product ions of 1.8CzP2A were detected at around m/z=243, m/z=329, and m/z=495. This result is characteristically derived from 1.8CzP2A and thus can be regarded as important data in identification of 1.8CzP2A contained in a mixture. (0469) Note that the product ion around m/z=243 is presumed to be a proton adduct of a radical of phenylcarbazole expressed as C18H13N·+; the product ion around m/z=329 is presumed to be a hydrogen ion adduct of a biradical expressed as C26H172·+ in the state where two carbazoles are dissociated; and the product ion around m/z=495 is presumed to be a hydrogen ion adduct of a radical expressed as C38H25N·+ in the state where one carbazole is dissociated. These indicate that a terminal of 1.8CzP2A includes two carbazole skeletons and a phenylcarbazole skeleton. Note that there is a possibility that the above m/z values±1 are detected as protonation or deprotonation products of the product ions.
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In ethanol; water; toluene; at 90℃; for 14h;
EXAMPLE 3 Synthesis Example 2 (0411) In this example, a synthesis method of an organic compound, 1,8-bis[4-(9H-carbazole-9-yl)phenyl]anthracene (abbreviation: 1,8CzP2A) is described. Note that chemical formula of 1,8CzP2A is shown below. (0412) In a 200 mL three-neck flask, a mixture of 1.2 g (3.7 mmol) of dibromoanthracene, 2.3 g (8.1 mmol) of 4-(9H-carbazol-9-yl)phenylboronic acid, 2.2 g (16 mmol) of potassium carbonate, 30 mL of toluene, and 10 mL of ethanol, 8 mL of water, and 85 mg (74 1mumol) of tetrakis(triphenylphosphine)palladium(0) was stirred under a nitrogen stream at 90 C. for 14 hours. (0413) After the stifling, the mixture was filtered, the obtained solid was washed with water and ethanol and then collected. This solid was purified by silica gel column chromatography (developing solvent: toluene). The purified solid was recrystallized by toluene, so that 2.3 g of a pale yellow solid was obtained in a yield of 93%. 2.0 g of the obtained solid was purified by a train sublimation method under a pressure of 2.7 Pa in an argon stream at 295 C. After the purification, 1.9 g of a pale yellow solid was obtained at a collection rate of 83%. Synthesis scheme (b) of the above synthesis method is shown below. (0414) The following shows analysis results by nuclear magnetic resonance (1H-NMR) spectroscopy of the pale yellow solid obtained by the above-described synthesis method. A 1H-NMR chart is shown in FIGS. 33A and 33B. The 1H-NMR charts revealed that 1,8CzP2A, the organic compound of one embodiment of the present invention represented by Structural Formula (110), was obtained in Synthesis Example 2. 1H NMR (CDCl3, 300 MHz) : delta=6.88 (t, J1=7.2 Hz, 4H), 7.08 (t, J1=7.8 Hz, 4H), 7.31(d, J1=8.1 Hz, 4H), 7.55 (dd, J1=1.5 Hz, J2=6.9 Hz, 2H), 7.60-7.68 (m, 6H), 7.80 (d, J1=8.1 Hz, 4H), 8.03 (d, J1=7.8 Hz, 4H), 8.14 (d, J1=7.8 Hz, 2H), 8.66 (s, 1H), 8.95 (s, 1H).
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; for 24h;Reflux;
To a 2 L round bottom flask was added 60 g (209 mmol) of (4- (9H-carbazol-9-yl) phenyl) boronic acid,54.3 g (230 mmol) of 1-bromo-4-chloro-2-nitrobenzene, 6 g (5.22 mmol) of tetrakistriphenylphosphine palladium & lt; RTI ID = 0.0 & gt;Was dissolved in 500 ml of tetrahydrofuranAnd potassium carbonate87 g (627 mmol) And the mixture was refluxed for 24 hours. The impurities were removed using chloroform and water, and then anhydrous magnesium sulfate was added to remove moisture.The solution was filtered and then recrystallized from chloroform and ethanol to obtain 79 g (94.7%) of intermediate 1.
2,3,5,6-tetrakis(4-(9H-carbazol-9-yl)phenyl)pyrazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89.7%
With potassium phosphate; tetrakis(triphenylphosphine) palladium(0) In ethanol; toluene at 80℃; for 20h; Inert atmosphere;
6 Synthesis of derivative M6
Weigh 1.0 g (3.48 mmol) of intermediate 2, 0.13 g (0.58 mmol) of 2,3,5,6-tetrachloropyrazine, 7.4 g (34.8 mmol) of tripotassium phosphate, 0.014 g (0.012 mmol) of tetrakis(triphenylphosphine)palladium in a 100 ml dry two-necked flask, Add 50 mL (toluene:ethanol=5:1) as the reaction solvent, under nitrogen, Heated to 80 °C for 20 hours. After the reaction, The reaction solution was extracted with dichloromethane (50 mL x 3) after solvent extraction, The extracts were combined and dried over anhydrous magnesium sulfate, dried to dry the solvent and dried. The crude product was treated with petroleum ether and dichloromethane (PE:DCM=15:1) And the residue was purified by column chromatography to obtain 0.54 g of a white crystalline powder, Yield 89.7%.
With potassium phosphate; tetrakis(triphenylphosphine) palladium(0); In ethanol; toluene; at 80℃; for 20h;Inert atmosphere;
Weigh 1.02 g (3.55 mmol) of intermediate 2, 0.13 g (0.59 mmol) of 3,4,5,6-<strong>[20074-67-3]tetrachloropyridazine</strong>, 7.6 g (35.5 mmol) of tripotassium phosphate, 0.023 g (0.012 mmol) of tetrakis(triphenylphosphine)palladium in 100 ml dry two-necked flask was added 50 mL (toluene:ethanol=5:1) As a reaction solvent, under nitrogen, and heated to 80 C for 20 hours. After completion of the reaction, the solvent was extracted under reduced pressure, and dichloromethane (50 mL x 3) The reaction solution was extracted and the extracts were combined and dried over anhydrous magnesium sulfate. The solvent was dried, drying. The crude product was treated with petroleum ether and dichloromethane (PE:DCM=15:1) And the residue was purified by column chromatography to obtain 0.53 g of a pale yellow crystalline powder, Yield 85.6%.
With C18H16F3NO3Pt; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃;Irradiation;
4- (9H-carbazol-9-yl) benzene boronic acid (0.5 mmol), diisopropylethylamine (1.0 mmol), complex Pt2 (2 mol%) and 4 mL DMF were added successively to the reaction flask.Room temperature, air and visible light conditions, the reaction process by thin layer chromatography.After completion of the reaction, 15 mL of saturated brine was added and extracted with ethyl acetate (3 * 15 mL). The organic phases were combined and concentrated. The column was separated by petroleum ether and ethyl acetate as eluant to give the title product 4- (9H-carbazol-9-yl) phenol in 88% yield.
71%
With oxygen; triethylamine; In 2-methyltetrahydrofuran; at 20℃; under 760.051 Torr; for 48h;UV-irradiation;
General procedure: [1,1'-Biphenyl] -4-phenylboronic acid (59.4mg, 0.3mmol, 1.0equiv) was added to a dried 20mL quartz test tube,Vacuum the quartz test tube while backfilling with oxygen three times.Under oxygen conditions, Et3N (62.5 L, 0.45 mmol, 1.5 equiv) and 2-methyltetrahydrofuran (4 ml) were sequentially added through a syringe.The resulting mixture was stirred for 5 minutes, then the quartz test tube was transferred to a photoreactor.The test tube was placed about 2 cm from the 15W UV lamp.The reaction mixture was stirred and illuminated for 24 h,After the specified time, the crude product was diluted with ethyl acetate, filtered through a pad of silica gel, and concentrated under reduced pressure.Flash chromatography on silica gel was then performed directly on silica gel (EtOAc / PE = 1/10) to give the desired product 1b (92% yield, white solid).
With bis-triphenylphosphine-palladium(II) chloride; sodium carbonate; In ethanol; water; toluene; at 100℃; for 12h;
To a three-necked flask was added 23 g (58 mmol) of the flask2-bromo-9,9-diphenylfluorene,10.6 g (87 mmol) of4- (9H-9-carbazole) phenylboronic acid,4.1 g (5.8 mmol) of bistriphenylphosphine palladium dichloride,150ml of 2M aqueous sodium carbonate solution, 300mlToluene and 100 ml of ethanol,The reaction system was heated to 100 C,The reaction was stirred for 12 hours.Cooled to room temperature,Then extracted with dichloromethane (200 ml)The organic phase was washed with distilled water (150 ml)Then dried over magnesium sulfate,And vacuum distillation.Column chromatography(N-hexane: dichloromethane: 20: 1) to give intermediate A-1a (13.2 g, 34 mmol).
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; at 80℃; for 5h;Inert atmosphere;
In a 250 mL three-necked flask,A solution of nitrogen was added, 0.02 mol of intermediate A6 was added,100 ml of THF,0.03 mol (4- (9-H-carbazol-9-yl) phenyl) boronic acid,0.0004 mol of tetrakis (triphenylphosphine) palladium, stirred,Then 0.06 mol of an aqueous K2CO3 solution (2M) was added,Heated to 80 C, refluxed for 5 hours,Sampling point plate, the reaction is complete. Natural cooling,Extracted with 200 ml of dichloromethane, and the layers were separated and the extract was dried over anhydrous sodium sulfate,The filtrate was steamed and purified by silica gel column,Intermediate B1 was obtained, HPLC purity 99.4%, yield 75.3%.
75.3%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; at 80℃; for 5h;Inert atmosphere;
In a 250mL three-neck flask, flush with nitrogen.Add 0.02 mole of intermediate A6, 100 ml of THF,0.03 mol (4-(9-H-carbazol-9-yl)phenyl)boronic acid,0.0004 mol of tetrakis(triphenylphosphine) palladium,Stirring,Then 0.06 mol K2CO3 aqueous solution (2M) was added and heated to 80C.The reaction was refluxed for 5 hours. Samples were taken and the reaction was complete. Natural cooling,Extract with 200 ml of dichloromethane, separate the layers, and dry the extract over anhydrous sodium sulfate.After filtration, the filtrate was evaporated and purified by silica gel column.Obtain intermediate B1,HPLC purity 99.4%,Yield 75.3%.
With bis-triphenylphosphine-palladium(II) chloride; potassium carbonate; In tetrahydrofuran; water; at 80℃; for 48h;Inert atmosphere;
To a two-neck flask were added 4-(9H-9-Carbazole)phenylboronic acid (1.52 g, 5.3 mmol), 4-bromo-2,6-difluorobenzylaldehyde (0.77 g, 3.5 mmol), THF (20.0 mL), and aqueous potassium carbonate solution (12.5 mL, 2.0 M). The mixture was degassed by the freeze-pump-thaw cycle two times. Pd(PPh3)2Cl2 (280.4 mg, 0.4 mmol) was added and degassed by freeze-pump-thaw once again. Then the bright yellow solution was slowly heated to 80 C under nitrogen atmosphere and kept for 48 h and then extracted with DCM and water. The organic layers were combined and dried with anhydrous Na2SO4. Finally, the crude product was concentrated under reduced pressure and further purified by silica gel column chromatography (PE/DCM = 1/4-1/2) to furnish the desired product Cz-24 (1.25 g, 93.0%). 1H NMR (400 MHz, CDCl3) delta 10.43 (s, 1H), 8.19 (d, J = 7.7 Hz, 2H), 7.85 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 8.5 Hz, 2H), 7.48 (dt, J = 8.1, 7.5 Hz, 4H), 7.38-7.32 (m, 4H). 13C NMR (100 MHz, CDCl3) delta 184.2, 164.9, 162.3, 148.6, 140.5, 139.3, 136.1, 128.6, 127.6, 126.2, 123.7, 120.4, 112.8, 110.7, 109.7. MS (EI-TOF) m/z: calculated for C25H15F2NO: 383.11 [M]; found: 383 [M].
With bis-triphenylphosphine-palladium(II) chloride; potassium carbonate; In tetrahydrofuran; water; at 80℃; for 48h;Inert atmosphere;
General procedure: To a two-neck flask were added 4-(9H-9-Carbazole)phenylboronic acid (1.52 g, 5.3 mmol), 4-bromo-2,6-difluorobenzylaldehyde (0.77 g, 3.5 mmol), THF (20.0 mL), and aqueous potassium carbonate solution (12.5 mL, 2.0 M). The mixture was degassed by the freeze-pump-thaw cycle two times. Pd(PPh3)2Cl2 (280.4 mg, 0.4 mmol) was added and degassed by freeze-pump-thaw once again. Then the bright yellow solution was slowly heated to 80 C under nitrogen atmosphere and kept for 48 h and then extracted with DCM and water. The organic layers were combined and dried with anhydrous Na2SO4. Finally, the crude product was concentrated under reduced pressure and further purified by silica gel column chromatography (PE/DCM = 1/4-1/2) to furnish the desired product Cz-24 (1.25 g, 93.0%).
2-(10-bromoanthracen-9-yl)-4,6-diphenyl-1,3,5-triazine[ No CAS ]
9-(4-(10-(4,6-diphenyl-1,3,5-triazin-2-yl)anthracen-9-yl)phenyl)-9H-carbazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90.05%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In ethanol; toluene; at 110℃; for 12h;Inert atmosphere;
1.29 g of 4-boronic acid-9-phenylcarbazole (4.50 mmol) and 2.00 g of 2-(9-bromoanthracene)-4,6-diphenyl-1,3,5-triazine (4.10 mmol ) Add to a clean three-necked flask and purge nitrogen three times.Then 0.14 g of tetra-triphenylphosphine palladium (0.12 mmol) was added to the flask, and nitrogen was pumped three times, and then 30 ml of a 2M potassium carbonate solution, 30 ml of ethanol, and 60 ml of toluene were added to the flask. nitrogen was pumped three times, and then the reaction was stirred at 110 degrees Celsius for 12 hours .Stop the reaction, reduce the temperature to room temperature, then use a rotary evaporator to evaporate the ethanol and toluene.The residue was extracted three times with water and dichloromethane. The organic phase was dried over anhydrous magnesium sulfate, filtered,Dichloromethane was then distilled off, and the crude product was recrystallized from toluene to obtain 2.40 g of the product. (Yield: 90.05%)
With potassium phosphate; tetrakis(triphenylphosphine) palladium(0) In ethanol; water; toluene for 12h; Inert atmosphere; Reflux;
1 1. Synthesis of Compound 1
Under an argon (Ar) atmosphere, to a 200 ml three-neck flask, 1.60 g of 4-(N-carbazolyl)phenylboronic acid, 1.13 g of 1,3-dibromo-7-tert-butylpyrene, 2.50 g of K3PO4, 0.35 g of Pd(PPh3)4, mixed solution of Toluene/EtOH/H2O5 were added, followed by heating and refluxing for about 12 hours. After cooling in the air, a solvent layer was separated and removed, solids precipitated in an organic layer were filtered, and 1.20 g (yield 60%) of Compound 1 as a yellow powder was obtained. The molecular weight of Compound 1 measured by FAB-MS was 741. In addition, chemical shift values of the compound measured by 1H-NMR (300 MHz, tetrahydrofuran-d8) were 8.38-8.42 (m,4H), 8.17-8.22 (m,7H), 8.03 (d,4H), 7.66 (d,4H), 7.60 (d,4H), 7.43 (t,4H), 7.27 (t,4H), 1.62 (s,9H). From the results, the compound of the yellow powder was identified as Compound 1.
48%
With potassium phosphate; tetrakis(triphenylphosphine) palladium(0) In tetrahydrofuran; water for 12h; Reflux;
4 4. Synthesis of Compound 2-1
1.6 g of 4-(9-carbazolyl)phenylboronic acid, 1.13 g of 1,3-dibromo-7-tert-butylpyrene, 0.35 g of tetrakis(triphenylphosphine)palladium(0), 2.5 g of potassium phosphate, 10 ml of water, and 150 ml of THF were refluxed for about 12 hours, and then stood until the temperature reached room temperature. Precipitated crystal (precipitate) was filtered, washed with ethanol, water and acetone one by one, and dried to obtain 1.2 g (yield 48%) of a lemon yellow crystal (solid). FAB-MS analysis result of the lemon yellow compound thus produced is as follows: M/z 740.3 (M+), 741.3 (MH+)
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; sodium hydroxide for 4h; Inert atmosphere; Reflux;
8 (8) Material number L8
Put 9,10-dibromoanthracene in a three-necked bottle.4-(9H-carbazole-9-)-benzeneboronic acid/4-(4-cyanobenzene)-benzeneboronic acid, solvent 50 ml, equipped with a mechanical stir bar,Under nitrogen for 10 min, the catalyst PdCl2 (dppf) 0.25 mol% to 3 mol%, 2M alkali solution 0.018 mol was added under the protection of nitrogen, and the mixture was heated under reflux for 4 hours.After the reaction, it was suction filtered, washed with toluene and washed with ethanol.After recrystallization from xylene, a powder L1 having a purity of 99% or more was obtained, and the product yield was 71%.
4-(10-(4-(9H-carbazol-9-yl)phenyl)anthracen-9-yl)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95.36%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In ethanol; toluene; at 110℃; for 12h;Inert atmosphere;
Add 2.00g of 4-boronic acid-9-phenylcarbazole (6.97 mmol) and 2.08 g of 4-(9-bromoanthracene)benzonitrile synthesis (5.80 mmol) were added to a clean three-necked flask, and nitrogen was pumped three times.Then 0.14 g of tetra-triphenylphosphine palladium (0.12 mmol) was added to the flask, and nitrogen was pumped three times, and then 30 ml of a 2M potassium carbonate solution, 30 ml of ethanol, and 60 ml of toluene were added to the flask, nitrogen was pumped three times, and then the reaction was stirred at 110 degrees Celsius for 12 hours.Stop the reaction, reduce the temperature to room temperature, then use a rotary evaporator to evaporate the ethanol and toluene.The residue was extracted three times with water and dichloromethane. The organic phase was dried over anhydrous magnesium sulfate, filtered,Dichloromethane was then distilled off, and the crude product was recrystallized from toluene to obtain 2.88 g of the product. (Yield: 95.36%)
[4'-(9-carbazolyl)-1,1'-biphenyl-4-yl]-[1,1'-biphenyl]-4-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
34.5 g
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,2-dimethoxyethane; water; toluene; for 10.0h;Reflux; Inert atmosphere;
Under argon atmosphere, into a mixture of 32.4 g (100.0 mmol) of <strong>[1160294-93-8]N-(4-bromophenyl)-4-phenylaniline</strong>, 30.1 g (105.0 mmol) of 4-(carbazole-9-yl)phenylboronic acid, and 2.31 g (2.00 mmol) of Pd[PPh3]4, 150 ml of toluene, 150 ml of dimethoxyethane, and 150 ml (300.0 mmol) of a 2 M aqueous solution of Na2CO3 were added. The resultant mixture was stirred for 10 h by refluxing under heating. After the reaction, the reaction liquid was cooled to room temperature and extracted with dichloromethane in a separating thnnel. The organic layer was dried over Mg504, filtered, and then concentrated. The residue was purified by silica gel column chromatography to obtain 34.5 g of a white solid, which was identified as the following intermediate 1 by FD-MS analysis (field desorption mass spectrometry) (yield: 7 1%).
2-(3-bromophenyl)-5-(pyridin-2-yl)-1,3,4-oxadiazole[ No CAS ]
2-(4'-(9H-carbazol-9-yl)-[1,1'-biphenyl]-3-yl)-5-(pyridin-2-yl)-1,3,4-oxadiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
82%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; at 80℃; for 12h;Inert atmosphere;
Weigh the intermediate A1.5g (5mmol), 4-(9-carbazolyl)benzeneboronic acid 1.44g (5mmol) in a two-necked bottle, Then add 4 ml of absolute ethanol in sequence. 5ml 2mol / L aqueous sodium carbonate solution, 20 ml of toluene, 173 mg of tetrakis(triphenylphosphine)palladium (0.15 mmol). The reaction was carried out at 80 C for 12 hours under argon gas protection. The crude product is purified by column chromatography. A white solid M2 1.9 g (yield 82%) was obtained.
9-(2',3',5',6'-tetrafluoro[1,1'-biphenyl]-4-yl)carbazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
59%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; at 80℃; for 13h;
A mixture of 2,3,5,6-tetrafluorobromobenzene (0.2 g, 0.873 mmol), 4-(9H-Carbazol-9-yl)phenylboronic acid (0.326 g, 1.13 mmol), 2M K2CO3 aq. (1.7 mL, 3.49 mmol) and Pd(PPh3)4 (0.1 g, 0.086 mmol) in THF (5 mL) was heated at 80 C. for 13 h. The reaction mixture was quenched with H2O, washed with brine and extracted with EtOAc. The crude was purified by silica gel column chromatography (hexane: toluene=5:1) to afford compound x (0.201 g, 0.513 mmol) in 59% yield as white powder. 1H-NMR (500MHz, CDCl3, delta): 8.16 (d, J=8.0 Hz, 2H), 7.74-7.70 (m, 4H), 7.53-7.51 (m, 2H), 7.47-7.43 (m, 2H), 7.35-7.30 (m, 2H), 7.16-7.09 (m, 1H), MS (ASAP): 391.2(M+). Calcd for C24H13F4N1: 391.1.
4''-(9H-carbazol-9-yl)-[1,1':4',1''-terphenyl]-4-yl trifluoromethanesulfonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
73%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; at 100℃;Inert atmosphere;
A solution of compound 4z (42.8 mg, 0.1 mmol), Pd(PPh3)4 ( 5.8 mg, 0.005 mmol), (4-(diphenylamino)phenyl)boronic acid (28.9 mg, 0.1 mmol), K2CO3 (1M in water, 1 mL)in THF (2 mL) was stirred at 100 C overnight. The reaction mixture was diluted withwater and extracted with ethyl acetate (5 mL x 3), and the combined organic phase was dried over anhydrous Na2SO4. Purification by silica gel column chromatography usingpetroleum ether as eluent to obtain the desired product S3, 39.7 mg, 73%.
With bis-triphenylphosphine-palladium(II) chloride; potassium carbonate; In tetrahydrofuran; water; at 78℃; for 12h;Inert atmosphere;
General procedure: (9-phenyl-9H-carbazol-3-yl)boronic acid (2.87g, 0.010 mol), 2,7-dibromo-9,9-dioctyl-9H-fluorene (6.58 g, 0.012 mol), PdCl2(PPh3)2 (0.1g), H2O (5 ml) and K2CO3 (6g, 0.043 mol) were added to a 250 ml flaskwith 40 ml THF. The mixture was heated at 78 C under argon atmospherefor 12h. After cooling down, the resulting mixture was extractedwith CH2Cl2, and the organic layer was collected and dried with MgSO4.The solvent was removed and purified by silica gel chromatography toobtain white solid, yield 95% (6.74 g).
With bis-triphenylphosphine-palladium(II) chloride; potassium carbonate; In tetrahydrofuran; water; at 78℃; for 12h;Inert atmosphere;
Add in 500 ml three-neck round bottom flask,4-(9H-carbazol-9-yl)benzeneboronic acid (2.87 g, 0.010 mol),2,7-dibromo-9,9-dioctylfluorene (6.58 g, 0.012 mol), Pd(PPh3)Cl2 (0.1 g),K2CO3 (6g, 0.043mol), THF (40ml), H2O (5ml),The mixture was evacuated under nitrogen and heated at 78 C for 12 h.After the reaction was completed, it was cooled to room temperature, extracted with DCM and dried over anhydrous magnesium sulfate.Over the elution column (DCM elution), remove the solvent by rotary evaporation.Add silica gel powder and sample through the column. A white solid was obtained.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 80℃; for 48h;Inert atmosphere;
A mixture of compound 1-1 (1.0mmol, 0.33g), (4-(9H-carbazol-9-yl)phenyl)boronic acid (2.2mmol, 0.63g), Na2CO3 (10mmol), Pd(PPh3)4 (0.2mmol) were stirred in THF (50ml) and H2O (5ml) for two days under an argon atmosphere at 80C. After completion of present reaction, the mixture was extracted with dichloromethane (3×60ml), and the combined organic layers were washed with brine, dried (Na2SO4), and concentrated in vacuo. The residues were purified by column chromatography using petroleum ether: dichloromethane (volume ratio=1:1) as eluent, affording the expected white solid product in a yield of 69.2%. 1H NMR (400MHz, CDCl3): delta (ppm)=8.18 (d, J=8Hz, 4H, Ar-H), 7.77 (d, J=8Hz, 2H, Ar-H), 7.72 (s, 8H, Ar-H), 7.59 (d, J=8Hz, 4H, Ar-H), 7.48 (t, J=8Hz, 4H, Ar-H), 7.34 (t, J=6Hz, 6H, Ar-H), 7.08 (t, J=8Hz, 2H, Ar-H), 6.86 (d, J=8Hz, 2H, Ar-H). 13C NMR (100MHz, CDCl3): delta (ppm)=143.2, 141.5, 140.8, 140.7, 138.5, 137.9, 135.7, 131.5, 128.2, 127.4, 126.7, 126.1, 124.9, 123.7, 120.5, 120.3, 119.6, 109.8. EI-MS: m/z=660.5[M]+. Anal. Calcd. For C50H32N2: C, 90.88; H, 4.88; N, 4.24. Found: C, 90.81; H, 4.85; N, 4.29.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 80℃; for 48h;Inert atmosphere;
A mixture of compound 2-1 (1.0mmol, 0.62g), (4-(9H-carbazol-9-yl)phenyl)boronic acid (2.2mmol, 0.63g), Na2CO3 (10mmol), Pd(PPh3)4 (0.2mmol) were stirred in THF (50ml) and H2O (5ml) for two days under an argon atmosphere at 80C. After completion of present reaction, the mixture was extracted with dichloromethane (3×60ml), and the combined organic layers were washed with brine, dried (Na2SO4), and concentrated in vacuo. The residues were purified by column chromatography using petroleum ether: dichloromethane (volume ratio=1:2) as eluent, affording the expected white solid product in a yield of 66.6%. 1H NMR (500MHz, CD2Cl2): delta (ppm)=8.06 (d, J=10Hz, 4H, Ar-H), 7.79 (d, J=10Hz, 2H, Ar-H), 7.68 (t, J=7.5Hz, 6H, Ar-H), 7.53 (d, J=10Hz, 4H, Ar-H), 7.45 (t, J=7.5Hz, 2H, Ar-H), 7.40 (t, J=7.5Hz, 2H, Ar-H), 7.23 (m, 12H, Ar-H), 7.08 (s, 4H, Ar-H), 6.85 (s, 2H, Ar-H). 13C NMR data of the compound were not obtained due to the poor solubility. HRMS-ESI (m/z): 948.796 [M]+. Anal. Calcd. For C64H38F6N2: C, 81.00; H, 4.04; N, 2.95. Found: C, 81.05; H, 4.01; N, 2.89.
With palladium diacetate; sodium carbonate; triphenylphosphine In 1,2-dimethoxyethane; water at 130℃; for 23h; Inert atmosphere;
4.3. General procedure for Suzuki-Miyaura cross-coupling reactions
General procedure: A mixture of 4-chloroquinazoline or 7-bromo-4-chloroquinazoline(1 mmol), the appropriate boronic acid (1.1 or 2.2 mmol, respectively),Pd(AcO)2 (5 mol%), PPh3 (10 mol%), aqueous Na2CO3 (5 mmol,dissolved in the minimum amount of water), and glyme (5 mL) wasdegassed by bubbling Ar through for 10 min. The reaction mixture wasstirred in a pressure tube at the indicated temperature (100-140 °C) for 22-23 h. After removing the glyme under reduced pressure, the reaction mixture was diluted with water and extracted with CH2Cl2 ( × 3). The combined organic layers were dried (MgSO4), filtered through a short pad of Celite, and concentrated under vacuum. The crude product was purified by flash chromatography on neutral alumina (eluent, mixturesof hexanes/EtAcO of increasing polarity and a few drops of Et3N) and/or washing with the indicated solvent.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In ethanol; water; toluene at 90℃; for 24h;
1 Example 1 Preparation of aniline
Add 4-(9H-carbazol-9-yl)phenylboronic acid (26.55g, 92.45mmol),2,6-Dibromo-tetramethylaniline (5.00g, 36.98mmol), sodium carbonate aqueous solution (150ml, 2M), a small amount of ethanol, tetrakis(triphenylphosphine)palladium (4.27g, 3.70mmol) in toluene (150ml )The solution was stirred and kept at 90°C for 24 hours, cooled to room temperature,Rotate and evaporate the solvent until a yellow solid appears. Add excess methanol to precipitate the product.The yellow solid is separated by filtration,Wash three times with methanol and dry under vacuum to obtain a yellow solid product(19.30g, 88.5% yield).
4-(4-(9H-carbazol-9-yl)phenyl)-7-nitrobenzo[c][1,2,5]oxadiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
86.22%
With bis(tri-tert-butylphosphine)palladium; caesium carbonate In tetrahydrofuran at 75℃; for 24h; Inert atmosphere;
1-7; 1-2
4-(9H-carbazol-9-yl)phenylboronic acid (1.00g, 3.49mmol),4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (0.66g, 3.31mmol), bis(tri-tert-butylphosphine)palladium (35.60mg, 0.07mol) and cesium fluoride (1.59 g, 10.49 mmol) was added to a 250 mL three-necked flask, and then tetrahydrofuran (18 mL) and water (2 mL) that had been ultrasonically degassed were added, and the mixed system was reacted at 75° C. for 24 hours under argon protection. The reaction was stopped, the system was cooled to room temperature, concentrated, dissolved in dichloromethane and filtered, the filtrate was further concentrated and the product was separated by column chromatography to obtain 1.22 g of a reddish brown product (photosensitizer) with a yield of 86.22%.
79.38%
With bis(tri-t-butylphosphine)palladium(0); cesium fluoride In tetrahydrofuran; methanol; water; toluene at 75℃; for 24h; Inert atmosphere; Schlenk technique;
1,1′-di[4-(9-carbazolyl)phenyl]ferrocene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In N,N-dimethyl-formamide at 120℃; for 168h; Inert atmosphere;
Synthesis of 1,1′-di[4-(9-carbazolyl))phenyl]ferrocene (DCPF).
80 mg FcBr, 180 mg NBAPC, 2 mL K2CO3 solution (450 mg/mL), 8 mLDMF and 30 mg tetrakis (triphenylphosphine) palladium(0) were addedinto a three-necked flask. The mixture was stirred at 120 C for 7 daysunder an argon atmosphere. The resulting mixture was cooled to roomtemperature and poured into deionized water. The mixture wasextracted with dichloromethane and the organic phase was dried using arotary evaporator. The crude product was further purified by columnchromatography on silica gel (petroleum ether and dichloromethane).Finally, the product was obtained orange-yellow powders after vacuumdrying. 1H NMR (CD2Cl2, 500 MHz): δ ppm 8.12 (d, J = 8.40 Hz, 4H),7.61 (d, J = 8.45 Hz, 4H), 7.48 (d, J = 8.40 Hz, 4H), 7.43 (d, J = 8.15 Hz,4H), 7.30 (t, J = 8.20 Hz, 4H), 7.25 (t, J = 7.55 Hz, 4H), 4.69 (s, 4H),4.40 (s, 4H). MS: m/z calculated for C46H32N2Fe: 668.6 Da; found: 668.2Da.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping