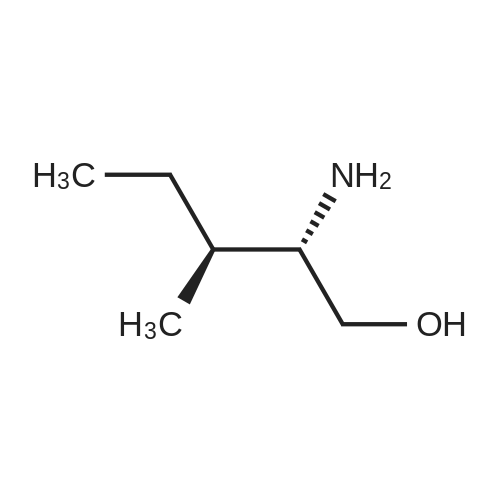
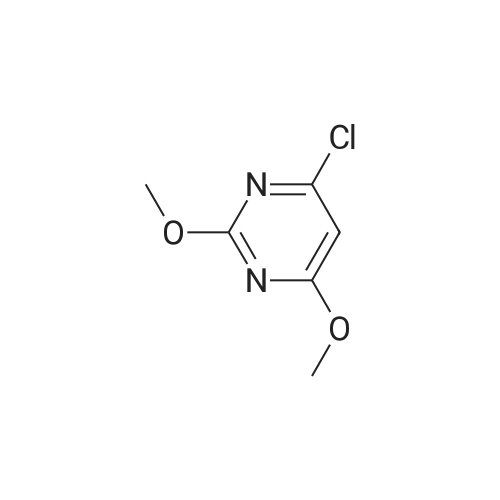
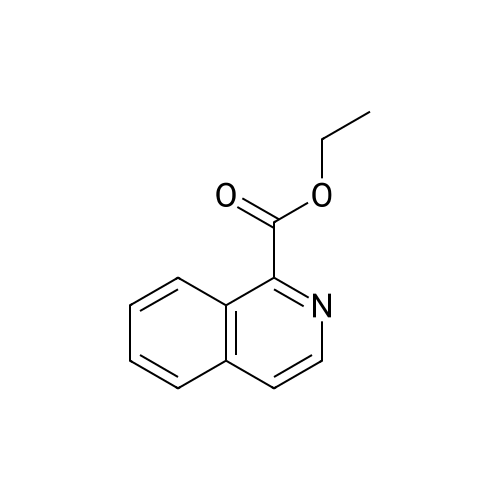
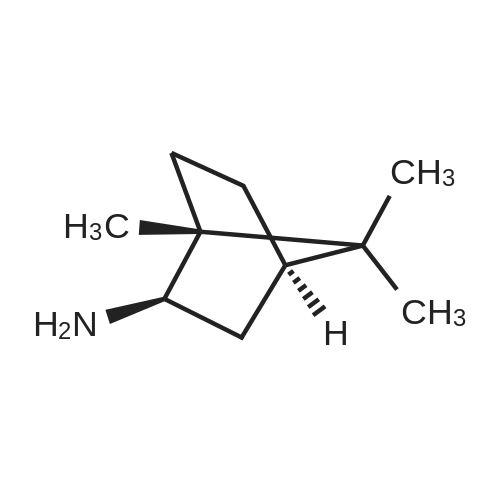
|
Stage #1: (1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-one With potassium <i>tert</i>-butylate In diethyl ether at -40 - 20℃;
Stage #2: With isopentyl nitrite In diethyl ether at -40 - 20℃; for 16.5h; |
4.5.6. (5aS,6R,9S,9aR)-6,11,11-Trimethyl-2-phenyl-5a,6,7,8,9,9a-hexahydro-4H-6,9-methano-benzo[b][1,2,4]triazolo[4,3-d][1,4]oxazin-2-ium tetrafluoroborate 46
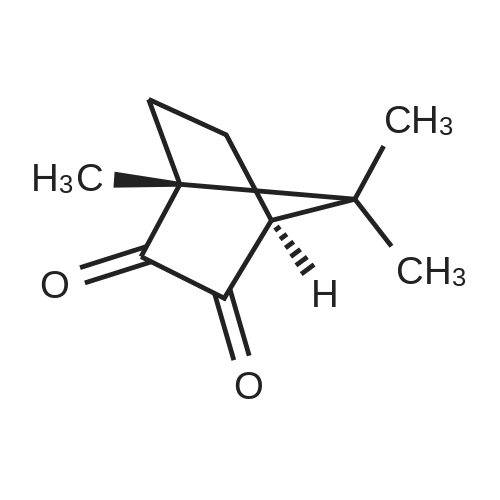
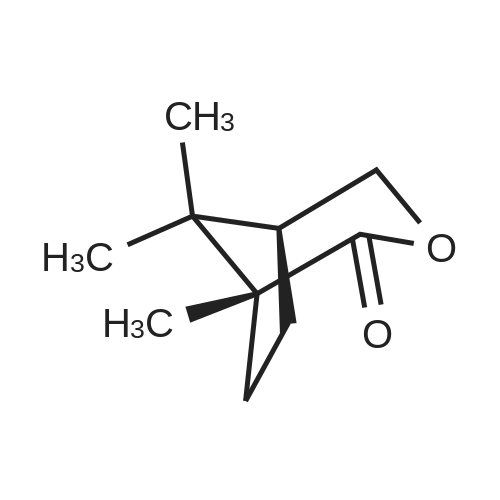
To a cooled (-40 °C) solution of potassium tert-butoxide (18.4 g, 103 mmol) in Et2O (125 mL) was added d-camphor (12.5 g, 82.0 mmol) in Et2O (40 mL) dropwise over 25 min. Once the addition was complete the mixture was stirred at ambient temperature for 1 h, recooled to -40 °C and isoamyl nitrite (22.0 mL, 164 mmol) was added dropwise over 30 min. The bright orange solution was warmed to ambient temperature over 16 h and then extracted with H2O (50 mL × 3). The combined aqueous phases were acidified to pH 2 with concd HCl (8 mL) and extracted with CH2Cl2 (50 mL × 3). The combined organic layers were washed successively with satd NaHCO3(aq) (20 mL), H2O (20 mL) and brine (20 mL) then dried (MgSO4), filtered and concentrated in vacuo to give the crude oxime as a yellow solid (19.9 g, 84%) as a 2:1 mixture of E/Z-diastereomers which was used immediately without further purification. To a cooled (0 °C) solution of oxime (12.1 g, 66.8 mmol) in THF (30 mL) was added LiAlH4 (50.0 mL of a 1 M solution in THF, 100 mmol) dropwise over 30 min. After H2 evolution had ceased, the solution was heated at reflux (80 °C) for 30 min. The solution was allowed to cool to ambient temperature, diluted with Et2O (65 mL) and quenched with H2O (4 mL), NaOH (10% w/v, 4 mL) and H2O (12 mL). The mixture was filtered through Celite and the filtrate was concentrated in vacuo to give the crude syn-amino alcohol product (8:1 dr) as a colourless solid (10.6 g, 94%), which was used immediately without further purification. To a cooled (0 °C) solution of the amino alcohol (2.30 g, 13.6 mmol) and Et3N (3.02 mL, 21.7 mmol) in CH2Cl2 (60 mL) was added chloroacetyl chloride (1.19 mL, 14.9 mmol) dropwise over 30 min. The solution was warmed to ambient temperature over 16 h then recooled to 0 °C and a solution of potassium tert-butoxide (6.40 g, 57.0 mmol) in isopropanol (50 mL) was added over 30 min. The mixture was allowed to warm to ambient temperature and stirred for 18 h before concentration in vacuo. The brown residue was taken up in EtOAc (20 mL) and H2O (30 mL) added. The product was extracted with EtOAc (20 mL × 3) and the combined organic fraction was dried (Na2SO4), filtered and concentrated in vacuo to give a brown solid. Chromatographic purification (50% EtOAc/petrol) gave the morpholinone as a pale yellow solid (640 mg, 44%). inlMMLBox (c 1.0, CHCl3), lit.36 +95.0 (c 1.0, CHCl3); mp§ 94-97 °C; δH (400 MHz, CDCl3) 5.93 (1H, br s, NH), 4.12 (1H, d, J 15.4, CHAHB-2), 3.78 (1H, d, J 15.4, CHAHB-2), 3.65 (1H, d, J 6.8 CH-8a), 3.37 (1H, d, J 6.8, CH-4a), 1.62-1.54 (4H, m, CH-5, CH2-7 and CHAHB-6), 1.13 (3H, s, (CH3)C-8), 1.08-1.02 (1H, m, CHAHB-6), 0.99 (3H, s, CH3) and 0.85 (3H, s, CH3). Data are in accordance with the literature.36 Y. Li, Z. Feng and S. You, Chem. Commun. (2008), pp. 2263-2265. Full Text via CrossRef | View Record in Scopus | Cited By in Scopus (21)36To a solution of morpholinone (200 mg, 0.956 mmol) in CH2Cl2 (20 mL) was added trimethyloxonium tetrafluoroborate (169 mg, 1.14 mmol) and the mixture was stirred for 16 h at ambient temperature. Phenylhydrazine (94.1 μL, 0.956 mmol) was added and the solution was stirred for 24 h. The mixture was then concentrated in vacuo and the residue triturated with Et2O (10 mL) to give a light brown solid that was dissolved in chlorobenzene (1 mL) and triethyl orthoformate (5 mL) and then heated at reflux (125 °C) for 12 h. The mixture was concentrated in vacuo then triturated with Et2O (10 mL) to afford the title product 46 as a colourless solid (75.9 mg, 20%). inlMMLBox (c 0.5, CHCl3), lit.36 +29.4 (c 1.0, CHCl3); mp§ 191-192 °C; δH (300 MHz, CDCl3) 10.28 (1H, s, NCHN), 7.91-7.89 (2H, m, ArH), 7.55-7.52 (3H, m, ArH), 5.07 (1H, d, J 15.1, CHO), 4.67 (1H, d, J 15.1, CHN), 4.48 (1H, d, J 7.0 CH), 4.08 (1H, d, J 7.0, CH2), 2.66 (1H, d, J 4.5 CH2), 1.96-1.84 (1H, m, CH2), 1.68-1.58 (1H, m, CH2), 1.39-1.24 (1H, m, CH2), 1.03 (3H, s, CH3), 0.88 (3H, s, CH3) and 0.66 (3H, s, CH3). Data are in accordance with the literature.36 |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping