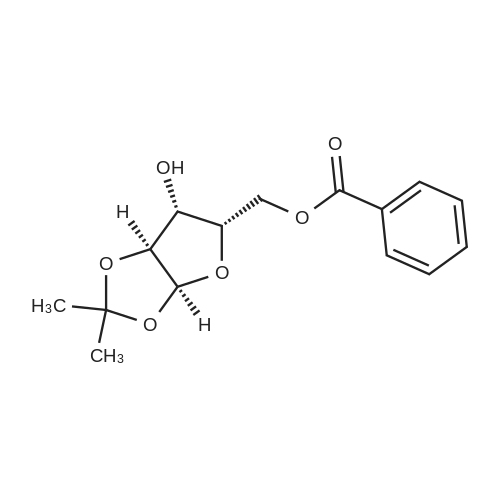
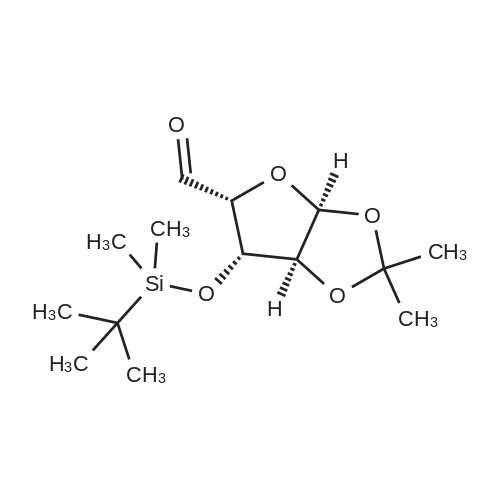
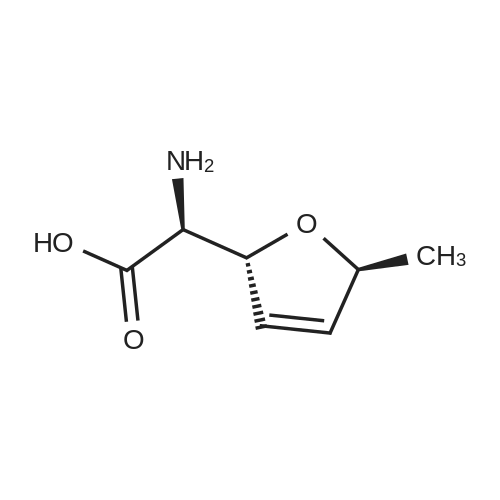
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: at 20 - 24℃; for 17.25 h; Industry scale Stage #2: With hydrogenchloride In water at 20℃; for 6 h;
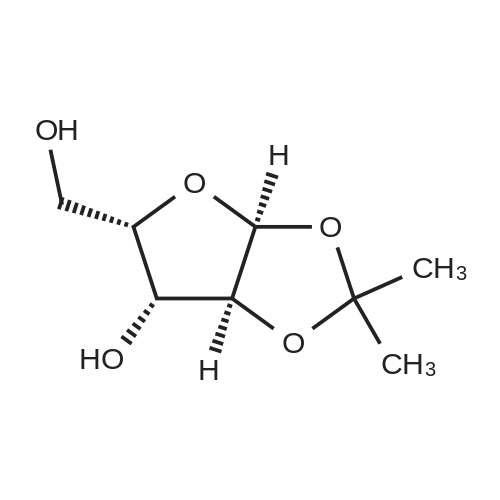
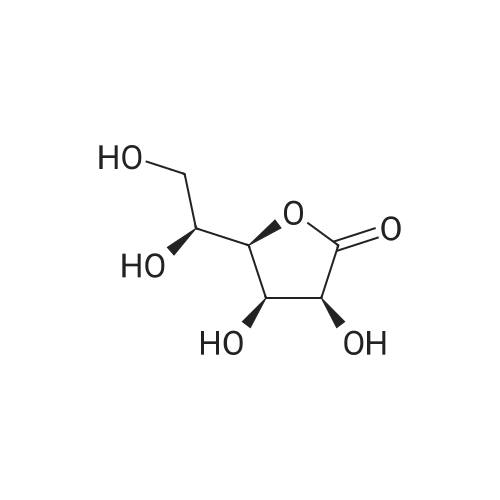
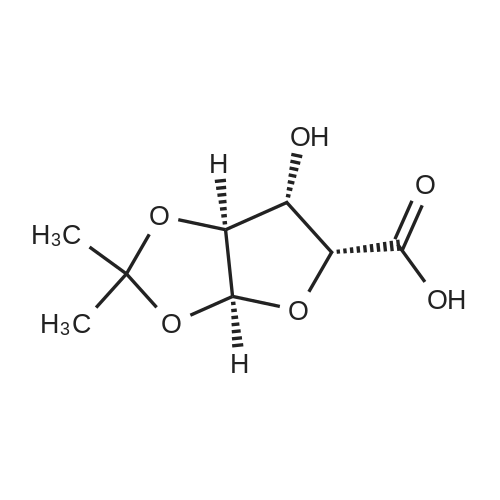
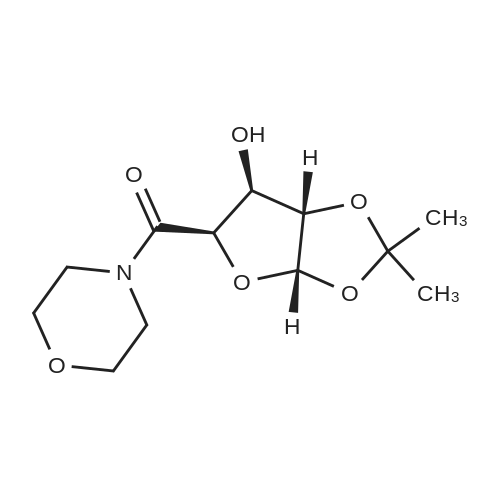
6. EXAMPLES Aspects of this invention can be understood from the following examples.6.1. Synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2.3-d][13]dioxol-5-yl)(morpholino)methanone To a 12L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged L-(-)-xylose (504.40 g, 3.360 mol), acetone (5L, reagent grade) and anhydrous MgSO4 powder (811.23g, 6.740 mol / 2.0 equiv). The suspension was set stirring at ambient and then concentrated H2SO4 (50 mL, 0.938 mol / 0.28 equiv) was added. A slow mild exotherm was noticed (temperature rose to 24°C over about 1 hr) and the reaction was allowed to stir at ambient overnight. After 16.25 hours, TLC suggested all L-xylose had been consumed, with the major product being the bis-acetonide along with some (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction mixture was filtered and the collected solids were washed twice with acetone (500 mL per wash). The stirring yellow filtrate was neutralized with concentrated NH4OH solution (39 mL) to pH = 8.7. After stirring for 10 min, the suspended solids were removed by filtration. The filtrate was concentrated to afford crude bis-acetonide intermediate as a yellow oil (725.23 g). The yellow oil was suspended in 2.5 L water stirring in a 5L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler. The pH was adjusted from 9 to 2 with 1N aq. HCl (142mL) and stirred at room temperature for 6 h until GC showed sufficient conversion of the bis-acetonide intermediate to (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction was neutralized by the addition of 50percent w/w aq. K2HPO4 until pH=7. The solvent was then evaporated and ethyl acetate (1.25L) was added to give a white suspension which was filtered. The filtrate was concentrated in vacuo to afford an orange oil which was dissolved in 1 L methyl tert-butyl ether. This solution had KF 0.23 wtpercent water and was concentrated to afford (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol as an orange oil (551.23g, 86percent yield, 96.7 areapercent pure by GC). 1H NMR (400 MHz, DMSO-d6)δ1.22 (s, 3 H) 1.37 (s, 3 H) 3.51 (dd, J=11.12, 5.81 Hz, 1 H) 3.61 (dd, J=11.12, 5.05 Hz, 1 H) 3.93 - 4.00 (m, 1 H) 3.96 (s, 1 H) 4.36 (d, J=3.79 Hz, 1 H) 4.86 (br. s., 2 H) 5.79 (d, J=3.54 Hz, 1 H). 13C NMR (101MHz, DMSO-d6) δ26.48, 27.02, 59.30, 73.88, 81.71, 85.48, 104.69, 110.73. To a solution of (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (25.0g, 131 mmol) in acetone (375 mL, 15X) and H2O (125 mL, 5X) was added NaHC03 (33.0g, 3.0 equiv), NaBr (2.8g, 20 molpercent) and TEMPO (0.40g, 2 molpercent) at 20°C. The mixture was cooled to 0-5°C and solid trichloroisocyanuric acid (TCCA, 30.5 g, 1.0 equiv) was then added in portions. The suspension was stirred at 20°C for 24h. Methanol (20 mL) was added and the mixture was stirred at 20°C for 1h. A white suspension was formed at this point. The mixture was filtered, washed with acetone (50 mL, 2X). The organic solvent was removed under vacuum and the aqueous layer was extracted with EtOAc (300 mL, 12X x3) and the combined organic layers were concentrated to afford an oily mixture with some solid residue. Acetone (125 mL, 5X) was added and the mixture was filtered. The acetone solution was then concentrated to afford the desired acid ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid) as a yellow solid (21.0g, 79percent). 1H NMR (methanol-d4), δ 6.00 (d, J= 3.2 Hz, 1H), 4.72 d, J= 3.2 Hz, 1H), 4.53 (d, J= 3.2 Hz, 1H), 4.38 (d, J= 3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H). To a solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (5.0g, 24.5 mmol) in THF (100 mL, 20X) was added TBTU (11.8g, 1.5 equiv), N-methylmorpholine (NMM, 4.1 mL, 1.5 equiv) and the mixture was stirred at 20°C for 30 min. Morpholine (3.2 mL, 1.5 equiv) was then added, and the reaction mixture was stirred at 20°C for an additional 6h. The solid was filtered off by filtration and the cake was washed with THF (10 mL, 2X x2). The organic solution was concentrated under vacuum and the residue was purified by silica gel column chromatography (hexanes:EtOAc, from 1:4 to 4:1) to afford 4.3 g of the desired morpholine amide (64percent) as a white solid. 1H NMR (CDCl3), 8 6.02 (d, J= 3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J= 3.2 Hz, 1H), 4.58 (d, J= 3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H).
Reference:
[1] Patent: EP2332947, 2011, A1, . Location in patent: Page/Page column 6-7
[2] Nucleosides and Nucleotides, 1995, vol. 14, # 3-5, p. 611 - 617
[3] Organic Letters, 2002, vol. 4, # 22, p. 3951 - 3953
[4] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 22, p. 9830 - 9836
Reference:
[1] Collection of Czechoslovak Chemical Communications, 2006, vol. 71, # 7, p. 1063 - 1087
[2] Nucleosides, Nucleotides and Nucleic Acids, 2001, vol. 20, # 4-7, p. 703 - 706
[3] Helvetica Chimica Acta, 1938, vol. 21, p. 263,267
[4] Patent: WO2012/140596, 2012, A1,
[5] Patent: CN106892948, 2017, A,
4
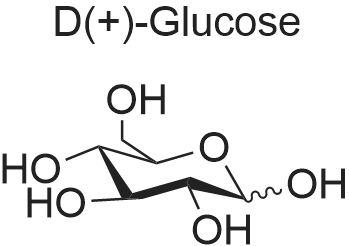
[ 609-06-3 ]
[ 67-64-1 ]
[ 131156-47-3 ]
Yield
Reaction Conditions
Operation in experiment
84.3%
at 20 - 30℃; for 12 h;
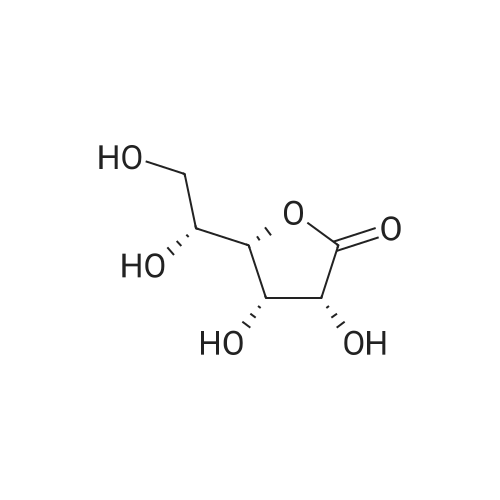
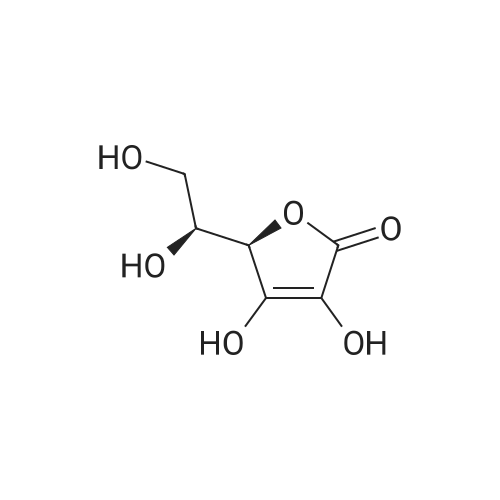
in room temperature,A solution of magnesium sulfate (160 g, 1.33 mol) and concentrated sulfuric acid (9.9 mL, 190 mmol)(2S, 3R, 4S) -2,3,4,5-tetrahydroxyvaler 1a (99.0 g, 659 mmol)In acetone (1.2 L)The resulting reaction was stirred for 12 hours.The reaction mixture was suction filtered,Washed with acetone (300 mL x 2)Combined filtrate,With 25percent ammonia to adjust the pH to 7,Solid precipitated, filtered again and washed with acetone (150 mL x 2). The filtrate was combined, concentrated under reduced pressure,The title compound Ib was obtained as a yellow oil (128.0 g, 84.3percent).
Reference:
[1] Patent: CN106892948, 2017, A, . Location in patent: Paragraph 0183; 0185; 0186; 0187
[2] Nucleosides, Nucleotides and Nucleic Acids, 2001, vol. 20, # 4-7, p. 703 - 706
[3] Collection of Czechoslovak Chemical Communications, 2006, vol. 71, # 7, p. 1063 - 1087
[4] Patent: WO2012/140596, 2012, A1, . Location in patent: Page/Page column 53-55
Reference:
[1] Helvetica Chimica Acta, 1938, vol. 21, p. 263,267
7
[ 74-90-8 ]
[ 609-06-3 ]
[ 6322-07-2 ]
Reference:
[1] Journal of Biological Chemistry, 1918, vol. 36, p. 348
[2] Justus Liebigs Annalen der Chemie, 1914, vol. 403, p. 249[3] Justus Liebigs Annalen der Chemie, 1910, vol. 376, p. 55 Anm.
8
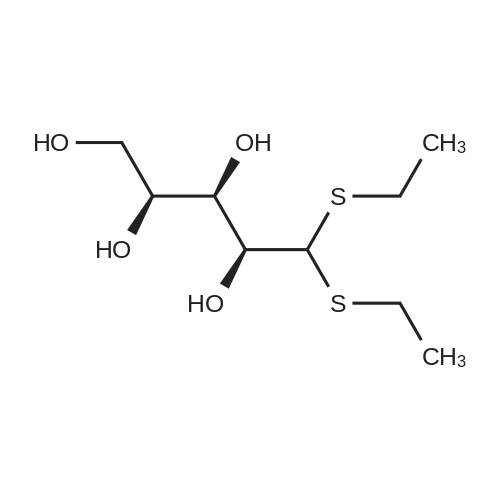
[ 609-06-3 ]
[ 3080-30-6 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 23, p. 7100 - 7110
9
[ 1195193-89-5 ]
[ 609-06-3 ]
Yield
Reaction Conditions
Operation in experiment
92%
at 20 - 30℃;
Compound 9 (10.0 g, 31.42 mmol) was dissolved in 30percent sodium ethoxide solution (22.7 g, 126.3 mmol), and the mixture was stirred at 20-30 degrees for 2-5 hours, after concentrating the reaction solution, 30 ml of dichloromethane was added, ρΗ=7 was adjusted with 3N hydrochloric acid, the organic phase was separated and the aqueous phase was extracted three times with dichloromethane, dried the organic phase with anhydrous sodium sulfate, after separation of the solid, the organic phase was concentrated to give white crystals L-xylose (4.4 g, yield 92percent);
Reference:
[1] Patent: CN108752396, 2018, A, . Location in patent: Paragraph 0068-0070
10
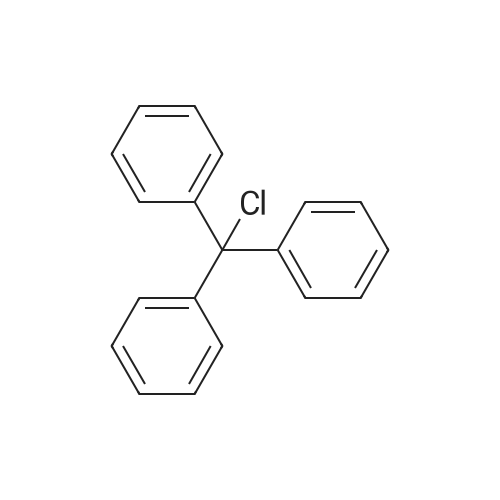
[ 124244-64-0 ]
[ 609-06-3 ]
Reference:
[1] Journal of the American Chemical Society, 1989, vol. 111, # 26, p. 9275 - 9276
11
[ 1128-23-0 ]
[ 609-06-3 ]
Reference:
[1] Patent: US2003/97029, 2003, A1,
12
[ 124244-63-9 ]
[ 609-06-3 ]
Reference:
[1] Journal of the American Chemical Society, 1989, vol. 111, # 26, p. 9275 - 9276
Reference:
[1] Journal of Organic Chemistry, 1992, vol. 57, # 22, p. 5899 - 5907
21
[ 87-99-0 ]
[ 609-06-3 ]
Reference:
[1] Advanced Synthesis and Catalysis, 2009, vol. 351, # 10, p. 1523 - 1530
22
[ 137491-74-8 ]
[ 609-06-3 ]
Reference:
[1] Liebigs Annalen der Chemie, 1992, # 1, p. 95 - 98
23
[ 7732-18-5 ]
[ 50-81-7 ]
[ 98-01-1 ]
[ 609-06-3 ]
[ 124-38-9 ]
Reference:
[1] Bulletin de la Societe Chimique de France, 1959, p. 74,75
[2] Bulletin de la Societe Chimique de France, 1959, p. 74,75
[3] Food Research, 1953, vol. 18, p. 633
Scheme 2 below shows an exemplary means for synthesizing the thioarabinitol reagents. Thioarabinitol 8 and 11 were synthesized from D-<strong>[609-06-3]xylose</strong> and <strong>[609-06-3]L-<strong>[609-06-3]xylose</strong></strong> respectively, following a similar strategy previously published by the inventors
Trifluoro-methanesulfonate2-[(S)-benzyloxy-((4R,5S,4'S)-2,2,2',2'-tetramethyl-[4,4']bi[[1,3]dioxolanyl]-5-yl)-methyl]-3-methyl-thiazol-3-ium;[ No CAS ]
Trifluoro-methanesulfonate2-[(S)-(tert-butyl-diphenyl-silanyloxy)-((4R,5S,4'S)-2,2,2',2'-tetramethyl-[4,4']bi[[1,3]dioxolanyl]-5-yl)-methyl]-3-methyl-thiazol-3-ium;[ No CAS ]
Trifluoro-methanesulfonate3-methyl-2-[(S)-((4R,5S,4'S)-2,2,2',2'-tetramethyl-[4,4']bi[[1,3]dioxolanyl]-5-yl)-triethylsilanyloxy-methyl]-thiazol-3-ium;[ No CAS ]






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping