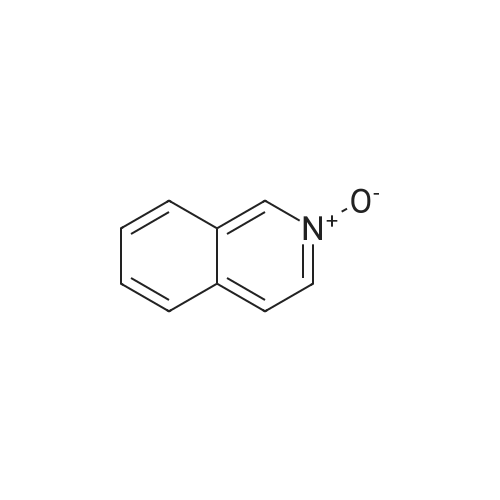
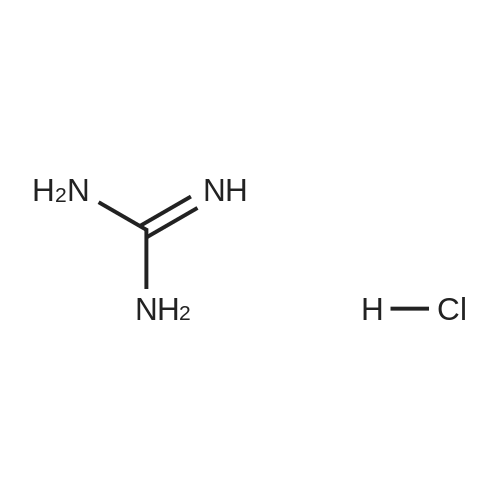
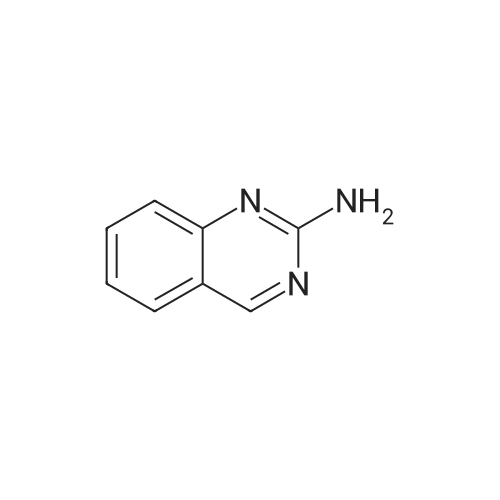
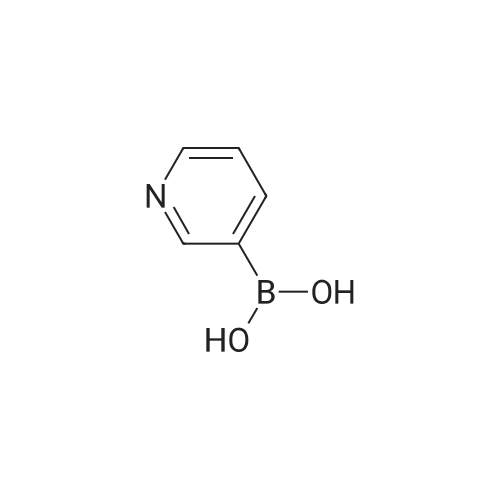
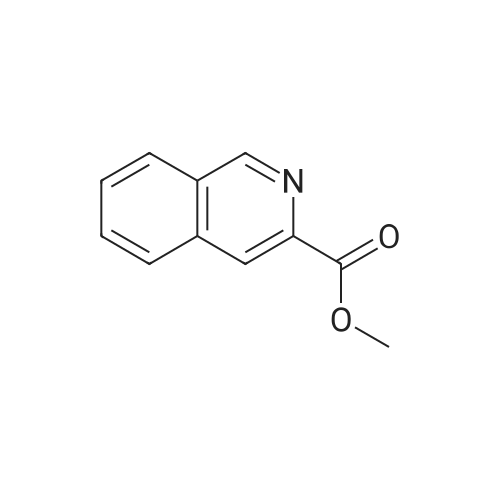
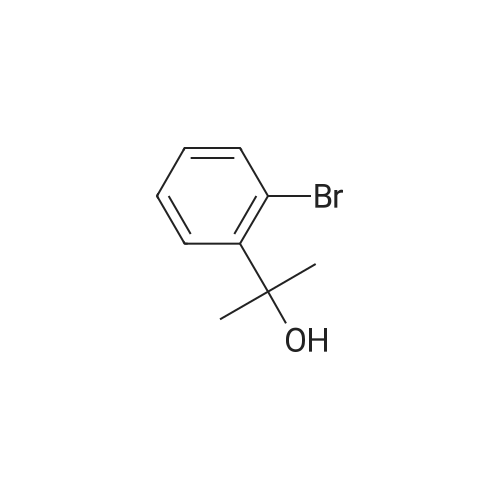
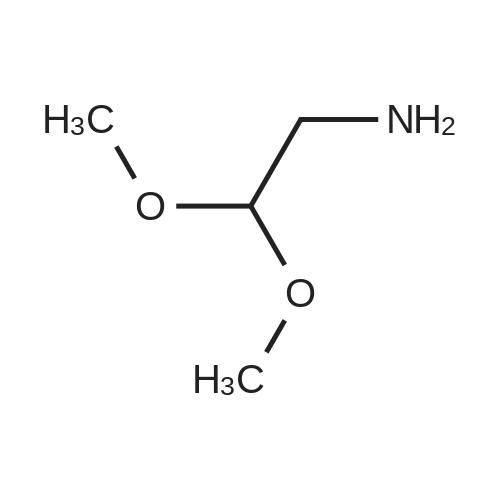
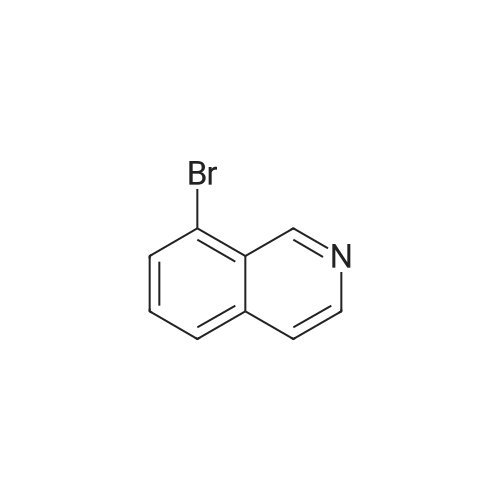
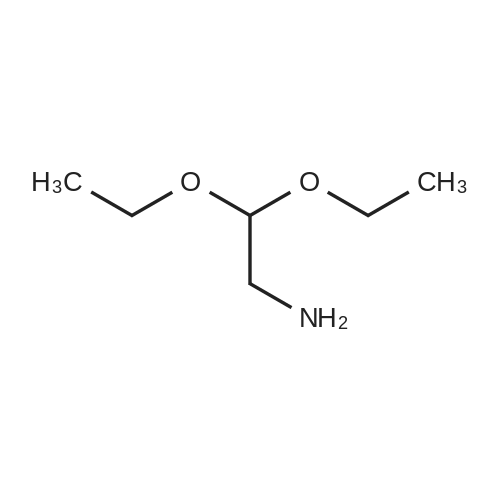
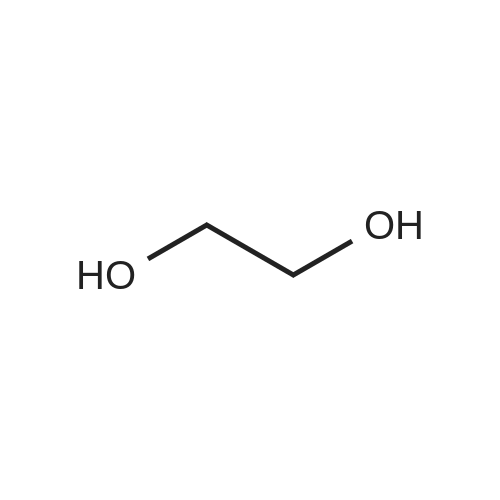
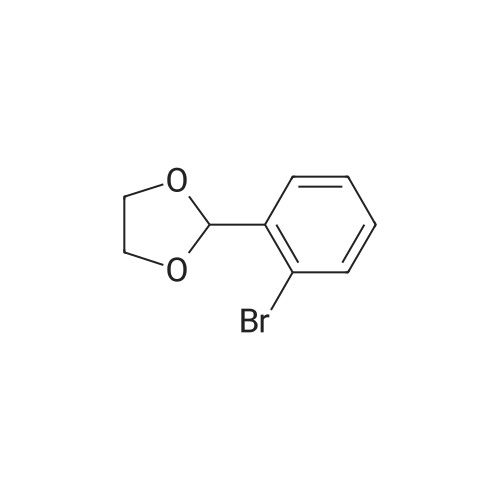
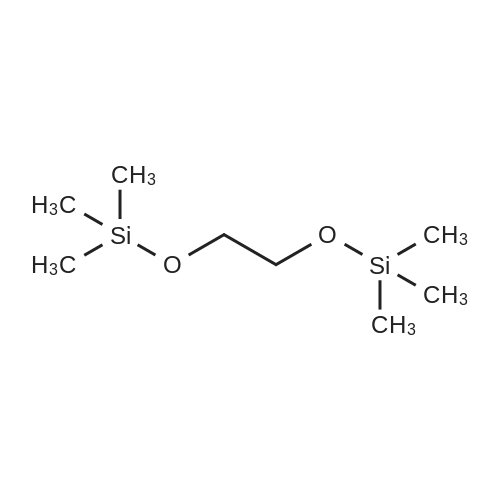
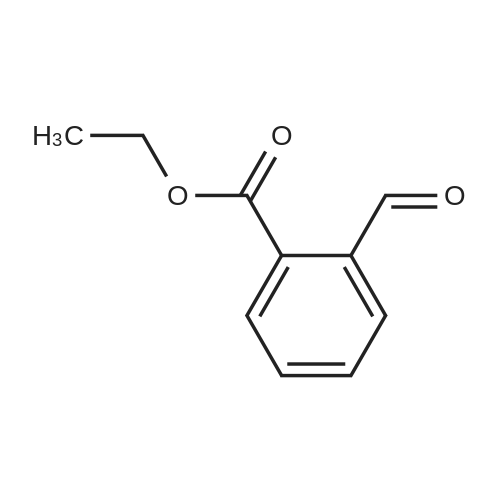
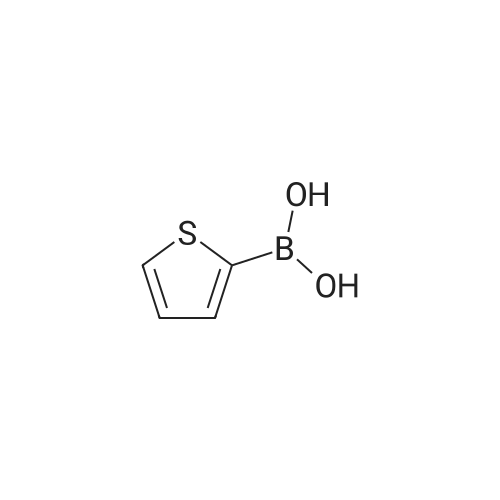
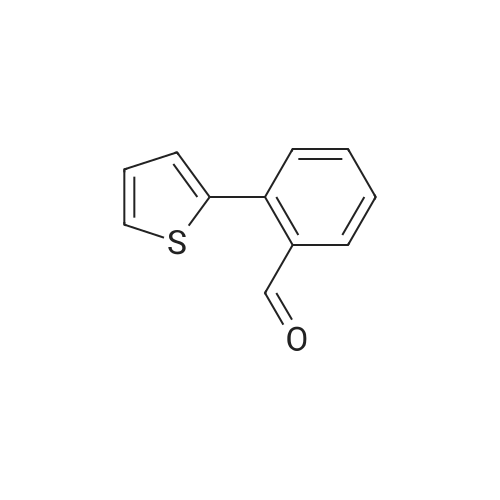
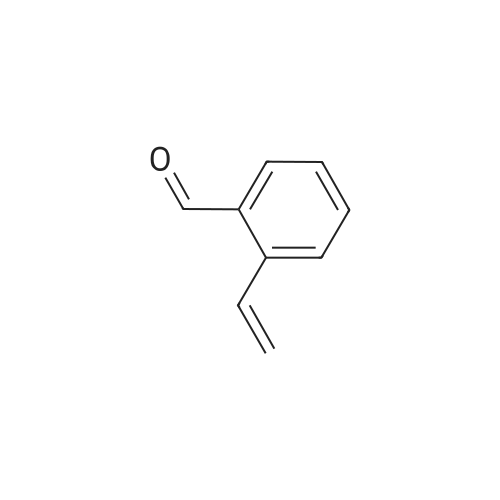
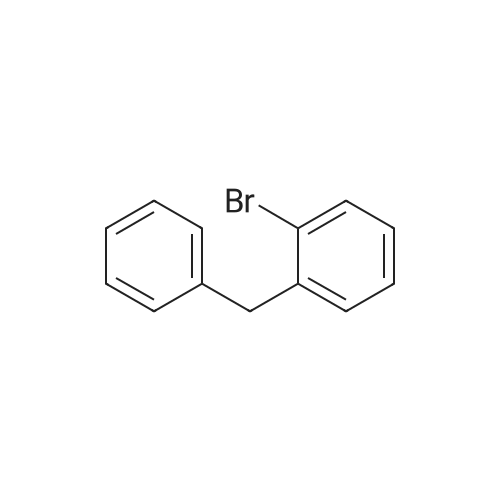
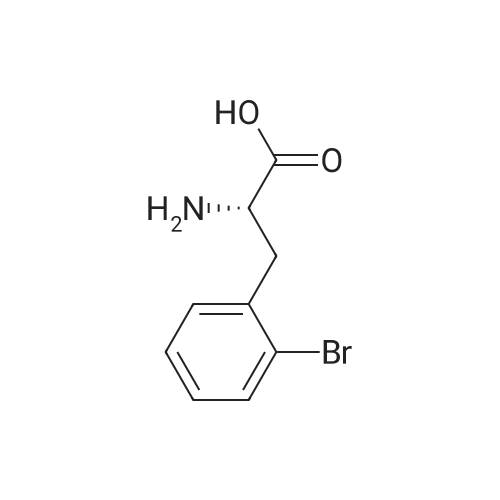
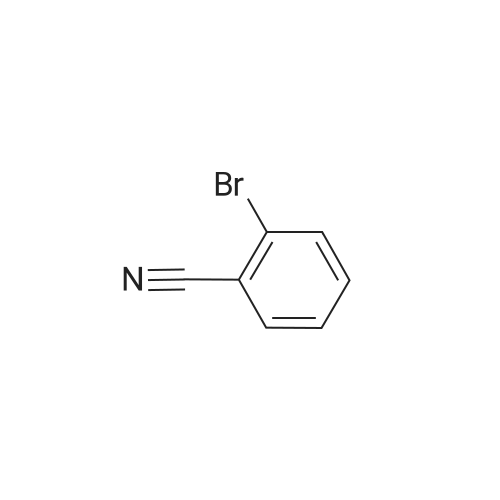
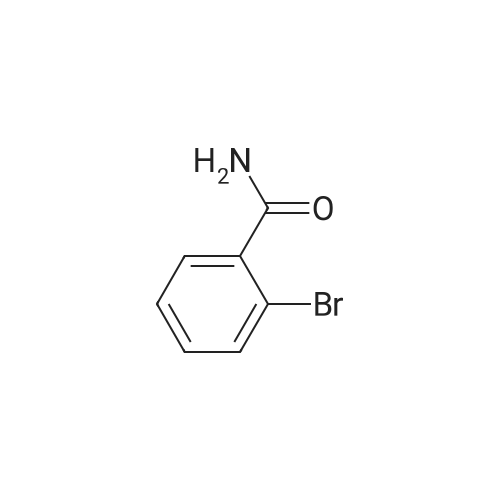
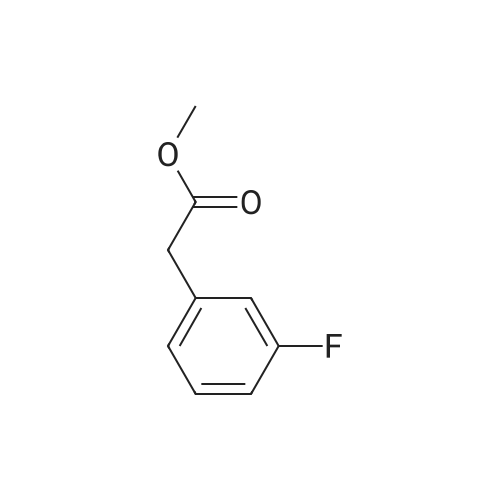
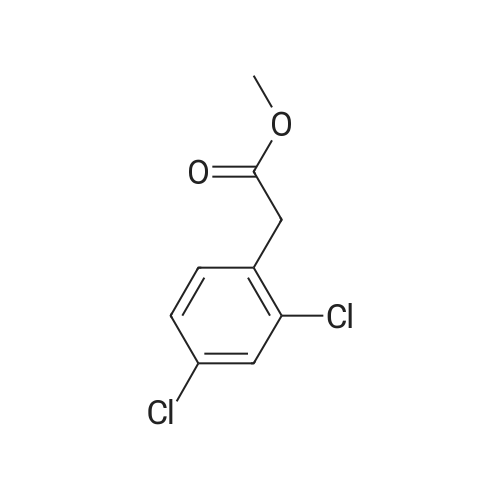
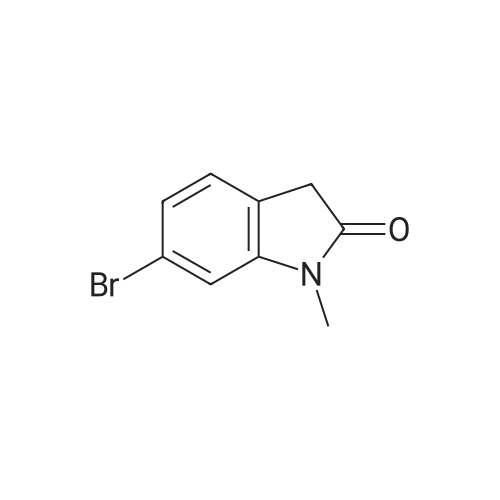
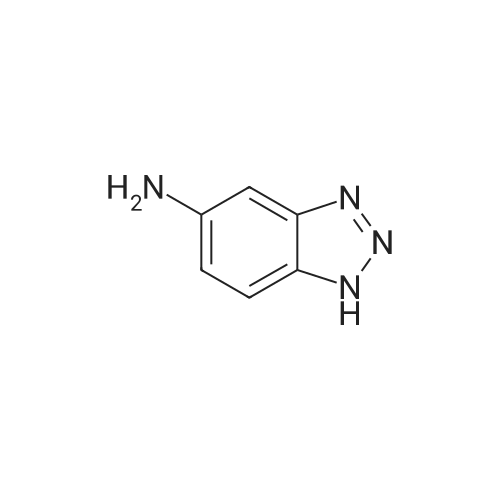
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Synthesis, 2009, # 16, p. 2679 - 2688
3
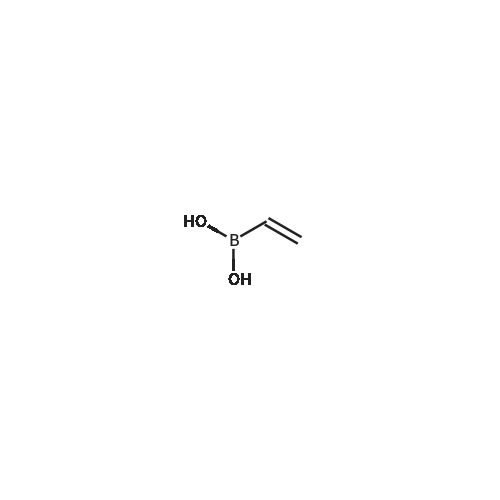
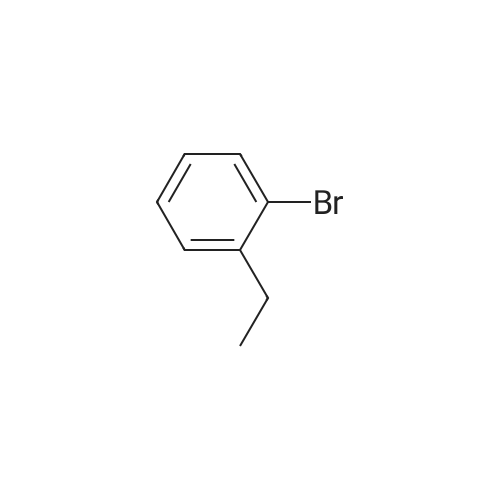
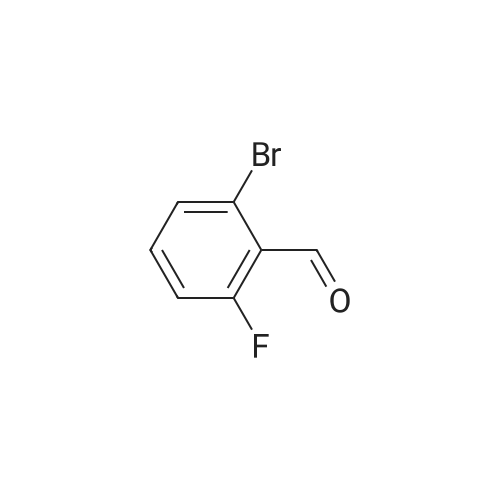
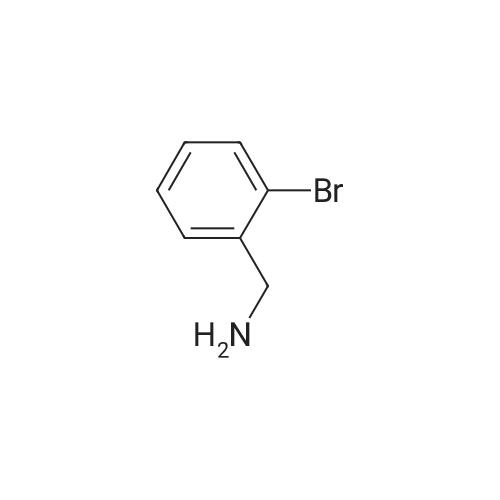
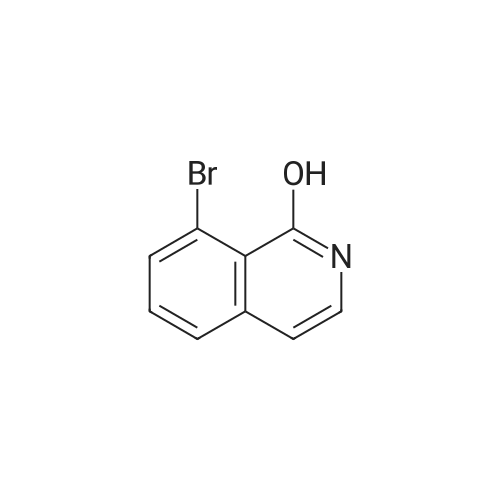
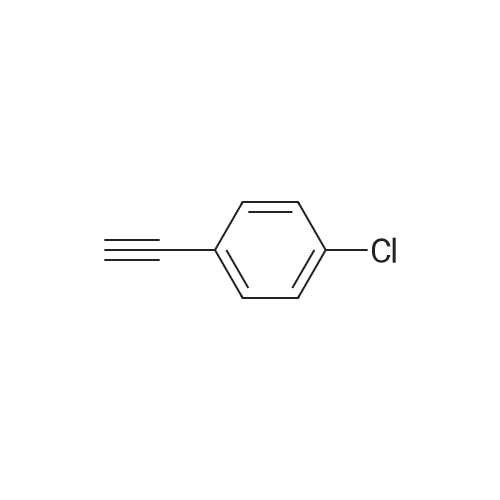
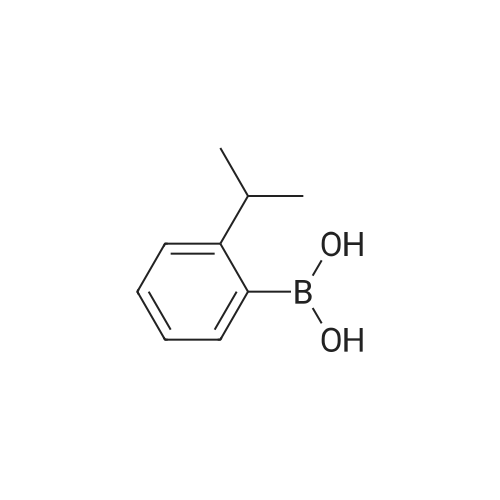
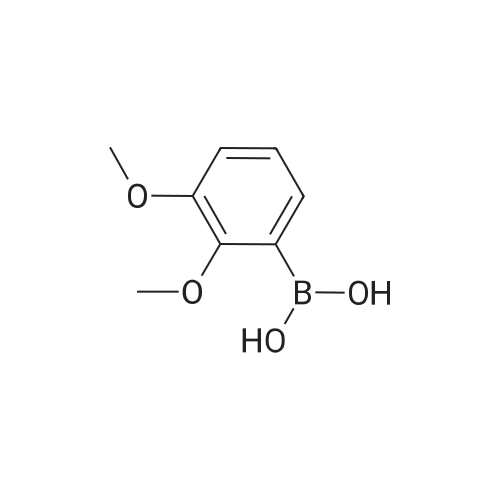
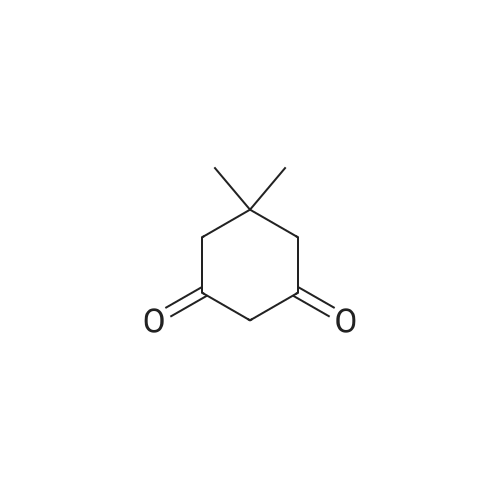
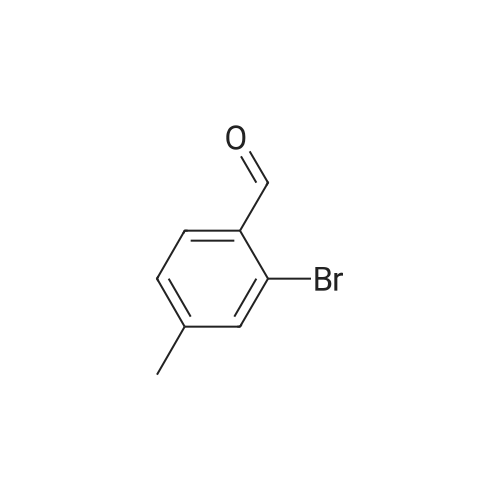
[ 1692-25-7 ]
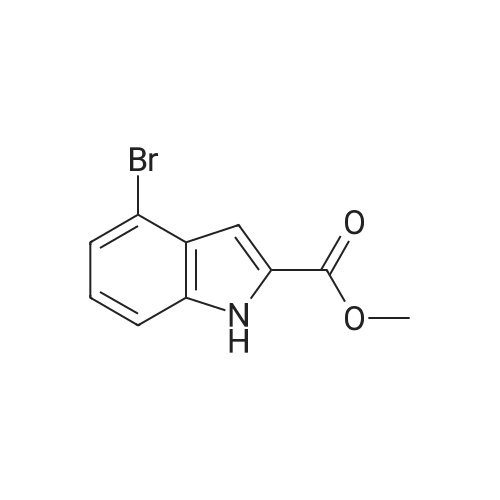
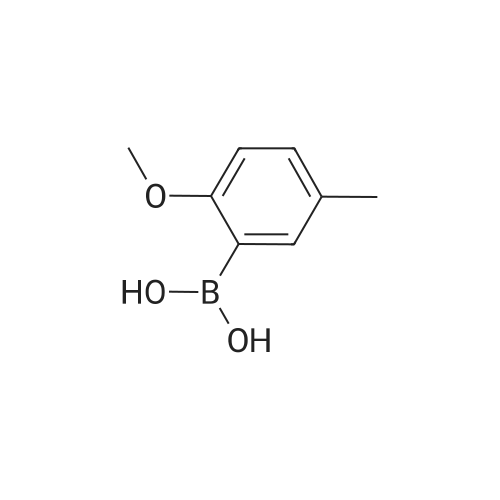
[ 6630-33-7 ]
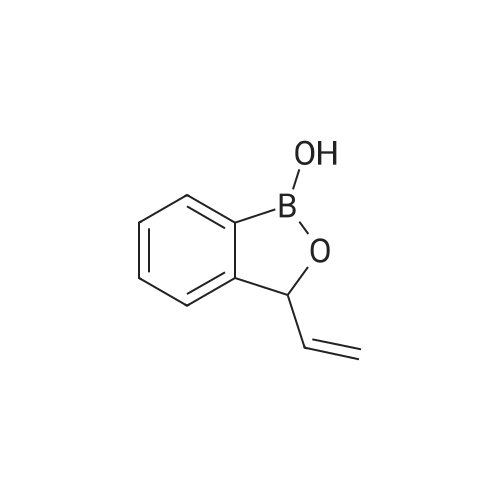
[ 176690-44-1 ]
Yield
Reaction Conditions
Operation in experiment
64%
With sodium carbonate In water; N,N-dimethyl-formamide at 110℃; Inert atmosphere
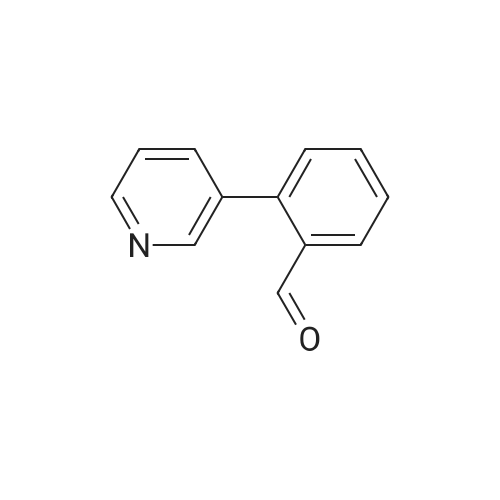
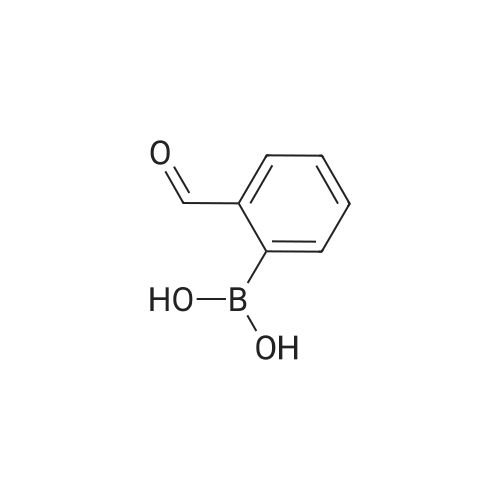
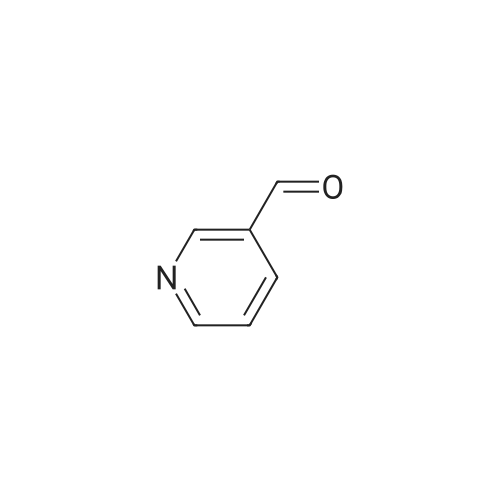
Example 380 : Synthesis of (1E,6E)-1-(4-hydroxyphenyl)-7-[2-(pyridin-3-yl)phenyl]hepta-1,6-diene-3,5-dione (CU513); (1) Synthesis of 2-(pyridin-3-yl)benzaldehyde; To a suspension of 1-bromobenzaldehyde (250 μL, 2.14 mmol), sodium carbonate (270 mg, 2.55 mmol), and 3-pyridineboronic acid (289 mg, 2.35 mmol) in 4.2 mL of N,N-dimethylformamide/water (2:1) were added palladium acetate (24 mg, 0.11 mmol) and triphenylphosphine (115 mg, 0.44 mmol) under nitrogen. After being stirred at 110°C overnight, the reaction mixture was filtered. The filtrate was diluted with chloroform, and the solution was washed with brine, and dried over MgSO4. After filtration, the filtrate was concentrated in vacuo, and the residue was purified by silica gel column chromatography (chloroform/methanol = 99/1 to 95/5) to obtain the title compound as a white powder (250 mg, 64percent).
Reference:
[1] Advanced Synthesis and Catalysis, 2014, vol. 356, # 16, p. 3415 - 3421
9
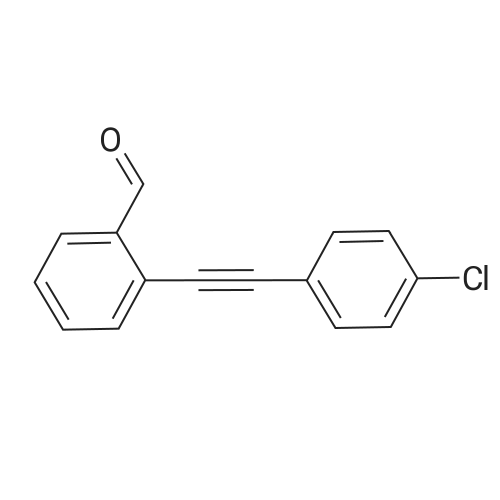
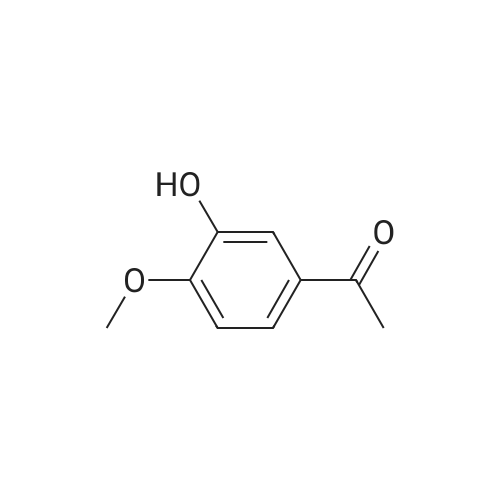
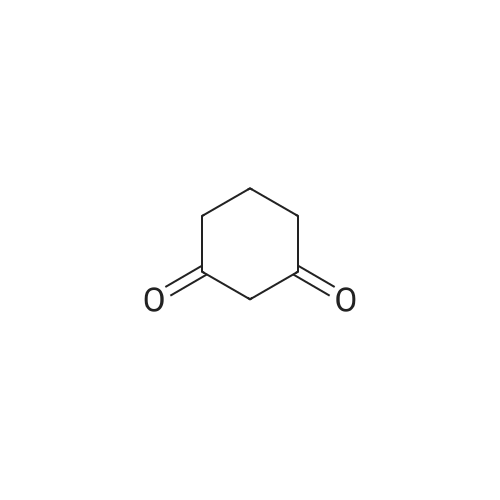
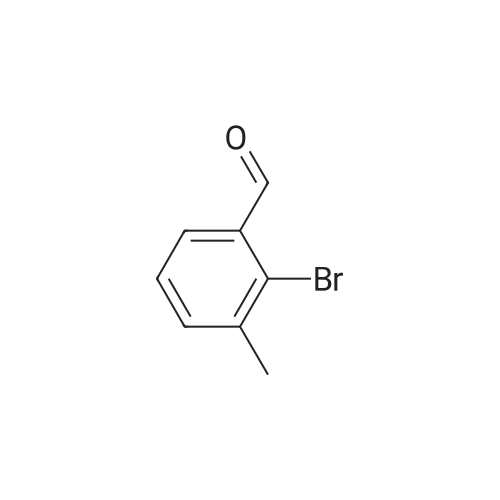
[ 6630-33-7 ]
[ 22483-09-6 ]
[ 63927-22-0 ]
Yield
Reaction Conditions
Operation in experiment
49.8%
Stage #1: for 3 h; Inert atmosphere; Reflux; Dean-Stark Stage #2: With aluminum (III) chloride In dichloromethane at 0 - 45℃; for 2 h; Inert atmosphere
A mixture of 2-bromobenzaldehyde (50 g, 270 mmol) , aminoacetaldehyde dimethyl acetal (28.4 g, 270 mmol) and toluene (400 mL) was refluxed under argon. Dehydration was carried out using dean stark for 2.0 hours. After removal of calculated amount of water, the reflux was continued for 1.0 hour. The toluene was evaporated under reduced pressure, the residue was dissolved in dichloromethane (600 mL) , and the solution was cooled to 0°C. To the cooled solution was slowly added aluminium chloride (118.9 g, 891.7 mmol) under argon. The reaction mixture was stirred at 45°C for 2.0 hours. After the completion of the reaction was confirmed by TLC, the mixture was cooled to room temperature and slowly poured into an ice water. The mixture was basified with 10percent sodium hydroxide solution, and the dichloromethane layer was separated. The aqueous layer was re-extracted with dichloromethane (2 x 100 mL) . The combined dichloromethane layers were washed with brine, and dried over sodium sulfate. The dichloromethane was evaporated, and the residue was purified by silica gel (100-200 mesh) column chromatography 5 with 8-12percent. ethyl acetate in hexane as a mobile phase to give the title compound as an off-white solid (28 g, 49.8percent). MS(ESI)m/z: 208 [M (79Br)+l] ,210 [M (81Br)+l]; XH NMR (400 MHz, DMSO-d5) : δ 7.17 (t, J= 7.8 Hz, 1H) ; 7.91 (d, J= 6.0 Hz, 1H) ; 8.02 (d, J = 8.4 Hz, 1H) ; 8.05 (d, J = 8.8 Hz, 1H) ; 8.65 10 (d, J = 5.2 Hz, 1H) 9.48 (s, IH) .
With toluene-4-sulfonic acid In benzene for 4 h; Heating / reflux
To a solution of 2-bromobenzaldehyde (5.30 mmol) in benzene (40 mL) was added ethylene glycol (1.26 mL, 22.6 mmol, 4.3 eq) and p-toluenesulfonic acid (70 mg, 0.37 mmol, 0.07 eq). The resulting mixture was heated at reflux in a Dean-Stark apparatus. After 4 h, the reaction was cooled to ambient temperature, quenched with triethylamine, and concentrated in vacuo. Purification was accomplished with chromatography on silica gel. Quantitative yield.
95%
With p-toluenesulfonic acid monohydrate In toluene
(181-1) 2-Bromobenzaldehyde (11.4 g) was dissolved in toluene (150 mL), and thereto were added ethyleneglycol (4.58 g) and p-toluenesulfonic acid monohydrate (135 mg), and the mixture was refluxed for 6 hours with azeotropic distillation. The reaction solution was washed with an aqueous sodium hydrogen carbonate solution, and the solvent was evaporated under reduced pressure to give 2-(2-bromophenyl)-1,3-dioxolane (13.4 g, 95percent). 1H NMR (CDCl3, 400 MHz) δ 7.60 (dd, 1H, J=7.6, 1.8 Hz), 7.57 (dd, 1H, J=7.6, 1.8 Hz), 7.34 (ddd, 1H, J=7.6, 7.6, 1.8 Hz), 7.22 (ddd, 1H, J=7.6, 7.6, 1.8 Hz), 6.10 (s, 1H), 4.15 (m, 2H), 4.09 (m, 2H).
95%
With p-toluenesulfonic acid monohydrate In toluene
(181-1) 2-Bromobenzaldehyde (11.4 g) was dissolved in toluene (150 mL), and thereto were added ethyleneglycol (4.58 g) and p-toluenesulfonic acid monohydrate (135 mg), and the mixture was refluxed for 6 hours with azeotropic distillation. The reaction solution was washed with an aqueous sodium hydrogen carbonate solution, and the solvent was evaporated under reduced pressure to give 2-(2-bromophenyl)-1,3-dioxolane (13.4 g, 95 percent). 1H NMR (CDCl3, 400MHz) δ 7.60 (dd, 1H, J=7.6, 1.8Hz), 7.57 (dd, 1H, J=7.6, 1.8Hz), 7.34 (ddd, 1H, J=7.6, 7.6, 1.8Hz), 7.22 (ddd, 1H, J=7.6, 7.6, 1.8Hz), 6.10 (s, 1H), 4.15 (m, 2H), 4.09 (m, 2H).
89%
With toluene-4-sulfonic acid In toluene for 48 h; Reflux
This was a modified procedure according to literature. [8] and [8a] To a round bottom flask, the2-bromobenzaldehyde (3.70 g, 20 mmol), ethylene glycol (1.73 mL, 25 mmol), p-toluenesulfonic acid (60.2 mg, 0.35 mmol) and toluene (30 mL) were added, and the mixture was heated at reflux with a Dean Stark Trap for 24 h. The reaction was monitored by TCL. After the completion of the reaction, the mixture was cooled to room temperature, and the KOH-EtOH solution was added until white solid appeared. Then the organic phase was washed with water and brine, dried over anhydrous K2CO3, and concentrated under vacuum. The crude product was purified by silica gel column chromatography using petroleum ether-EtOAc 100:1 (v/v) as eluent. The product was obtained as yellow oil (4.08 g, 89percent yield).
82%
Reflux
Toluene (1 L), 2-bromobenzaldehyde (190 g, 1.05 mole) and ethylene glycol (76 ml, 1.3 mole) were heated to reflux with azeotropical distillation of water (-‘10 h). 600 ml of toluene was distilled off and the residual solution was cooled to RT and washed with sat. NaHCO3 solution (200 ml), water (200 ml), dried over Na2SO4, and evaporated under reduced pressure. The crude compound was distilled under high vacuum. 190 g (82percent) of 2-(2-bromophenyl)-1,3-dioxolane as a colorless liquid was received, purity by GC: 98percent.
82%
With toluene-4-sulfonic acid In benzene for 24 h; Reflux; Dean-Stark
In a 100 mL two-necked flask, 4.97 g (26.9 mmol) of 2-bromobenzaldehyde, 3.49 g (56.2 mmol) of ethylene glycol and 470 mg (2.47 mmol) of p-toluenesulfonic acid monohydrate were added, 10 mL of benzene was added, and the resulting mixture was refluxed with a Dean-Stark apparatus for 24 hours. After cooling to room temperature, the reaction was terminated with saturated aqueous sodium hydrogen carbonate solution, and extracted with dichloromethane. The organic layer was dried with sodium sulfate, and the solvent was distilled off under reduced pressure to obtain a crude product. The crude product was purified by silica gel column chromatography (dichloromethane) to obtain 5.01 g (22.1 mmol) of 2- (2-bromophenyl)-1,3-dioxolane in a yield of 82percent. The measurement results of NMR are shown below.
77%
With toluene-4-sulfonic acid In toluene at 118 - 120℃; for 2 h; Dean-Stark; Microwave irradiation
A solution of 2-bromobenzaldehyde (12a, 37.1 g, 0.20 mol), ethylene glycol (50 mL, 55.6 g, 0.90 mol) and p-toluenesulfonic acid (1.0 g, 0.005 mol) in toluene (200 mL) was refluxed in a Dean-Stark apparatus (bp 118-120 °C) in a microwave reactor under irradiation with a constant 650 W energy for 2 h.29 The reaction mixture was washed with aqueous sodium hydrogen carbonate solution (5 w/wpercent, 200 mL) and water (200 mL), dried over MgSO4 and the solvent was evaporated. The crude pale yellow oil was distilled under reduced pressure (0.04 mbar) at 86-88 °C to give 13a (35.15 g, 77percent) as a colorless oil. 1H NMR (DMSO-d6, 500 MHz) δ 7.63 (1H, dd, J=8.1, 1.2 Hz), 7.58 (1H, dd, J=7.3, 1.8 Hz), 7.43 (1H, td, J=8.1, 1.2 Hz), 7.34 (1H, td, J=7.3, 1.8 Hz), 5.95 (1H, s), 4.08 (2H, m), 3.98 (2H, m). 13C NMR (DMSO-d6, 100 MHz) δ 136.7, 132.9, 131.2, 128.3, 127.8, 122.3, 101.9, 65.1. IR (KBr, cm-1) 2889, 1472, 1352. Anal. Calcd for C9H9BrO2 (229.07): C 47.19, H 3.96, Br 34.88percent. Found: C 46.69, H 4.07, Br 34.83percent.
Reference:
[1] Patent: US2004/147401, 2004, A1, . Location in patent: Page 33
[2] Organic and Biomolecular Chemistry, 2009, vol. 7, # 6, p. 1106 - 1114
[3] Organic Letters, 2013, vol. 15, # 18, p. 4718 - 4721
[4] European Journal of Organic Chemistry, 2014, vol. 2014, # 10, p. 2084 - 2091
[5] Organic Process Research and Development, 2016, vol. 20, # 2, p. 253 - 261
[6] Journal of Organic Chemistry, 2000, vol. 65, # 2, p. 432 - 437
[7] ACS Combinatorial Science, 2013, vol. 15, # 4, p. 202 - 207
[8] Indian Journal of Chemistry - Section A Inorganic, Physical, Theoretical and Analytical Chemistry, 2004, vol. 43, # 11, p. 2287 - 2293
[9] Patent: US2003/181496, 2003, A1,
[10] Patent: EP1479384, 2004, A1,
[11] Organic Letters, 2013, vol. 15, # 24, p. 6190 - 6193
[12] Chemistry - An Asian Journal, 2018,
[13] Journal of Organometallic Chemistry, 1997, vol. 529, # 1-2, p. 35 - 50
[14] Organic Letters, 2004, vol. 6, # 9, p. 1515 - 1517
[15] Journal of Organic Chemistry, 2010, vol. 75, # 22, p. 7573 - 7579
[16] Angewandte Chemie - International Edition, 2017, vol. 56, # 9, p. 2464 - 2468[17] Angew. Chem., 2017, vol. 129, # 9, p. 2504 - 2508,5
[18] Journal of Organic Chemistry, 2008, vol. 73, # 16, p. 6330 - 6340
[19] Organic and Biomolecular Chemistry, 2014, vol. 12, # 40, p. 8019 - 8030
[20] Journal of Organic Chemistry, 1984, vol. 49, # 4, p. 621 - 625
[21] Organic Letters, 2003, vol. 5, # 4, p. 545 - 548
[22] Angewandte Chemie - International Edition, 2012, vol. 51, # 43, p. 10861 - 10865[23] Angew. Chem., 2012, vol. 124, # 43, p. 11019 - 11023
[24] Catalysis Science and Technology, 2014, vol. 4, # 8, p. 2618 - 2625
[25] Dalton Transactions, 2017, vol. 46, # 20, p. 6570 - 6579
[26] Angewandte Chemie - International Edition, 2018, vol. 57, # 1, p. 140 - 145[27] Angew. Chem., 2018, vol. 130, # 1, p. 146 - 151,6
[28] Advanced Synthesis and Catalysis, 2018, vol. 360, # 6, p. 1111 - 1115
[29] Dalton Transactions, 2011, vol. 40, # 14, p. 3695 - 3702
[30] Tetrahedron, 2012, vol. 68, # 18, p. 3633 - 3640
[31] Chemistry - A European Journal, 2013, vol. 19, # 19, p. 6034 - 6043
[32] Advanced Synthesis and Catalysis, 2016, vol. 358, # 18, p. 2940 - 2948
[33] Journal of Organic Chemistry, 1992, vol. 57, # 3, p. 1015 - 1018
[34] Journal of Labelled Compounds and Radiopharmaceuticals, 2017, vol. 60, # 1, p. 30 - 35
[35] European Journal of Organic Chemistry, 2015, vol. 2015, # 23, p. 5167 - 5182
[36] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1999, # 1, p. 49 - 64
[37] Journal of Organic Chemistry, 2005, vol. 70, # 7, p. 2470 - 2475
[38] Organic Letters, 2012, vol. 14, # 1, p. 194 - 197
[39] Patent: WO2016/132343, 2016, A1, . Location in patent: Page/Page column 25
[40] Patent: JP2015/189765, 2015, A, . Location in patent: Paragraph 0031-0032
[41] Journal of the American Chemical Society, 1984, vol. 106, p. 3964
[42] Organic and Biomolecular Chemistry, 2016, vol. 14, # 30, p. 7268 - 7274
[43] Tetrahedron, 2014, vol. 70, # 2, p. 286 - 293
[44] Journal of the American Chemical Society, 2018, vol. 140, # 20, p. 6432 - 6440
[45] Tetrahedron Letters, 2005, vol. 46, # 13, p. 2341 - 2343
[46] Journal of Organic Chemistry, 1987, vol. 52, # 18, p. 4079 - 4085
[47] Journal of Organometallic Chemistry, 2001, vol. 623, # 1-2, p. 87 - 94
[48] Bioorganic and Medicinal Chemistry, 2006, vol. 14, # 8, p. 2620 - 2626
[49] Chemical Communications, 2006, # 21, p. 2260 - 2261
[50] Journal of Organic Chemistry, 2006, vol. 71, # 20, p. 7840 - 7845
[51] Organic Letters, 2000, vol. 2, # 8, p. 1117 - 1120
[52] Patent: EP1867634, 2007, A1, . Location in patent: Page/Page column 13
[53] Patent: WO2008/37784, 2008, A1, . Location in patent: Page/Page column 33
[54] Organometallics, 2010, vol. 29, # 17, p. 3955 - 3965
[55] Advanced Synthesis and Catalysis, 2011, vol. 353, # 4, p. 557 - 562
[56] European Journal of Organic Chemistry, 2011, # 9, p. 1736 - 1742
[57] Journal of Polymer Science, Part A: Polymer Chemistry, 2010, vol. 48, # 23, p. 5339 - 5347
[58] Chinese Journal of Chemistry, 2012, vol. 30, # 10, p. 2495 - 2500
[59] Organic and Biomolecular Chemistry, 2014, vol. 12, # 15, p. 2388 - 2393
[60] Chemical Communications, 2014, vol. 50, # 62, p. 8468 - 8471
[61] RSC Advances, 2015, vol. 5, # 15, p. 11405 - 11422
[62] Chemistry - A European Journal, 2015, vol. 21, # 37, p. 12908 - 12913
[63] Angewandte Chemie - International Edition, 2016, vol. 55, # 9, p. 3135 - 3139[64] Angew. Chem., 2016, vol. 128, # 9, p. 3187 - 3191,5
[65] Organic Letters, 2014, vol. 16, # 21, p. 5580 - 5583
[66] Organic and Biomolecular Chemistry, 2016, vol. 14, # 44, p. 10415 - 10426
[67] Advanced Synthesis and Catalysis, 2018, vol. 360, # 8, p. 1628 - 1633
[68] Organic Letters, 2018,
13
[ 6630-33-7 ]
[ 34824-58-3 ]
Yield
Reaction Conditions
Operation in experiment
94%
With toluene-4-sulfonic acid In ethylene glycol; toluene
Example 4 Synthesis of N-methyl-2-{2-(1,3-dioxolan-2-yl)phenyl}-2-methoxyiminoacetamide (III) A mixture of 50.0 g (0.270 mol) of 2-bromobenzaldehyde, 33.5 g (0.54 mol) of ethylene glycol, 0.51 g (2.7 mmol) of p-toluenesulfonic acid and 300 ml of toluene was heated under reflux for 2 hours while removing water. The reaction mixture was cooled and 300 ml of toluene was added. The mixture was washed with saturated sodium bicarbonate aqueous solution, water and saturated brine in that order and then dried over anhydrous sodium sulfate. After evaporation of the solvent, the residue was distilled under a reduced pressure to give 66.0 g of 2-(2-bromophenyl)-1,3-dioxolan. The yield was 94percent. Boiling point; 104 - 108°C/1.5 mm 1H-NMR δ (CDCl3); 4.05 - 4.2 (4H, m), 6.11 (1H, s), 7.2 - 7.6 (4H, m)
With toluene-4-sulfonic acid In ethylene glycol; toluene
C. 2-(2-Bromophenyl)-1,3-dioxolane A 12 L 3-necked flask fitted with an overhead stirrer was charged with 2-bromobenzaldehyde (800 g, 4.324 moles), ethylene glycol (402.6 g, 6.485 moles), p-toluenesulfonic acid. H2 O (3.95 g, 0.021 moles) and toluene (3.785 kg, 41.074 moles). One side of the flask was stoppered (glass) and a Dean-Stark separator/condenser/N2 port was attached to the other side. The heterogeneous yellow reaction mixture was stirred under a nitrogen atmosphere and heated to reflux for about 45 minutes. Water was collected via the Dean-Stark separator and the residue was cooled to room temperature and washed with 1.2 L of saturated aqueous NaHCO3 followed by 1.2 L of saturated aqueous NaCl. The combined organic layers were dried over anhydrous MgSO4, filtered, concentrated on a rotary evaporator and dried under high vacuum to provide title compound in the form of an oil. The so-formed oil was vacuum distilled to provide 52 g and 876.2 g of title compound (88.9percent yield).
Reference:
[1] European Journal of Organic Chemistry, 2018, vol. 2018, # 7, p. 926 - 931
[2] European Journal of Organic Chemistry, 2011, # 20-21, p. 3904 - 3910
[3] Organic and Biomolecular Chemistry, 2017, vol. 15, # 39, p. 8308 - 8312
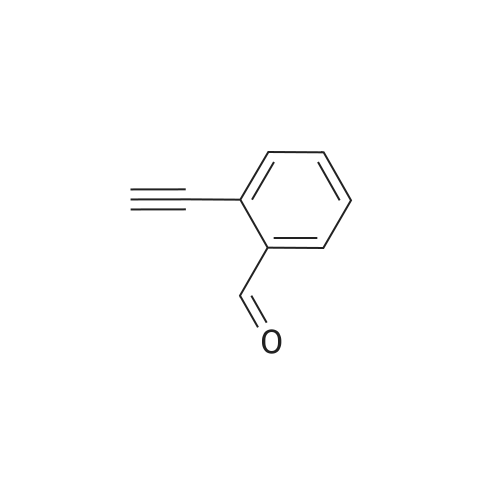
27
[ 6630-33-7 ]
[ 28272-96-0 ]
Reference:
[1] Journal of Organic Chemistry, 2002, vol. 67, # 14, p. 4968 - 4971
28
[ 2554-06-5 ]
[ 6630-33-7 ]
[ 28272-96-0 ]
Reference:
[1] Chemistry - A European Journal, 2013, vol. 19, # 6, p. 2150 - 2157
29
[ 6630-33-7 ]
[ 28272-96-0 ]
Reference:
[1] Synlett, 2006, # 17, p. 2771 - 2776
[2] Heterocycles, 1989, vol. 28, # 1, p. 275 - 282
[3] Journal of Organic Chemistry, 2011, vol. 76, # 7, p. 1979 - 1991
[4] Heterocycles, 2012, vol. 86, # 1, p. 469 - 485
[5] Angewandte Chemie - International Edition, 2017, vol. 56, # 9, p. 2473 - 2477[6] Angew. Chem., 2017, vol. 129, p. 2513 - 2517,5
[7] Patent: US2017/37038, 2017, A1,
30
[ 6630-33-7 ]
[ 26260-02-6 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1999, # 1, p. 49 - 64
[2] Synlett, 2016, vol. 27, # 12, p. 1794 - 1797
31
[ 6630-33-7 ]
[ 1973-22-4 ]
Reference:
[1] Journal of the American Chemical Society, 2013, vol. 135, # 22, p. 8157 - 8160
32
[ 6630-33-7 ]
[ 27326-43-8 ]
Reference:
[1] Chemistry - A European Journal, 2014, vol. 20, # 45, p. 14633 - 14636
33
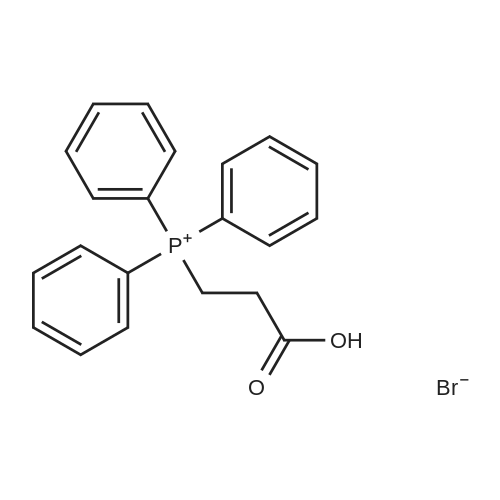
[ 2136-75-6 ]
[ 6630-33-7 ]
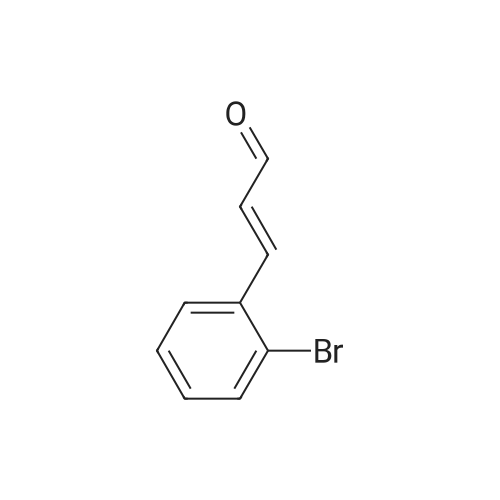
[ 138555-58-5 ]
Yield
Reaction Conditions
Operation in experiment
78%
at 0 - 25℃; Inert atmosphere
General procedure: To a solution of triphenylphosphoranylidene (1.5 mmol) in 10 ml of solvent (CH2Cl2 or THF) was added dropwise a solution of the aldehyde (1.0 mmol) in 4 ml of solvent (CH2Cl2 or THF) at 0 °C under an inert argon atmosphere. The resulting mixture was stirred at 25 °C until completion of the reaction, which was monitored by TLC. The solvent was then evaporated under reduced pressure to give the crude product which was purified by silica gel (60-120 mesh) column chromatography using petroleum ether/ethyl acetate as the eluent to afford the desired product.
Reference:
[1] Tetrahedron Letters, 2011, vol. 52, # 31, p. 4051 - 4055
[2] Organic and Biomolecular Chemistry, 2014, vol. 12, # 43, p. 8588 - 8592
[3] Organic Letters, 2016, vol. 18, # 4, p. 752 - 755
[4] Chemistry - A European Journal, 2018, vol. 24, # 22, p. 5765 - 5769
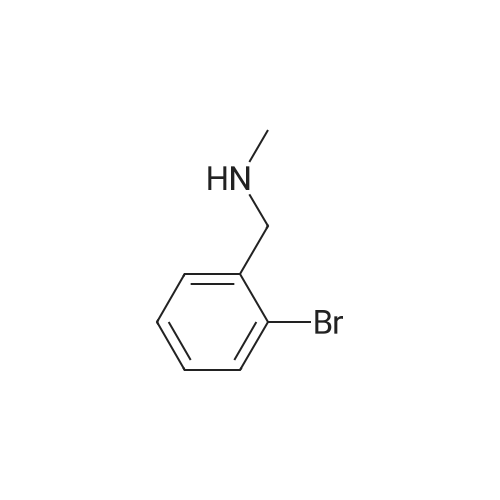
With sodium tetrahydroborate; methylamine In tetrahydrofuran; methanol
EXAMPLE (Compound 22) Preparation of a: 29.4 g of o-Bromobenzaldehyde and 81 ml of aqueous, 40percent methylamine solution are combined with 238 ml of THF and at RT 19 g of NaBH4 are added in batches within 25 minutes. The mixture is left to stand overnight at RT, concentrated using a rotary evaporator, and the residue is stirred into ice water. The aqueous phase is extracted twice with ether and the combined ether phases are evaporated down under reduced pressure. After chromatography on silica gel with ethyl acetate or ethyl acetate/methanol (4:1) as eluant, 18.5 g of N-methyl-2-bromobenzylamine (a) are obtained in the form of a yellowish liquid. Yield: 58percent.
Reference:
[1] Journal of the American Chemical Society, 2017, vol. 139, # 2, p. 888 - 896
68
[ 1670-14-0 ]
[ 6630-33-7 ]
[ 77989-15-2 ]
Yield
Reaction Conditions
Operation in experiment
59%
With copper(II) acetate monohydrate; sodium carbonate In toluene at 100℃; for 24 h;
General procedure: A mixture ofaldehyde 1 (6.8 mmol), amidine hydrochloride 2 (2 g,11.4 mmol), Na2CO3 (1.21 g, 11.4 mmol, 1.0 equiv) andCu(OAc)2 (10 molpercent) was stirred in toluene (20 mL) under100 °C in air for 24 h. After completion of the reaction, themixture was cooled to room temperature. The water wasadded to the reaction system and atmospheric distillation untiltoluene was evaporated. The resulting solution was filteredand residue with hot water washed 3 times. The crude productwas purified by column chromatography on silica gel usingpetroleum ether/EtOAc (100:1) as an eluent to give the correspondingproducts 7a-7x.
Reference:
[1] Journal of Fluorescence, 2018, vol. 28, # 2, p. 707 - 723
69
[ 6630-33-7 ]
[ 84459-32-5 ]
Yield
Reaction Conditions
Operation in experiment
94%
at 0℃; for 4.66667 h;
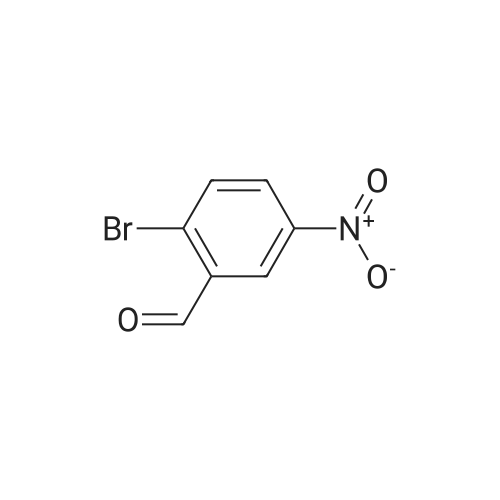
To a solution of 2-bromobenzaldehyde (10.0 g, 53.7 mmol) in H2SO4 (100 mL) was added K O3 (5.43 g, 53.7 mmol) in portions over 1 h at 0°C. The mixture was stirred for 40 min and additional K O3 (0.72 g) was added. The reaction mixture was stirred at 0°C for 3 h then poured into ice water. The resulting precipitate was collected by filtration, rinsed with water and recrystallized from EtOAc/Pentane to give 2-bromo-5-nitrobenzaldehyde (11.7 g, 94percent yield) as a white solid. MS (ESI) calcd for C7H4BrN03: 228.9
94%
at 0℃; for 3.67 h;
To a solution of 2-bromobenzaldehyde (10.0 g, 53.7 mmol) in H2SO4 (100 mL) was added KNO3 (5.43 g, 53.7 mmol) in portions over 1 h at 0° C. The mixture was stirred for 40 min and additional KNO3 (0.72 g) was added. The reaction mixture was stirred at 0° C. for 3 h then poured into ice water. The resulting precipitate was collected by filtration, rinsed with water and recrystallized from EtOAc/Pentane to give 2-bromo-5-nitrobenzaldehyde (11.7 g, 94percent yield) as a white solid. MS (ESI) calcd for C7H4BrNO3: 228.9.
83%
at 0℃; for 3 h;
KNO3 (1.09 g, 10.8 mmol) was added slowly to a stirred red solution of 2-bromobenzaldehyde (2.0 g, 10.8 mmol) in H2SO4 (10 mL) at 0 °C. After 30 minutes extra portion of KNO3 (0.16 g, 1.6 mmol) was added, and the reaction mixture was stirred for 2.5 hours at 0 °C. Then the reaction mixture was poured over ice water (50 mL), extracted with Et2O (3×25 mL). Combined organic layers were washed with water (30 mL), brine (40 mL) and dried over Na2SO4. Solvents were evaporated in vacuo. The residual yellow solid was recrystallized from EtOAc/hexane (1/2, 40 mL) to give pure nitroaldehyde SI-1e (1.32 g, 5.7 mmol, 53percent). Further recrystallization of evaporated mother liquor from EtOAc/hexane (1/3, 15 mL) gives additional nitroaldehyde SI-1e (0.75 g, 3.2 mmol, 30percent) of the same purity.
55%
at 5 - 20℃; for 1.5 h;
A mixture of concentrated sulfuric acid (17.01 mL) andfuming nitric acid (2.250 mL) was cooled to 5 °C followedby the dropwise addition of 2—bromobenzaldehyde (3.15 mL,27.0 mmol) over a period of 30 minutes. The mixture wasthen allowed to warm to room temperature and stirred for60 minutes. The reaction mixture was poured intoice/water (200 mL) and the precipitated solid isolated by filtration, washed with water and sucked dry to give a pale yellow solid. This was recrystallised from 50:50 cyclohexane:ethyl acetate (30 mL) to give the title compound as an off—white solid (3.39 g, 55percent) . ‘H NMR(300 MHz, CDC13) : 3 10.39 (s, 1H) , 8.72 (d, 1H) , 8.29 (dd,1H) , 7.89 (d, 1H)
54.6%
at 0℃; for 4.66667 h;
Intermediate 17: l-(2-bromo-5-nitrophenyl)-N-methylmethanamine; Intermediate 17A:; [00237] Potassium nitrate (2.59 mL, 54.0 mmol) was added portionwise to a stirred and chilled (ice bath) solution of 2-bromobenzaldehyde (10 g, 54.0 mmol) in sulfuric acid (50 mL, 938 mmol) over 1 h. After 40 min an additional portion of KNO3 (0.72 g) was added. After 3 h stirring at 0 0C, the mixture was poured over ice water, and the product was filtered and washed with water. The crude pale yellow solid was recrystallized from 1 : 1 ethyl acetate/hexane (-60 mL) to give Intermediate 17A (6.789 g, 29.5 mmol, 54.6 percent yield). MS (ESI) m/z 230, 232 (M+H)+.
Reference:
[1] Patent: WO2014/186313, 2014, A1, . Location in patent: Page/Page column 173-174
[2] Patent: US2015/152108, 2015, A1, . Location in patent: Paragraph 0881; 0882
[3] Tetrahedron Letters, 2016, vol. 57, # 1, p. 11 - 14
[4] Organic and Biomolecular Chemistry, 2017, vol. 15, # 6, p. 1355 - 1362
[5] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 20, p. 5051 - 5057
[6] Patent: WO2015/92431, 2015, A1, . Location in patent: Page/Page column 303
[7] Patent: WO2008/79836, 2008, A2, . Location in patent: Page/Page column 106
[8] European Journal of Organic Chemistry, 2011, # 28, p. 5626 - 5635
[9] Molecules, 2000, vol. 5, # 3, p. 227 - 239
[10] Patent: US5062884, 1991, A,
70
[ 122-51-0 ]
[ 6630-33-7 ]
[ 35822-58-3 ]
Reference:
[1] ACS Medicinal Chemistry Letters, 2018, vol. 9, # 9, p. 947 - 951
[2] Journal of the Chemical Society, Dalton Transactions: Inorganic Chemistry (1972-1999), 1989, p. 1697 - 1704
[3] Green Chemistry, 2017, vol. 19, # 8, p. 1990 - 1998
[4] Journal of the Chemical Society, Chemical Communications, 1986, # 6, p. 453 - 455
[5] Patent: US6080773, 2000, A,
[6] Patent: EP898566, 2002, B1,
71
[ 998-30-1 ]
[ 6630-33-7 ]
[ 35822-58-3 ]
Reference:
[1] Green Chemistry, 2017, vol. 19, # 8, p. 1990 - 1998
72
[ 107-21-1 ]
[ 6630-33-7 ]
[ 35822-58-3 ]
Reference:
[1] Chinese Journal of Chemistry, 2015, vol. 33, # 4, p. 479 - 485
73
[ 6630-33-7 ]
[ 154701-60-7 ]
Reference:
[1] Journal of Organic Chemistry, 2012, vol. 77, # 3, p. 1477 - 1488
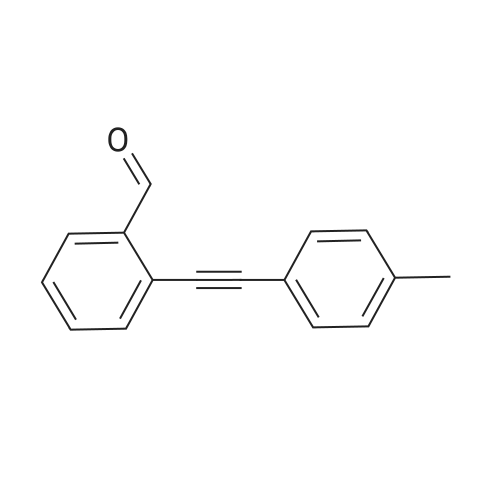
To a solution of 2-bromobenzaldehyde (1.85 g, 10 mmol) and 4- methoxyphenyl acetylene (1.58 g, 12 mmol) in 40 mL of triethylamine were added dichlorobis(triphenylphosphine) palladium(ll) (140 mg, 2 molpercent) and copper(l) iodide (20 mg, 1 molpercent). The reaction mixture was heated at 5O0C under nitrogen for 3 hours. The reaction mixture was cooled to room temperature and the ammonium salt was removed by filtration. The filtrate was concentrated under reduced pressure. Purification of the crude compound by column chromatography (SilicaGel 230-400 mesh; 10percent ethyl acetate in hexanes as eluent) afforded of 2- (4-methoxy phenylethynyl) benzaldehyde (2.1 g, 89percent).
89%
at 50℃; for 3 h;
To a solution of 2-bromobenzaldehyde (1.85 g, 10 mmol) and 4-methoxyphenyl acetylene (1.58 g, 12 mmol) in 40 mL of triethylamine were added dichlorobis(triphenylphosphine)palladium(II) (140 mg, 2 mol percent) and copper (I) iodide (20 mg, 1 mol percent). The reaction mixture was heated at 50° C. under nitrogen for 3 h. The reaction mixture was cooled to room temperature and the ammonium salt was removed by filtration. The filtrate was concentrated under reduced pressure. Purification of the crude compound by column chromatography (silica gel 230-400 mesh; 10percent ethyl acetate in hexanes as eluent) afforded 2-(4-methoxyphenylethynyl)benzaldehyde (2.1 g, 89percent). The above compound (2.06 g, 8.73 mmol) and t-butylamine (3.83 g, 52.4 mmol) were stirred under nitrogen for 24 h at room temperature. The resulting mixture was extracted with ether and the organic layer was dried over anhydrous Na2SO4, and concentrated to give the imine (2.4 g, 94percent), which was used in the next step without further purification. To a solution of the above imine (2.39 g, 8.2 mmol) in 100 mL anhydrous DMF was added (0.156 g, 0.82 mmol) copper (I) iodide, and the solution was flushed with nitrogen. The reaction mixture was heated at 100° C. for 4 h. The mixture was cooled to room temperature, and diluted with ether (200 mL). The organic layer was washed with saturated aqueous ammonium chloride (3.x.100 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated to give the crude compound as a dark colored solid. Purification by column chromatography (silica gel 230-400 mesh; 10percent ethylacetate in hexanes as eluent) afforded 3-(4-methoxyphenyl)isoquinoline (1.064 g, 55percent), as a white solid. The 3-(4-methoxyphenyl)isoquinoline (1.05 g, 4.47 mmol) was suspended in 30 mL hydroiodic acid and 12 mL of acetic acid was added. The reaction mixture was stirred at 110° C. for 2 h, then cooled to room temperature. The precipitate formed was filtered off, washed with acetic acid (2.x.5 mL) and dried under vacuum, to give a yellow solid. The crude compound was purified by triturating with 5percent methanol in ether to give 4-isoquinolin-3-yl-phenol (0.83 g, 84percent) as a white powder. Selected data: MS (ES) m/z: 222.89, 221.86; MP 218-219° C.
84%
With potassium carbonate In N,N-dimethyl-formamide at 100℃; for 13 h; Green chemistry
General procedure: A mixture of aryl halide (1mmol), terminal alkyne (1mmol), K2CO3 (2mmol) and MNPFemTriazNHCAg complex (6) (100mg) in DMF (5mL) was stirred at 100°C. The progress of reaction was monitored by TLC. After completion, the reaction mixture was quenched in ice cold water and 6 was separated by external magnet. The reaction mixture was extracted with ethyl acetate (3×25mL). Evaporation of solvent in vaccuo followed by column chromatography over silica gel using petroleum ether/ethyl acetate afforded desired Sonogashira coupling products.
Reference:
[1] Chemical Communications, 2012, vol. 48, # 61, p. 7634 - 7636
[2] ACS Combinatorial Science, 2011, vol. 13, # 3, p. 265 - 271
[3] Journal of Organic Chemistry, 2014, vol. 79, # 13, p. 6113 - 6122
[4] Advanced Synthesis and Catalysis, 2012, vol. 354, # 4, p. 555 - 562
[5] Advanced Synthesis and Catalysis, 2016, vol. 358, # 16, p. 2684 - 2691
[6] European Journal of Organic Chemistry, 2017, vol. 2017, # 11, p. 1425 - 1433
[7] Organic and Biomolecular Chemistry, 2017, vol. 15, # 33, p. 6913 - 6920
[8] Tetrahedron, 1999, vol. 55, # 1, p. 29 - 62
[9] Organic letters, 2001, vol. 3, # 25, p. 4035 - 4038
[10] Journal of Organic Chemistry, 2002, vol. 67, # 10, p. 3437 - 3444
[11] Patent: WO2007/16525, 2007, A2, . Location in patent: Page/Page column 62-63
[12] Patent: US2008/188467, 2008, A1, . Location in patent: Page/Page column 22
[13] Organic Letters, 2015, vol. 17, # 24, p. 6126 - 6129
[14] Journal of Organometallic Chemistry, 2018, vol. 866, p. 112 - 122
[15] Tetrahedron Letters, 2009, vol. 50, # 33, p. 4706 - 4709
[16] Organic Letters, 2016, vol. 18, # 19, p. 5150 - 5153
[17] Synthesis (Germany), 2013, vol. 45, # 11, p. 1553 - 1563
[18] Journal of Organic Chemistry, 2005, vol. 70, # 3, p. 892 - 897
[19] Journal of Organic Chemistry, 2003, vol. 68, # 3, p. 920 - 928
[20] Journal of Organic Chemistry, 2002, vol. 67, # 20, p. 7042 - 7047
[21] Journal of Organic Chemistry, 2007, vol. 72, # 12, p. 4462 - 4468
[22] Chemical Communications, 2010, vol. 46, # 39, p. 7427 - 7429
[23] Organic Letters, 2011, vol. 13, # 4, p. 640 - 643
[24] Chemistry - A European Journal, 2011, vol. 17, # 18, p. 4981 - 4985
[25] Advanced Synthesis and Catalysis, 2012, vol. 354, # 10, p. 1890 - 1896
[26] European Journal of Organic Chemistry, 2012, # 24, p. 4590 - 4602
[27] Angewandte Chemie - International Edition, 2012, vol. 51, # 43, p. 10861 - 10865[28] Angew. Chem., 2012, vol. 124, # 43, p. 11019 - 11023
[29] Organic Letters, 2013, vol. 15, # 4, p. 874 - 877
[30] Chemistry - A European Journal, 2013, vol. 19, # 15, p. 4695 - 4700
[31] Chemistry - A European Journal, 2013, vol. 19, # 46, p. 15682 - 15688
[32] Chemistry - A European Journal, 2014, vol. 20, # 9, p. 2425 - 2430
[33] Chemistry - A European Journal, 2014, vol. 20, # 2, p. 390 - 393
[34] Journal of Organic Chemistry, 2014, vol. 79, # 8, p. 3494 - 3505
[35] Chemistry - A European Journal, 2015, vol. 21, # 7, p. 3042 - 3052
[36] Organic Letters, 2015, vol. 17, # 16, p. 4018 - 4021
[37] Journal of Organic Chemistry, 2014, vol. 79, # 21, p. 10674 - 10681
[38] Chemistry - A European Journal, 2016, vol. 22, # 27, p. 9125 - 9129
[39] Angewandte Chemie - International Edition, 2016, vol. 55, # 39, p. 11882 - 11886[40] Angew. Chem., 2016, vol. 128, p. 12061 - 12065,5
[41] RSC Advances, 2016, vol. 6, # 99, p. 97404 - 97419
[42] European Journal of Organic Chemistry, 2016, vol. 2016, # 29, p. 4961 - 4964
[43] Chemical Communications, 2017, vol. 53, # 23, p. 3369 - 3372
[44] Organic Letters, 2017, vol. 19, # 19, p. 5070 - 5073
[45] Heterocycles, 2017, vol. 94, # 10, p. 1847 - 1855
[46] Organic Letters, 2017, vol. 19, # 21, p. 5856 - 5859
[47] Angewandte Chemie - International Edition, 2017, vol. 56, # 49, p. 15570 - 15574[48] Angew. Chem., 2017, vol. 129, p. 15776 - 15780,5
[49] Journal of Organic Chemistry, 2017, vol. 82, # 20, p. 11238 - 11246
[50] Organic Letters, 2018, vol. 20, # 16, p. 4815 - 4818
76
[ 6630-33-7 ]
[ 176910-67-1 ]
Reference:
[1] Angewandte Chemie - International Edition, 2017, vol. 56, # 36, p. 10928 - 10932[2] Angew. Chem., 2017, vol. 129, # 36, p. 11068 - 11072,5
Reference:
[1] European Journal of Organic Chemistry, 2011, # 20-21, p. 3904 - 3910
[2] European Journal of Organic Chemistry, 2011, # 20-21, p. 3904 - 3910
89
[ 873-73-4 ]
[ 6630-33-7 ]
[ 1251832-81-1 ]
Reference:
[1] Journal of Organic Chemistry, 2014, vol. 79, # 13, p. 6113 - 6122
[2] Molecules, 2013, vol. 18, # 1, p. 814 - 831
[3] Organic Letters, 2016, vol. 18, # 19, p. 5150 - 5153
[4] Organic and Biomolecular Chemistry, 2017, vol. 15, # 33, p. 6913 - 6920
[5] Advanced Synthesis and Catalysis, 2016, vol. 358, # 16, p. 2684 - 2691
[6] Advanced Synthesis and Catalysis, 2009, vol. 351, # 17, p. 2833 - 2838
[7] Organic Letters, 2011, vol. 13, # 4, p. 640 - 643
[8] Advanced Synthesis and Catalysis, 2011, vol. 353, # 2-3, p. 392 - 400
[9] Chemistry - A European Journal, 2011, vol. 17, # 18, p. 4981 - 4985
[10] Advanced Synthesis and Catalysis, 2012, vol. 354, # 10, p. 1890 - 1896
[11] Angewandte Chemie - International Edition, 2012, vol. 51, # 43, p. 10861 - 10865[12] Angew. Chem., 2012, vol. 124, # 43, p. 11019 - 11023
[13] Chemistry - A European Journal, 2013, vol. 19, # 15, p. 4695 - 4700
[14] Chemistry - A European Journal, 2014, vol. 20, # 9, p. 2425 - 2430
[15] Chemistry - A European Journal, 2015, vol. 21, # 7, p. 3042 - 3052
[16] Journal of Organic Chemistry, 2015, vol. 80, # 15, p. 7635 - 7641
[17] Organic Letters, 2015, vol. 17, # 16, p. 4018 - 4021
[18] Advanced Synthesis and Catalysis, 2015, vol. 357, # 14-15, p. 3255 - 3261
[19] Journal of Organic Chemistry, 2014, vol. 79, # 21, p. 10674 - 10681
[20] Patent: CN105330663, 2016, A, . Location in patent: Paragraph 0038; 0039; 0040
[21] Angewandte Chemie - International Edition, 2016, vol. 55, # 39, p. 11882 - 11886[22] Angew. Chem., 2016, vol. 128, p. 12061 - 12065,5
[23] Organic Letters, 2017, vol. 19, # 16, p. 4387 - 4390
[24] Organic Letters, 2017, vol. 19, # 19, p. 5070 - 5073
[25] Organic Letters, 2017, vol. 19, # 21, p. 5856 - 5859
[26] Angewandte Chemie - International Edition, 2017, vol. 56, # 49, p. 15570 - 15574[27] Angew. Chem., 2017, vol. 129, p. 15776 - 15780,5
90
[ 6630-33-7 ]
[ 1251832-81-1 ]
Reference:
[1] European Journal of Organic Chemistry, 2017, vol. 2017, # 11, p. 1425 - 1433
[2] Angewandte Chemie - International Edition, 2017, vol. 56, # 36, p. 10928 - 10932[3] Angew. Chem., 2017, vol. 129, # 36, p. 11068 - 11072,5
91
[ 6630-33-7 ]
[ 1158503-82-2 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 6, p. 1740 - 1742
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In N,N-dimethyl-formamide; for 6h;Inert atmosphere; Reflux;
General procedure: To a solution of 2-bromobenzaldehyde (315 mul, 2.70 mmol) in DMF (20 ml) was added phenyl boronic acid (395 mg, 3.24 mmol), tetrakis-(triphenylphosphine)-palladium (31 mg, 0.027 mmol) and Na2CO3 (430 mg, 4.05 mmol). The result solution was stirred and refluxed for 6 h. After the reaction completed, the solution was cooled to room temperature, then partitioned between satd NaHCO3 and EtOAc, and the mixture was extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/diethyl ether = 8:1) to afford the product (369 mg, 2.03 mmol, 75% yield).
With dichloro[1,1'-bis(di-t-butylphosphino)ferrocene]palladium(II); potassium carbonate; In water; acetonitrile; at 60℃;Inert atmosphere; Sealed vessel;
Typical preparative procedures were performed on 10.0 mmol scale using a Mettler-Toledo FlexiWeigh 30 automated solid handling unit to pre-weigh the aryl halide (10.0 mmol, 1.0 equiv), aryl boronic acid (12.0 mmol, 1.2 equiv) and Pd-118 (65.2 mg, 0.1 mmol, 1.0 mol %). Acetonitrile (10.0 mL) was added, followed by K2CO3 added as a stock aqueous solution (10.0 mL water containing 2.07 g, 15.0 mmol, 1.5 equiv). NB. No internal standard was added for preparative reactions. Reaction mixtures were sealed under a N2 atmosphere and heated to 60 C with magnetic stirring. HPLC analysis showed that reactions using aryl bromides were complete within 1 h but that aryl chlorides typically required 24 h.After reaction mixtures had cooled to room temperature, stirring was stopped and the phases were allowed to separate. The lower aqueous phase was removed and discarded (cutting away any interfacial catalyst residues if present) and the acetonitrile phase concentrated to dryness to give typically a light to dark brown solid or dark-coloured gum. Solids were triturated with methanol (20 mL) for 1-2 h, then isolated by filtration, washed once or twice with methanol (4 mL each wash) and dried under vacuum. Oils were purified by flash silica gel chromatography as noted below. All isolated compounds gave 1H NMR data in agreement with published values. Literature data is given for mp values for comparison.
8-bromoisoquinoline (24). To 7.0 mL (60.0 mmol) 2-bromobenzaldehyde (23) was added 10.0 mL (69.0 mmol) aminoacetaldehyde diethyl acetal. After 3 h at 100° C., the reaction mixture was cooled to room temperature and the layers separated. The organic layer was purified by vaccum distillation to give 15.89 g bromobenzalaminoacetal (b.p. 141-148° C. at approximately 1 mm Hg). To 143 g concentrated sulfuric acid at 0° C. was added 15.89 g bromobenzalaminoacetal. With mechanical stirring, the resulting mixture was added in portions over 5 min to 20 g phosphoric anhydride in 10 g concentrated sulfuric acid maintained at 160° C. After 25 min at 160° C., the reaction mixture was cooled, poured onto ice and washed with 300 mL ethyl ether. The aqueous layer was basified with solid NaOH to pH=10 and extracted with EtOAc repeatedly. The combined EtOAc layers were washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. Purification by flash chromatography (75*110 mm silica gel, linear gradient 0.5-3percent (10percent NH4H:MeOH):CH2CI2) produced 24. 1H NMR (CDCl3, 400 MHz) 89.627 (s, 1H, ArH); 8.622 (d, 1H, J=5.67 Hz, ArH); 7.858 (dd, 1H, J=0.87, 7.45 Hz, ArH); 7.799 (d, 1H, J=8.32 Hz, ArH); 7.631 (d, 1H, J=5.76 Hz, ArH); 7.538 (dd, 1H, J=7.50, 8.23 Hz, ArH); MS (Electrospray): m/z 207.9, 209.0 (M+H, 79Br, 81Br).
With sodium hydroxide; triethylamine; In ethanol; dichloromethane; isopropyl alcohol;
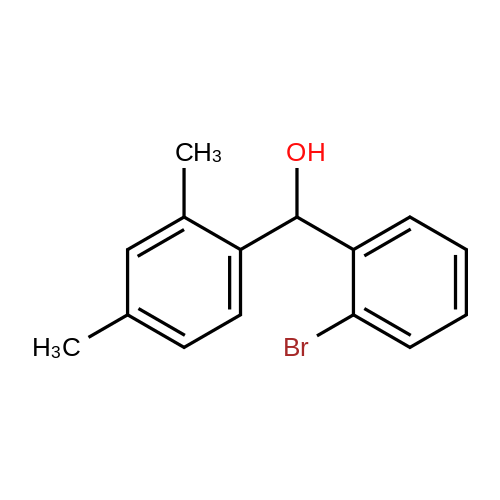
1-(Pyrid-3-yl)-2-(2-bromophenyl)-2-hydroxyethanone 146 g (0.8 mol) of 2-bromobenzaldehyde, 85.6 g (0.8 mol) of 3-pyridinealdehyde, 20 g (0.08 mol) of 3-ethyl-5-(2-hydroxyethyl)-4-methylthiazoliumbromide and 40.4 g (0.4 mol) of triethylamine in 500 ml of ethanol were heated at 75° C. for 16 hours. Thereafter, the ethanol was distilled off and the residue was taken up in methylene chloride. The solution was washed twice with water and then extracted twice with 300 ml of 4N hydrochloric acid. The acidic aqueous phase was then rendered slightly alkaline with 4N sodium hydroxide solution and extracted three times with methylene chloride. After the solvent had been distilled off, the crude product was stirred with isopropanol, filtered off under suction and dried to give a product of melting point 105°-108° C.
With sodium carbonate;palladium diacetate; triphenylphosphine; In water; N,N-dimethyl-formamide; at 110℃;Inert atmosphere;
Example 380 : Synthesis of (1E,6E)-1-(4-hydroxyphenyl)-7-[2-(pyridin-3-yl)phenyl]hepta-1,6-diene-3,5-dione (CU513); (1) Synthesis of 2-(pyridin-3-yl)benzaldehyde; To a suspension of 1-bromobenzaldehyde (250 muL, 2.14 mmol), sodium carbonate (270 mg, 2.55 mmol), and 3-pyridineboronic acid (289 mg, 2.35 mmol) in 4.2 mL of N,N-dimethylformamide/water (2:1) were added palladium acetate (24 mg, 0.11 mmol) and triphenylphosphine (115 mg, 0.44 mmol) under nitrogen. After being stirred at 110C overnight, the reaction mixture was filtered. The filtrate was diluted with chloroform, and the solution was washed with brine, and dried over MgSO4. After filtration, the filtrate was concentrated in vacuo, and the residue was purified by silica gel column chromatography (chloroform/methanol = 99/1 to 95/5) to obtain the title compound as a white powder (250 mg, 64%).
With tetrakis(triphenylphosphine) palladium(0); potassium phosphate monohydrate; In 1,2-dimethoxyethane; at 150℃; under 4500.45 Torr; for 1.5h;Microwave irradiation;
With palladium diacetate; caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 85℃; for 16h;Inert atmosphere;
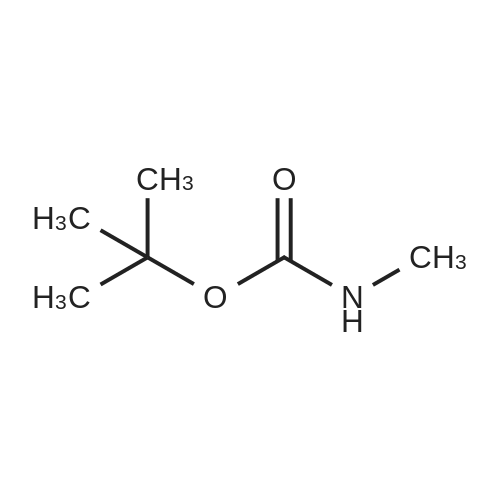
2-bromobenzaldehyde (2.560 g, 13.84 mmol), N-methyl tert-butyl carbamate (1.996 g, 15.22 mmol), and Cs2CO3 (6.760 g, 20.75 mmol) were added to 1,4-dioxane (100 mL). The solution was degassed and flushed with nitrogen three times. Pd(OAc)2 (0.155 g, 0.962 mmol) and Xantphos (0.801 g, 1.384 mmol) were added. The solution was degassed and flushed with nitrogen one additional time and then heated at 85 C for 16 hours. The solution was cooled, and the pH was adjusted to 7 using 1M HCl. The solution was extracted with 50% EtOAc (hexanes), and the extract was washed with brine and dried on anhydrous Na2SO4. The solution was concentrated and purified by flash column chromatography on silica gel using 10% EtOAc (hexanes) to give 2.77 g of (2-formyl-phenyl)-methyl-carbamic acid tert-butyl ester (85%). 1H NMR (300MHz, dimethylsulfoxide-d6) d 9.97 (s, 1H), 7.80 (dd, 1H, J1 = 7.53 Hz, J2 = 1.98 Hz), 7.71 (td, 1H, J1 = 7.54 Hz, J2 = 1.98 Hz), 7.45 (t, 2H, J = 8.33 Hz), 3.23 (s, 3H), 1.26 (bs, 9H). MS (ESI) M+: 236.
General procedure: A mixture of appropriate heterocyclic amine (1.0 mmol) (3) and aromatic aldehyde (1.2 mmol) (4) were stirred in tetrahydrofuran under ice-cold condition for 5 min, followed by addition of the thioglycolic acid (2.0 mmol). After 5 min, dicyclohexylcarbodiimide(1.2 mmol) was added to the reaction mixture at 0 C and the reaction mixture was stirred for an additional 3-5 h at room temperature to complete the reaction. Progresses of the reactions were monitored by TLC using acetone:chloroform (7:3) as mobile phase. The precipitated dicyclohexylurea was filtered off; the filtrate was concentrated to drynessunder reduced pressure. Deionized water was added to the residueand extracted with ethylacetate. The organic layer was successively washed with 5% aqueous sodium hydrogen carbonate and sodium chloride. Finally organic layer was dried over anhydrous sodium sulfate. The crude solid obtained on evaporation of the solvent under reduced pressure was subjected for column chromatography using silica gel 100-200 mesh using n-hexane:ethyl acetate (7:3) as the solvent system and further recrystallized in acetone to yield white/yellowish-white products[42].
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; for 2h;Inert atmosphere;
General procedure: A round-bottomed flask was charged with Pd2(dba)3 (5 mol percent ), ligand (10 molpercent), aryl halide (1mmol), appropriate isoquinolinamine (1 mmol), base (1.5 mmol) and dry solvent (5 mL). Theflask was flushed with argon for 5 min. The mixture was heated at reflux under magnetic stirring.After cooling down to room temperature, the reaction mixture was concentrated and the residuewas purified by flash column chromatography on silica gel.
With 1,1'-bis-(diphenylphosphino)ferrocene; palladium diacetate; 1,8-diazabicyclo[5.4.0]undec-7-ene; In N,N-dimethyl-formamide; at 90℃; for 3h;Sealed tube; Molecular sieve;
General procedure: To a stirred mixture of 2-bromobenzaldehyde (0.5 mmol) and the corresponding hydrazinehydrochloride (0.6 mmol) in DMF taken in a 25 mL sealed tube, was added Pd(OAc)2 (5 mol %), dppf (6 mol %), molecular sieves (W/W), DBU (1.25 mmol), and Co2(CO)8 (0.15 mmol). The reaction vessel was closed immediately and heated at 90 C for 3 h. The reaction mixture was cooled to room temperature, filtered through celite bed. The filtrate was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with water, brine solution, dried over MgSO4, evaporated in vacuum and purified using column chromatography on silica gel (60-120 mesh) to afford the pure products.
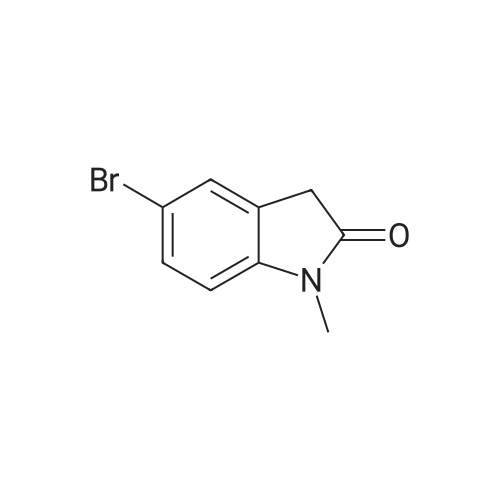
(E)-5-bromo-3-(2-bromobenzylidene)-1-methylindolin-2-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
73%
With piperidine; In ethanol; for 4h;Reflux; Inert atmosphere;
General procedure: To a solution of the corresponding oxindole (1.0 equiv) in EtOH (0.7 M) at room temperature was added sequentially 2-bromobenzaldehyde (1.2 equiv) and piperidine (0.1 equiv). The reaction mixture was heated to reflux and stirred until the starting oxindole was consumed as judged by TLC (ca. ~4 h). The reaction was allowed to cool to room temperature by removal of the oil bath, and then concentrated under reduced pressure. The crude mixture was purified by flash chromatography eluting with hexanes/EtOAc at the indicated ratio to afford the title compound.
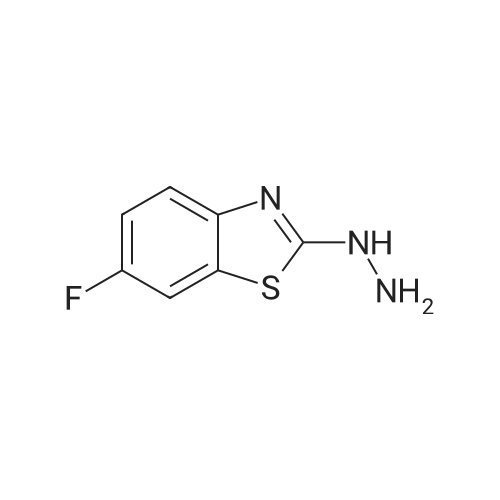
2-(2-(2-bromobenzylidene)hydrazino)-6-fluorobenzothiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76%
With acetic acid; In ethanol; at 80℃; for 0.166667h;Microwave irradiation;
General procedure: 2-(2-Arylidenehydrazino)-6-fluorobenzothiazoles 6a-r. General Procedure D. A mixture of compound 2 (0.0549 g, 0.0003 mol), the appropriate aromatic aldehyde (0.00033 mol) and glacial acetic acid (0.1 mL) in ethanol (5 mL) was heated under microwave (20 W) at 80 °C for 10 min. On cooling, the precipitated solid was collected by filtration, washed with water, dried and crystallized from the appropriate solvent to give the desired compounds 6a-r.
A mixture of 2-bromobenzaldehyde (50 g, 270 mmol) , aminoacetaldehyde dimethyl acetal (28.4 g, 270 mmol) and toluene (400 mL) was refluxed under argon. Dehydration was carried out using dean stark for 2.0 hours. After removal of calculated amount of water, the reflux was continued for 1.0 hour. The toluene was evaporated under reduced pressure, the residue was dissolved in dichloromethane (600 mL) , and the solution was cooled to 0°C. To the cooled solution was slowly added aluminium chloride (118.9 g, 891.7 mmol) under argon. The reaction mixture was stirred at 45°C for 2.0 hours. After the completion of the reaction was confirmed by TLC, the mixture was cooled to room temperature and slowly poured into an ice water. The mixture was basified with 10percent sodium hydroxide solution, and the dichloromethane layer was separated. The aqueous layer was re-extracted with dichloromethane (2 x 100 mL) . The combined dichloromethane layers were washed with brine, and dried over sodium sulfate. The dichloromethane was evaporated, and the residue was purified by silica gel (100-200 mesh) column chromatography 5 with 8-12percent. ethyl acetate in hexane as a mobile phase to give the title compound as an off-white solid (28 g, 49.8percent). MS(ESI)m/z: 208 [M (79Br)+l] ,210 [M (81Br)+l]; XH NMR (400 MHz, DMSO-d5) : delta 7.17 (t, J= 7.8 Hz, 1H) ; 7.91 (d, J= 6.0 Hz, 1H) ; 8.02 (d, J = 8.4 Hz, 1H) ; 8.05 (d, J = 8.8 Hz, 1H) ; 8.65 10 (d, J = 5.2 Hz, 1H) 9.48 (s, IH) .
With copper(II) acetate monohydrate; sodium carbonate; In toluene; at 100.0℃; for 24.0h;
General procedure: A mixture ofaldehyde 1 (6.8 mmol), amidine hydrochloride 2 (2 g,11.4 mmol), Na2CO3 (1.21 g, 11.4 mmol, 1.0 equiv) andCu(OAc)2 (10 molpercent) was stirred in toluene (20 mL) under100 °C in air for 24 h. After completion of the reaction, themixture was cooled to room temperature. The water wasadded to the reaction system and atmospheric distillation untiltoluene was evaporated. The resulting solution was filteredand residue with hot water washed 3 times. The crude productwas purified by column chromatography on silica gel usingpetroleum ether/EtOAc (100:1) as an eluent to give the correspondingproducts 7a-7x.
With potassium carbonate; copper(II) oxide; In N,N-dimethyl-formamide; at 50℃; for 24h;Reflux;
Intermediate 1-5 (16.0 g of 59.45 mmol) and o-bromobenzeneboronic acid (12.54 g 62.43 mmol) were added to dry DMF (300 mL), and anhydrous potassium carbonate (24.69 g 178.62 mmol) was added and gradually stirred at 50 At C, copper oxide (0.24 g of 3 mmol) was added and the mixture was heated to reflux for 24 h.After completion of the reaction, the mixture was cooled to room temperature, and the excess inorganic salt was filtered off, and then added with 2M aqueous hydrochloric acid (360 mL), heated to 60 C, stirred for 4 h, cooled to room temperature, washed with water, extracted with ethyl acetate, dried organic Separation and purification by chromatography gave Intermediate 1-7 (17.75 g, 80%), MW: 373.64.
General procedure: A mixture of compound A (5 mmol) and corresponding benzaldehyde (5 mmol) in 10 mL ethanol was refluxed for 24 hours in a reaction flask wrapped up by silver paper. Upon the reactions completed (monitored by TLC), tawny solids were obtained through suction filtration. The precipitates were washed with ethanol (5 mLx3) and recrystallized from ethanol with 50-84% yields.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping