* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Spectrochimica Acta - Part A: Molecular and Biomolecular Spectroscopy, 2010, vol. 77, # 2, p. 473 - 477
6
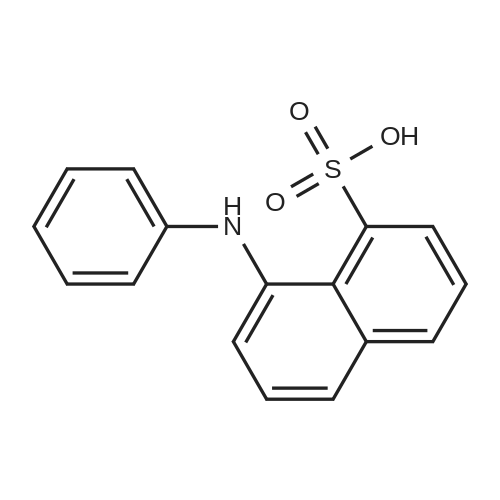
[ 155021-30-0 ]
[ 10238-21-8 ]
[ 7585-39-9 ]
Reference:
[1] Journal of Inclusion Phenomena and Macrocyclic Chemistry, 2010, vol. 68, # 3-4, p. 417 - 421
7
[ 2232-08-8 ]
[ 7585-39-9 ]
[ 67217-55-4 ]
Yield
Reaction Conditions
Operation in experiment
45.39%
Stage #1: at 60℃; for 2 h; Stage #2: With sodium hydroxide In water at 20℃; for 0.666667 h;
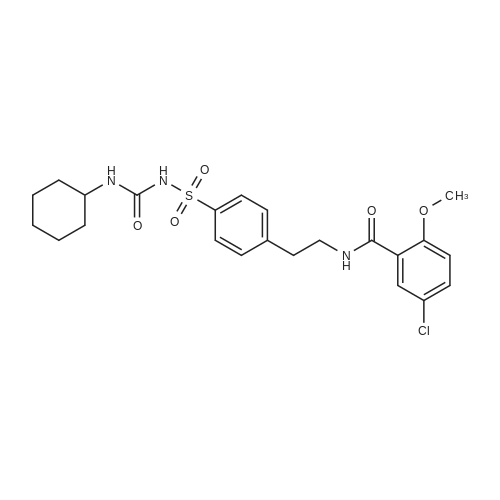
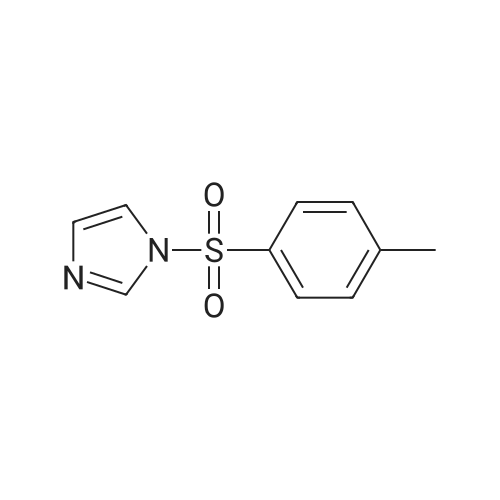
Synthesis was carried out as described in.1 In a 1L- double-necked, round-bottomed flask, β-CD (40.0 g, 35.2 mmol) was dissolved in deionized water (450 mL) by heating at 60 °C with vigorous stirring. To the resulting milky suspension, 1-(p-toluenesulfonyl)-imidazole (12 g, 54 mmol) was added. After 2 hrs stirring, a solution of sodium hydroxide (9 g, 225 mmol) in 25 mL of water was added slowly (in about 20 minutes). After further 20 minutes stirring, unreacted 1-(p-toluenesulfonyl)imidazole was separated by filtration through a sintered glass funnel. The reaction was quenched by the addition of ammonium chloride (24.1 g, 900 mmol) with swirling to dissolve all the solids. The resulting mixture was concentrated to about half of its originalvolume by blowing a stream of air over its surface. The resulting suspension was filtered througha large sintered-glass funnel and collected solid was washed with ice water (2x100 mL) andacetone (1x200 mL) and dried over CaCl2 in vacuum desiccators until constant weight. 20.63 g,16 mmol, 45.39percent.
45.39%
Stage #1: for 2 h; Stage #2: With sodium hydroxide In water for 0.666667 h;
Synthesis was carried out as described in [1]. In a 1L- double-necked, round-bottomed flask, β-CD (40.0 g, 35.2 mmol) was dissolved in deionized water (450 mL) by heating at 60 °C with vigorous stirring. To the resulting milky suspension, 1-(p-toluenesulfonyl)-imidazole (12 g, 54 mmol) was added. After 2 hrs stirring, a solution of sodium hydroxide (9 g, 225 mmol) in 25 mL of water was added slowly (in about 20 minutes). After further 20 minutes stirring, unreacted 1-(p-toluenesulfonyl)imidazole was separated by filtration through a sintered glass funnel. The reaction was quenched by the addition of ammonium chloride (24.1 g, 900 mmol) with swirling to dissolve all the solids. The resulting mixture was concentrated to about half of its original volume by blowing a stream of air over its surface. The resulting suspension was filtered through a large sintered-glass funnel and collected solid was washed with ice water (2x100 mL) and acetone (1x200 mL) and dried over CaCl2 in vacuum desiccators until constant weight. 20.63 g, 16 mmol, 45.39percent.TLC. Rf: 0.88 (i-Pr-OH:H2O:EtOAc:NH4OH/5:5:1:1), 1-H NMR (500 MHz, DMSO-d6) δ: 2.42 (s, 3 H), 3.20-3.65 (overlap with HDO, m, 40 H), 4.15-4.23 (m, 1 H), 4.30-4.38 (m, 2 H), 4.40-4.54 (m, 2 H), 4.50 (br s, 3 H), 4.76 (br s, 2 H), 4.84 (br s, 4 H), 5.62-5.84 (m, 14 H), 7.43 (d, 2 H, J = 7.75), 7.73 (d, 2 H, J = 7.75).
40.26%
Stage #1: at 20℃; for 2 h; Stage #2: With sodium hydroxide In water
The synthesis of mono-6-(p-toluenesulphonyl)-6-deoxy-β-cyclodextrin was carried out using a previously reportedprocedure.43,44 In a 1 L double necked round-bottomed flask,β-cyclodextrin (40.0 g, 35.2 mmol) was dissolved in deionized water(450 mL) at 60 °C with vigorous stirring. 1-(p-Toluenesulphonyl)-imidazole (12 g, 54 mmol) was added to the resulting milkysuspension at room temperature and stirred for 2 h. After that,a solution of sodium hydroxide 9 g (225 mmol, in 25 mL water)was added dropwise over 20 min. After addition of thesodium hydroxide solution, the separation of unreacted 1-(ptoluenesulphonyl)imidazole was carried out by simple filtrationusing a sintered glass funnel. Finally the reaction was quenchedby the addition of ammonium chloride (24.1 g, 900 mmol), withagitation to dissolve the added ammonium chloride. The resultingmixture was concentrated to about half of its original volume byblowing a stream of air over its surface. The resulting suspensionwas filtered using a sintered-glass funnel and the collected solidwas washed twice using 200 mL ice-cold water and 100 mL ofacetone in one portion. The collected solid was well dried overnightin a vacuum at 60 °C and kept over CaCl2 in vacuum desiccatorsyielding 18.30 g, or 40.26percent.
35.1%
Stage #1: at 20℃; for 4 h; Stage #2: With sodium hydroxide In water for 0.166667 h;
β-Cyclodextrin (35.0 g, 30.8 mmol) was dissolved in 350 mL H2O with 1-(p-toluenesulfonyl)imidazole (8.9 g, 40.0 mmol) and stirred at 20° C. for 4 h. NaOH (50 mL, 20percent w/v) was then added to the mixture and stirred for 10 min, inducing a precipitate. The precipitate was filtered off and the filtrate was collected, then neutralized to pH 7 with 25 g NH4Cl to form another precipitate. This precipitate was collected by filtration and washed with 100 mL of H2O three times and 100 mL of acetone twice and dried overnight under vacuum. Since both mono- and ditosylate forms existed along with unreacted β-CD, an HP20 (C 18) column was run. The mixture was loaded in bulk water and eluted with water until no more β-CD-OH emerged, at which point methanol was used as eluent and fractions were collected. Fractions were confirmed via TLC using isopropanol:H2O:EtOAc:30percent NH4OH (3:2:1:1) as solvent and acid stain (20percent H2SO4) visualization. Yield: 12.3 g (35.1percent). 1H NMR (300 MHz, DMSO-D6, δ): 7.80-7.66 (d, 2H, S-Benzene), 7.50-7.33 (d, 2H, Benz-CH3), 5.93-5.48 (b, 14H, OH on C2, C3 of CD), 4.87-4.70 (s, 7H, C1H of CD), 4.63-4.08 (b, 6H, OH on C6 of CD), 3.75-3.43 (m, 28H, C2H, C3H, C4H, and C5H of CD, overlap with HDO), 3.43-3.11 (m, 14H, C6H of CD), 2.43-2.34 (s, 3H, CH3 on OTs).
28.2%
at 20 - 60℃; for 6 h;
In a 250-mL three-necked round-bottomed flask, bCD (5.0 g,4.4 mmol) was dissolved in 112.5 mL of water by heating to60 C under vigorous stirring.28 After cooling to room temperature,finely powdered 1-(p-toluenesulfonyl)imidazole (3.9 g, 17.7 mmol)was added to the suspension. After 6 h, a solution of sodium hydroxide (2.3 g, 56.3 mmol) in 6.3 mL of water was added over20 min. After 10 min, unreacted 1-(p-toluenesulfonyl)imidazolewas removed by filtration. To the filtrate was added ammoniumchloride (6.1 g, 112.5 mmol) to quench the reaction. The resultingmixture was concentrated by blowing a stream of air across themixture and the product began to precipitate out of the solution.The suspension was filtered, and the solid was washed with icewater and acetone. The vacuum-dried tosyl bCD was obtained in28.2percent yield. The mono-tosylated bCD was treated with an equivalentamount of sodium azide in 16 mL of water at 80 C for 5 h.29After cooling, the solution was precipitated with acetone and thenlyophilized. Lyophilized azido bCD and triphenylphosphine (PPh3)(224 mg, 848 lmol) were dissolved in DMF (8 mL) and stirred for2 h at room temperature. After adding 1.6 mL of water, the solutionwas stirred for 3 h at 90 C, and the resulting product was precipitatedwith acetone. Amino bCD was purified by cation-exchangechromatography (CM-Sephadex C25) using 0.5-M ammoniumbicarbonate as a solvent and desalted with Bio-gel P2. The productwas confirmed using 1H NMR spectra.
28.2%
at 20℃; for 6 h;
Tosyl -CD was obtained from the Microbial CarbohydrateResource Bank (MCRB) at Konkuk University, Korea. Tosyl -CDwas prepared according to the method in Byun (Byun, Zhong, &Bittman, 2000). In a 1 L three-necked round-bottom flask, -CD(10.0 g, 8.8 mmol) was dissolved in 250 mL of water by heating it to60C under vigorous stirring. After cooling to room temperature,finely powdered 1-(p-toluenesulfonyl)imidazole (6 g, 27 mmol)was added to the suspension. After 6 h, a solution of sodium hydrox-ide (4.5 g, 112.5 mmol) in 12.5 mL of water was added over 20 min.After 10 min, the unreacted 1-(p-toluenesulfonyl)imidazole wasseparated by filtration. Ammonium chloride (12.05 g, 225 mmol)was then added to the reaction mixture to quench the reaction.The resulting mixture was concentrated by blowing a stream of aironto the surface overnight, after which the product precipitatedfrom the solution. The suspension was filtered, and the solid waswashed with ice water and acetone twice. The vacuum-dried tosyl-CD was obtained at a 28.2percent yield.
28.2%
Stage #1: at 20℃; for 6 h; Stage #2: With sodium hydroxide In water at 20℃; for 0.5 h;
In a 1L three-necked round-bottom flask, β-CD (10.0g, 8.8mmol) was dissolved in 250mL of water by heating it to 60°C under vigorous stirring. After cooling to room temperature, finely powdered 1-(p-toluenesulfonyl)imidazole (6g, 27mmol) was added to the suspension. After 6h, a solution of sodiumhydroxide (4.5g, 112.5mmol) in 12.5mL of water was added over 20min. After 10min, the unreacted 1-(p-toluenesulfonyl)imidazole was separated by filtration. Ammonium chloride (12.05g, 225mmol) was then added to the reaction mixture to quench the reaction. The resulting mixture was concentrated by blowing a stream of air onto the surface overnight, after which the product precipitated from the solution. The suspension was filtered, and the solid was washed with ice water and acetone twice. The vacuum-dried tosyl β-CD was obtained at a 28.2percent yield.
28%
Stage #1: at 20℃; for 6 h; Stage #2: With sodium hydroxide In water for 0.333333 h;
General procedure: Biotin tethering to cyclooligosaccharides starts from monotosylation on the primary hydroxylgroup, but the monotosylation method of both cyclooligosaccharides slightly differed from each other.For monotosylation of β-CD, β-CD (10.0 g, 8.8 mmol) was dissolved in water (250 mL) and heated to60 °C. After cooling to room temperature, 1-(p-toluenesulfonyl) imidazole (6 g, 27 mmol) was addedand mixed for 6 h. Sodium hydroxide (4.5 g, 112.5 mmol) solution in water (12.5 mL) was addeddropwise for 20 min. After filtration, the filtrate was quenched using ammonium chloride (12.05 g,225 mmol), and the resulting mixture was subjected to air flow for drying. The precipitated productwas washed with water and acetone. The yield of monotosylated β-CD was 28percent. For monotosylatedCyS, CyS (1 g, 0.28 mmol) was dissolved in water (50 mL), and copper sulfate (1 g, 4 mmol) in water(100 mL), was added. NaOH (1 g, 25 mmol) solution in water (50 mL) was mixed and stirredfor 10 min. The suspension became dark blue, and p-toluenesulfonyl chloride (2.5 g, 13.2 mmol)dissolved in 0.1 mL of acetonitrile was added in a dropwise manner for 1 h. The suspension wasstirred at room temperature for 4 h then neutralized with ammonium chloride. After removing the precipitate, the filtrate was concentrated and desalted on a Bio-Gel P2 column. The sample wassubjected to semi-preparative HPLC (Agilent Technologies 1200 series, Santa Clara, CA, USA) on areverse-phase column (Eclipse XDB-C18, 9.4 x 250 mm, 5 μm) at room temperature. The yieldof monotosylated CyS was 25percent. Further azidation and amination were performed, as describedin our previous report [43,44]. The mono-6-amino-β-CD or mono-6-amino-CyS were dissolved inDMF and reacted with biotinamidohexanoic acid N-hydroxysuccinimide ester for 24 h at 40 °C.After acetone precipitation, the samples were desalted and lyophilized. The overall synthetic yield fromoriginal cyclooligosaccharide to biotinylated one is about 8percent. The structures of the resulting biotinylcyclooligosaccharides were analyzed by NMR spectroscopy and MALDI-TOF mass spectrometry.
21%
With sodium hydroxide In water for 1 h;
25 g (0.022 mol; 1 eq.) of β-cyclodextrin (Roquette Freres SA) are suspended in 200 ml of distilled water, in a 500 ml Erlenmeyer flask, and then sodium hydroxide chips (8.8 g) are added in a single fraction. The reaction medium becomes clear. Compound 1 (5 g; 0.022 mol; 1 eq.) is rapidly added to the reaction medium (the tosylimidazole remains in suspension). After one hour, the pH is acidified to pH 6 with concentrated HCl. The white precipitate formed is filtered off and then washed with hot distilled water (2*100 ml) and with acetone (3*100 ml), and then recrystallized from water. After filtration and washing with acetone, compound 2 is obtained with a 21percent yield. TLC: Rf=0.4 eluent: 6percent NH4OH/EtOH/BuOH 5/5/4 (v/v/v) M.p.: 180° C. (decomposition point between 175° C. and 210° C.) 1H NMR DMSO-d6 δ (ppm): 7.75 (d, 2H, Hb/b', 3Ja-b=9 Hz); 7.4 (d, 2H, Hc/c', 3Jb-a=9 Hz); 5.8-5.5 (m, OH); 4.8 (m, 7H, H1-CD); 4.5-4.3 (m, 2H, H6I-CD/H6'I-CD); 3.8-3.5 (m, 20H, H5-CD/H6II-VII-CD/H6II-VII-CD/H3-CD); 3.3 (m, 14H, H2-CD/H4-CD); 2.4 (s, 3H, CH3) ESI-MS+: m/z measured at 1290.2 for [M+H]+, calculated at 1290.2 for C49H77O37S
Reference:
[1] Tetrahedron Letters, 2013, vol. 54, # 25, p. 3268 - 3273
[2] Synthetic Communications, 2014, vol. 44, # 5, p. 589 - 599
[3] Green Chemistry, 2014, vol. 16, # 6, p. 3117 - 3124
[4] Carbohydrate Research, 2016, vol. 430, p. 85 - 94
[5] Organic Syntheses, 2000, vol. 77, p. 225 - 225
[6] Organic and Biomolecular Chemistry, 2011, vol. 9, # 7, p. 2209 - 2218
[7] Journal of the American Chemical Society, 2012, vol. 134, # 18, p. 7596 - 7599
[8] Patent: US2015/202323, 2015, A1, . Location in patent: Paragraph 0112
[9] Journal of the American Chemical Society, 2018, vol. 140, # 7, p. 2426 - 2429
[10] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 6, p. 1855 - 1858
[11] Carbohydrate Research, 2014, vol. 391, # 1, p. 37 - 42
[12] Carbohydrate Polymers, 2017, vol. 163, p. 118 - 128
[13] Catalysis Communications, 2018, vol. 103, p. 83 - 87
[14] Molecules, 2018, vol. 23, # 1,
[15] Tetrahedron, 2006, vol. 62, # 51, p. 11963 - 11971
[16] Biomacromolecules, 2010, vol. 11, # 7, p. 1710 - 1715
[17] Patent: US2007/142324, 2007, A1, . Location in patent: Page/Page column 6-7
[18] Letters in Drug Design and Discovery, 2010, vol. 7, # 9, p. 665 - 673
[19] Macromolecular Bioscience, 2012, vol. 12, # 11, p. 1452 - 1458
[20] Chemistry - A European Journal, 2014, vol. 20, # 35, p. 10944 - 10952
[21] Bulletin of the Korean Chemical Society, 2014, vol. 35, # 8, p. 2487 - 2493
[22] Molecules, 2016, vol. 21, # 6,
[23] RSC Advances, 2017, vol. 7, # 24, p. 14477 - 14480
[24] Patent: US2017/231915, 2017, A1, . Location in patent: Paragraph 0115
[25] Organic Letters, 2018, vol. 20, # 3, p. 736 - 739
8
[ 98-59-9 ]
[ 7585-39-9 ]
[ 67217-55-4 ]
Yield
Reaction Conditions
Operation in experiment
88%
at 0 - 20℃;
A 500 mL round-bottom flask equipped with a magnetic stirbar, a vacuum adapter and a septum was charged with a solution of dry β-cyclodextrin (8.530 g, 7.51 mmol) and 200 mL of dry pyridine. The solution was cooled to 0 °C before 1.29g (6.76 mmol) of tosyl chloride was added. The resulting solution was allowed to warm to room temperature overnight The pyridine was removed as much as possible in vacuo. The resulting residue was then recrystallized twice from 40 mL of hot water to yield 7.54 (88percent) of a white crystalline solid.
88%
at 0 - 20℃;
Example 8 Synthesis of β-cyclodextrin-Tosylate, 8 (Melton, L. D., and Slessor, K. N., Carbohydrate Research, 18, p. 29 (1971)) A 500 mL round-bottom flask equipped with a magnetic stirbar, a vacuum adapter and a septum was charged with a solution of dry β-cyclodextrin (8.530 g, 7.51 mmol) and 200 mL of dry pyridine. The solution was cooled to 0° C. before 1.29 g (6.76 mmol) of tosyl chloride was added. The resulting solution was allowed to warm to room temperature overnight. The pyridine was removed as much as possible in vacuo. The resulting residue was then recrystallized twice from 40 mL of hot water to yield 7.54 (88percent) of a white crystalline solid 8.
88%
at 0 - 20℃;
Example 18 Synthesis of β-cyclodextrin-Tosylate, 16 (Melton, L. D., and Slessor, K. N., Carbohydrate Research, 18, p. 29 (1971))Example 18 Synthesis of β-cyclodextrin-Tosylate, 16 (Melton, L. D., and Slessor, K. N., Carbohydrate Research, 18, p. 29 (1971)) A 500 mL round-bottom flask equipped with a magnetic stirbar, a vacuum adapter and a septum was charged with a solution of dry β-cyclodextrin (8.530 g, 7.51 mmol) and 200 mL of dry pyridine. The solution was cooled to 0° C. before 1.29 g (6.76 mmol) of tosyl chloride was added. The resulting solution was allowed to warm to room temperature overnight. The pyridine was removed as much as possible in vacuo. The resulting residue was then recrystallized twice from 40 mL of hot water to yield 7.54 (88percent) of a white crystalline solid.
84.6%
With sodium hydroxide In water; acetonitrile at 0℃; for 4 h; Inert atmosphere
Synthesis of mono-6-deoxy-6-(p-tolylsulfonyl)^-cyclodextrin (CDOTs): A solution of p-toluenesulfonyl chloride (846.3 mg, 4.44 mmol) in 5 ml ACN was added to 80 ml aqueous solution of β-CD (5.0 g, 4.44 mmol) and NaOH (434.8 mg, 10.8 mmol) dropwise over 15 min. After stirring for 4 hrs at °C in N2 atmosphere, the solution was neutralized by adding 0.6 ml of 2.0 N aqueoushydrochloric acid and the product was recrystallized at 4 °C overnight, and then washed with acetone. CDO-Ts was obtained in a yield of 84.6percent. The product were tracked by TLC (1-propanol: ethyl acetate: water: ΝΗ320 = 3: 1 :2:1 (v/v)) visualized with cerium-ammonium-molybdate (CAM) solution.
46%
With copper(I) sulfate pentahydrate; sodium hydroxide In water; acetonitrile at 20℃; for 5 h;
To a solution of β-CD (2.5 g, 2.2 mmol) in water (110 mL), copper sulfate (1.65 g, 6.6 mmol) in water (165 mL) and sodium hydroxide (2.2 g, 55 mmol) in water (55 mL) were sequentially added. After 10 min, p-toluenesulfonyl chloride (3.3 g, 17.4 mmol) in acetonitrile (22 mL) was added drop by drop during 1 h. The reaction mixture was stirred for 4 h at ambient temperature, and then neutralized (pH = 12.5) with hydrochloric acid, the salts were eliminated by filtering and the volume of solution was decreased to 2/3 of its initial volume by lyophilization. _he β-CD-OTs was crystallized (three times) by dissolving in boiling water and washed with acetone (2* 7 mL), ether (2* 9 mL) and dried. After recrystallizations, pure β-CD-OTs (1.34 g, 46percent) was achieved (Baussanne et al., 2000).
43%
With sodium hydroxide In water at 4℃; for 0.583333 h;
β-CD (50.0 g, 44.0 mmol) was dissolved in 500 mL of a 0,4 M aqueous sodium hydroxide solution and cooled to 4 °C. While stirring vigorously, p-toluenesulfonyl chloride (35.0 g,184 mmol) was added within 5 minutes. The resulting suspension was stirred for additional 30 minutes and was then filtered. The filtrate was neutralized by addition of hydrochloric acid and stirred for an hour. The precipitate that was formed thereby was isolated via filtration, washed with water (3x) and dried at 60 °C overnight.Yield: 24.68 g (19.16 mmol, 43percent)71H-NMR (500 MHz, DMSO-d6): δ [ppm]= 7.77 (d, 2H), 7.45 (d, 2H), 5.74 (m, 14H), 4.79 (t,7H), 4.50 (t, 7H), 3.18-3.78 (m, 42H), 2.45 (s, 3H).FT-IR [cm-1]: 3301 (b), 2924 (w), 1641 (w), 1554 (w), 1358 (w), 1021 (s), 836 (w).MALDI-TOF-MS (m/z)= 1311.4 [M+Na]+, 1327.5 [M+K]+, 1465.5 [disubst. CD + Na]+.
38.3%
at 0 - 5℃; for 5 h;
To a NaOH solution (3.6 g, 0.09mol of NaOH in water120mL)was added β-CD (6.0 g, 5.3mmol)at 0-5. p-Tolylsulfonyl chloride (TsCl,1.2 g, 6.3mmol)was added into the solution with vigorous stirring at 0-5. After5h at 0-5, the precipitate wasremoved by filtration. 10percent HCl wasadded into the filtrate, and thepH was adjusted to pH=7. The mixture was kept in the refrigerator overnight to afford a whitesolid product. The white solid was recrystallized in hot water to afford 2.3 gof product (yield 38.3percent).
38.3%
With sodium hydroxide In water at 0 - 5℃; for 5 h;
According to the published method [Journal of Chromatography A, 2009, 1216(2),257-63], β-CD (6.0 g, 5.3 mmol) and NaOH (3.6 g, 0.09 mol) were stirred in a water solution (120 mL) at 0-5. Then p-tolylsulfonyl chloride (TsCl, 1.2 g, 6.3 mmol) was added into the solution at 0-5. After 5 h at 0-5, the precipitate was removed by filtration. 10percent HCl was added into the filtrate, and the pH was adjusted to pH=7. The mixture was kept in the refrigerator overnight to afford a white solid product. The white solid was recrystallized in hot water to afford 2.3 g of product (yield 38.3percent). The characteristic data were in accordance with literature [Journal of Chromatography A,2009, 1216(2),257-63]
35%
Stage #1: With sodium hydroxide In water for 0.0833333 h; Stage #2: at 25℃; for 1 h;
A solution of bCD was prepared by slowly adding 35.2 mL(88 mmol) of aqueous 2.5 N NaOH to 10 g (8.8 mmol) of bCD beingsuspended in 90 mL water. After 5 min, 2.52 g (13.22 mmol) of ptoluenesulfonylchloride was added in five portions and the formedsuspension was continuously stirred for 1 h at 25 C (400 rpm,magnetic stirrer). After filtration and washing of the residue(2 10 mL water), a total of 91 mL of pre-swollen cation exchangeresin 50W 4 20–50 mesh in the H+ form (100 meq, exchangecapacity 1.1 meq/mL) was added to the filtered solution. After20 min, the mixture was first filtered through a Büchner funnelprovided with glass wool to collect the exchange resin. Second,the precipitated white product was collected on filter paper. Afterwashing with 3 30 mL water, the product was lyophilized.Yield: 35percent (based on starting b-cyclodextrin).Melting point: 160–162 C.FTIR: 1600 cm1 (Ph-SO2–), 1230 cm1 (Ph-SO2-R), 815 cm1(Ph-SO2-O-R), 665 cm1 (Ph-SO2–).1H NMR (DMSO-d6): d = 7.77 (mc, 2H Ph), 7.43 (mc, 2H Ph),5.83–5.63 (m, 14H, OH1 and OH3 Cyd), 4.85–4.77 (m, 7H, H1Cyd), 4.51–4.45 (m, 6H, OH6 CyD), 4.35 (mc, 2H, H60 CyD), 4.20(mc, 1H, H50 CyD), 3.75–3.40 (m, 25H, H3, H5 and H6 CyD), 3.40–3.15 (m, H2, H4 overlap with water), 2.44 (s, 3H, Ph-CH3) ppm.Derivatization Degree (DD): >98 mol percent.13C NMR (DMSO-d6): d = 144.77 (C Ts); 132.846 (C Ts); 129.98(C Ts); 127.55 (C Ts); 101.89 (C1); 98.14 (C10); 81.47(C4); 81.15(C40); 73.02, 72.40, 72.00 (C2, C3, C5); 69.67 (C50); 68.87 (C60);59.88 (C6); 21.17 (CH3 Ts) ppm.m/z (ESI): calculated 1289.1970, found 1289.3770 [M+H]+.Elemental analysis: calculated—C: 45.65; H: 5.94; S: 2.49; detected—C: 45.71; H: 6.29; S: 2.84.
35%
With sodium hydroxide In water at 0℃; for 8 h;
11.35 g (10 mmol) of β-CD were dissolved in 200 mL of 0.6 M NaOH(aq) at 0 °C. After 30 min,2.3 g (12.1 mmol) of 4-Toluenesulfonyl chloride (TsCl), finely powdered with a mortar and pestle, wereadded to the aqueous solution. The reaction mixture was magnetically stirred for 8 h at 0 °C. Theunreacted TsCl was removed by filtration onto a sintered glass funnel. The aqueous phase was acidifiedat 0 °C with 10 mL of HCl (37percent) added dropwise. The product precipitated as white solid. The mixturewas kept at 4 °C overnight, filtered on paper and washed with deionized water until neutral pH. In orderto reduce the content of unreacted β-CD, the solid was dispersed in 100 mL of hot water (65 °C),stirred for 10 min, and finally filtered. This procedure was qualitatively followed by TLC (eluent:2-propanol:H2O:EtOAc:NH4OH = 5:3:1:0.5) and repeated for 3 times until the β-CD spot on TLC (Rf:0.25) was negligible compared to the CD1 spot (Rf: 0.47). The final product was washed with acetone(100 mL × 3 times), dried in air for 24 h and under vacuum (<5 mbar) at 25 °C for 3 h. Yield: 35percent(4.50 g). The tosylation procedure gave also the formation of very small amounts of ditosylates (Rf = 0.58),according to the literature [Brady2000]. Due to the small ΔRf between the mono- and the di-tosylate noattempt of further separation was made. ESI-MS analysis performed on the final product confirmed thepresence of negligible amounts of pristine CD and di-tosylate (see Figure SI2).
30%
With sodium hydroxide In water at 0 - 5℃; for 1 h;
2.3 Synthesis of mono-(6-O-(p-tolylsulfonyl))-β-CD The synthesis was done following the reported method [ 48 ]. β-CD (3.0 g, 2.64 mmol) was dissolved in 30 mL of a 0.4 M aqueous sodium hydroxide solution and cooled down to 0 °C. p-toluene sulfonyl chloride (2.1 g, 11.01 mmol) was added to the solution in small portions under vigorous stirring over 5 min. The suspension was kept under stirring at 5 °C for 1 h and then filtered off immediately. The filtrate was cooled at 0-5 °C and neutralized with hydrochloric acid and stirred for 1 h. The resultant precipitate was filtered off, washed three times with water and dried under vacuum at 60 °C for 20 h. Yield: 0.9 g (30percent).
25%
Stage #1: With sodium hydroxide In water at 0 - 5℃; for 5 h; Stage #3: at 0℃;
A three-liter, three-necked, round-bottomed flask equipped with a mechanical stirring and thermometer was charged with β-cyclodextrin (1) hydrate (50 g, 44 mmol) and a solution of 25 g (625 mmol) of sodium hydroxide in 1.0 liter of water. The solution was stirred at 0-5° C. in an ice-water bath and p-toluenesulfonyl chloride (TsCl) (20 g, 105 mmol) was added in one portion. The reaction mixture was stirred vigorously for 2 h at 0-5° C., and then another portion of TsCl (30 g, 157 mmol) was added and the reaction mixture stirred at this temperature for further 3 h. The reaction mixture was filtered in a fritted glass funnel to separate unreacted TsCl. The filtrate was cooled at 0-5° C. while 10percent aqueous hydrochloric acid (HCl, 150 ml) was added. The resulting solution was stored overnight in a refrigerator at 0° C., and then filtered. The product was dried and recrystallized from boiling water. Storage provided 14.0 g (25percent) of 2 as a white solid. TLC analysis of 2 performed on silica plates (EtOAc:2-propanol:conc. NH4OH:water-7:7:5:4) showed one major spot (Rf=0.45, Rf for β-CD=0.21).1H NMR (DMSO-d6) δ: 2.42 (s, 3H), 3.20-3.67 (m, 40H), 4.16-4.20 (m, 1H), 4.32 (d, 1H, J=9), 4.37-4.39 (m, 1H), 4.45-4.48 (m, 2H), 4.52-4.53 (m, 3H), 4.77 (d, 2H, J=3.4), 4.83-4.84 (m, 5H), 5.64-5.85 (m, 14H), 7.42 (d, 2H, J=8.2), and 7.75 (d, 2H, J=8.2). HPLC (Luna 5u NH2 100 A, size 250-4.6 mm, mobile phase 65percent acetonitrile-35percent H2O, flow 1.2 ml/min), Rt (2)=6.4 min., Rt (1)=12.3 min.
21.4%
Stage #1: With sodium hydroxide In water at 0℃; for 1 h; Stage #2: at 20℃; for 3 h;
β-CD (24 g) was suspended in 180 mL of water, NaOH (2.623 g) in 20 mL of water was added dropwise over 30 min, and reacted under vigorous agitation at 0 °C for a period of 1 h then p-toluenesulfonyl chloride (4.032 g) in 20 mL of acetonitrile was added dropwise over 1 h, causing immediate formation of a white precipitate. After 3 h of stirring at 20 °C filtered off unreacted toluenesulfonyl chloride, the solution was neutralized with a pH of 7-8 by hydrochloric acid, and the filtrate refrigerated overnight at 4 °C. The white precipitate was recovered by suction filtration and recrystallization in water for at least three times. The sample obtained was dried at 60 °C for 48 h in a vacuum oven, and OTs-CD was obtained as a white solid (5.14 g, yield: 21.40percent). IR (KBr, cm-1): 3388 (s, OH), 2928 (w, CH2), 1597 (s, Ph). 1H NMR (400 MHz, DMSO-d6): δ 7.42 (2H), 7.74 (2H), 4.75 (7H), 3.49-3.64 (28H), 3.28-3.36 (14H), 2.42 (3H). 13C NMR (DMSO): δ 144.9 (s), 132.7 (s), 129.9 (s), 127.6 (s), 102.3 (m), 81.5 (m), 71.9-73.1 (m), 69.8, 69.0, 59.5 (t), 21.3 (s) ppm. MALDI-TOF on CCA matrix (OTs-CD + Na+) m/z calcd for C49H76NaO37S (OTs-CD + Na+) 1311.37, measured 1311.13.
15%
With sodium hydroxide In water; acetonitrile at 20℃; for 2 h;
The detailed preparation for the picolinamide-modified β-cyclodextrin/Pd(II) complex (Pd(II)PCA-β-CD) is illustrated in Scheme 1. First, mono-6-tosyl β-cyclodextrin (1, Tos-β-CD) was obtained by the method of Khan etc (Khan & Pitchumani, 2016). Then, Tos-β-CD was reacted with NaN3 to form 6-monodeoxy-6-monoazido-β-CD (2, N3-β-CD), which was further reduced by triphenylphosphine to give a key intermediate: 6-monodeoxy-6-amino-β-CD (3). PCA-β-CD (4) was prepared by condensation of PCA (2-pyridinecarboxylic acid) with NH2-β-CD using N,N’-dicyclohexylcarbo-diimide (DCC) and 1-hydroxy-1H-benzotriazole (HOBt) in N,N’-dimethylformamide (DMF). The synthesis of PCA-(CH2)nNH-β-CD (6) was achieved in two simple steps: nucleophilic substitution (from 1 to 5) and amidation (from 5 to 6). Finally, all the obtained ligands were stirred with Pd(OAc)2 at room temperature for 24 h in toluene, and the target complexes Pd(II)PCA-β-CDs (C1-C3) were obtained as light yellow powders. The water solubility of the 4, 6 ligands and C1, C2, C3 complexes in different temperatures were studied. The results showed the complexes have high solubility in the aqueous solution (see Table 1S in SI). Reaction conditions: (a) TsCl, NaOH, CH3CN, H2O, rt, 2h, 15percent yield; (b) NaN3, DMF, 80°C, 12h, 93percent yield; (c) PPh3, DMF, rt, 10h, 91percent yield; (d) DCC, HOBt, DMF, 4h at 0°C, 18h at rt, 72–83percent yield; (e) Pd(OAc)2, toluene, rt, 24h, 60–70percent yield; (f) diamine, 80°C, 12h, 30percent yield.
12.68%
With sodium hydroxide In water; acetonitrile at 10 - 15℃; for 4 h;
A solution of sodium hydroxide (6.0000g, 150mmol) in water (20mL) was added dropwise to a solution of β-CD (56.7490g, 50mmol) in water (500mL) with magnetic stirring at 10–15°C over about 15min. The solution became homogeneous, and then a solution of p-toluenesulfonyl chloride (11.4384g, 60mmol) in acetonitrile (30mL) was added dropwise at 10–15°C over about 45min forming white precipitate immediately. The resultant solution was kept stirring for 3.0h, and rose to room temperature. The precipitate formed was collected by suction filtration and then suspended in water (300mL) with magnetic stirring at room temperature for 3.0h. The precipitate collected by suction filtration was washed successively with acetone (100mL) and water (160mL), and then dried in vacuum at 80°C for 8.0h to afford white solid powder 8.1746g in 12.68percent yield. (0016) [α]D25 +124.15(c 0.8044, DMF); mp>160°C (decomp.); 1H NMR (400MHz, DMSO-d6): δ=7.76 (d, J=8.3Hz, 2H), 7.44 (d, J=8.2Hz, 2H), 5.83–5.63 (m, 14H), 4.85–4.76 (m, 7H), 4.50–4.44 (m, 5H), 4.37–4.32 (m, 2H), 4.21–4.17 (m, 1H), 3.70–3.43 (m, 26H), 3.40–3.19 (m, 14H, overlaps with H2O), 2.43ppm (s, 3H); 13C NMR (400MHz, DMSO-d6): δ=144.67, 132.53, 129.75, 127.44, 102.09–101.14 (m), 81.51–80.63 (m), 72.88–71.69 (m), 69.56, 68.76, 59.74–59.04 (m), 21.06ppm; MS (ESI): m/z: 1311.3 [M+Na]+, 1289.0 [M+H]+.
10%
With sodium hydroxide In water; acetonitrile at 20℃; for 4 h;
β-cyclodextrin (120.0 g, 105.8 mmol) was suspended in 800 ml of water. NaOH (13.14 g, 328 mmol) in 40 ml water was added dropwise. The suspension became homogeneous before the addition was complete. p- Toluenesulfonyl chloride (20.16 g, 105.8 mmol) in 60 ml of acetonitrile was added dropwise. After 4 hours of reaction at room temperature the precipitate was removed by filtration and 8 mmol diluted HCl was added into the filtrate. The filtrate was then refrigerated overnight at 4°C. The resulting white precipitate was collected by filtration and dried, yielding the crude product. The pure product was obtained by recrystallization in hot water. Yield: 10percent. 1H NMR (500 Hz, DMSO-d6) δ 7.75 (d, J=8.3 Hz, 2 H), 7.43 (d, J=8.3 Hz, 2 H), 5.83- 5.63 (m, 14H), 4.85-4.77 (m, 7H); 4.52-4.17 (m, 6 H), 3.70-3.42 (m, 28 H), 3.39-3.20 (m, overlaps with HOD), 2.43 (s, 3 H) ppm.
8%
Stage #1: With sodium hydroxide In water at 20℃; for 1.5 h; Stage #2: for 2 h;
Take 210 g of β-cyclodextrin recrystallized from water, then added in portions with 1300 mL of water in a three-necked flask, and stirred at room temperature. Weigh 17.2 g of sodium hydroxide dissolved in 50 mL of water, and then slowly the aqueous sodium hydroxide solution was added dropwise to the β-cyclodextrin suspension until the reaction solution was gradually clarifies, and stirring was continued for 1.5 h. After the cyclodextrin was completely dissolved and clarified in the reaction solution, a solution of 26.0 g of p-toluenesulfonyl chloride in acetonitrile (80 mL) was slowly added dropwise to the reaction solution (dropwise over about 30 minutes). After stirring was continued for 2 hours, the solution was filtered off Insoluble matter, the filtrate was adjusted to pH = 7.5 with 2 mol / L hydrochloric acid, a large number of white precipitate precipitated. The precipitate was collected by suction filtration and the precipitate was dissolved in 450 mL of distilled water under heating. The insoluble material was filtered while hot and the filtrate was allowed to stand at room temperature for 12 h. The precipitate was collected and recrystallized several times with water until thin layer chromatography showed the product is Pure. Obtained 17g of final product, yield 8percent;
Reference:
[1] Patent: EP1133318, 2007, B1, . Location in patent: Page/Page column 32
[2] Patent: US9550860, 2017, B2, . Location in patent: Page/Page column 66
[3] Patent: US2017/49903, 2017, A1, . Location in patent: Paragraph 0162; 0163
[4] Patent: WO2016/126910, 2016, A1, . Location in patent: Page/Page column 29
[5] Journal of the American Chemical Society, 2017, vol. 139, # 36, p. 12354 - 12357
[6] Journal of the American Chemical Society, 2011, vol. 133, # 35, p. 13918 - 13921
[7] Carbohydrate Polymers, 2015, vol. 132, p. 205 - 213
[8] Journal of the American Chemical Society, 2006, vol. 128, # 46, p. 14750 - 14751
[9] Macromolecules, 2016, vol. 49, # 15, p. 5587 - 5598
[10] Carbohydrate Research, 1995, vol. 267, # 1, p. 49 - 63
[11] Macromolecules, 2010, vol. 43, # 13, p. 5538 - 5543
[12] Beilstein Journal of Organic Chemistry, 2014, vol. 10, p. 2263 - 2269
[13] Chemistry Letters, 2001, # 8, p. 838 - 839
[14] New Journal of Chemistry, 2002, vol. 26, # 9, p. 1130 - 1136
[15] Chemistry - A European Journal, 2016, vol. 22, # 51, p. 18435 - 18441
[16] Chinese Journal of Chemistry, 2017, vol. 35, # 7, p. 1125 - 1132
[17] Macromolecules, 2012, vol. 45, # 15, p. 5941 - 5947
[18] Synthetic Communications, 1995, vol. 25, # 5, p. 703 - 710
[19] Tetrahedron Letters, 2010, vol. 51, # 5, p. 800 - 803
[20] Tetrahedron Letters, 2013, vol. 54, # 36, p. 4953 - 4956
[21] New Journal of Chemistry, 2013, vol. 37, # 8, p. 2275 - 2279
[22] Synthetic Communications, 2015, vol. 45, # 3, p. 338 - 347
[23] Russian Journal of Physical Chemistry A, 2008, vol. 82, # 8, p. 1357 - 1362
[24] Carbohydrate Research, 2013, vol. 381, p. 59 - 63
[25] Molecules, 2015, vol. 20, # 9, p. 15881 - 15892
[26] Chemical Communications, 2017, vol. 53, # 95, p. 12782 - 12785
[27] Carbohydrate Research, 2011, vol. 346, # 2, p. 285 - 293
[28] Journal of Medicinal Chemistry, 2004, vol. 47, # 1, p. 233 - 239
[29] Beilstein Journal of Organic Chemistry, 2014, vol. 10, p. 814 - 824
[30] Langmuir, 2010, vol. 26, # 6, p. 4529 - 4534
[31] Journal of Materials Chemistry B, 2014, vol. 2, # 47, p. 8418 - 8426
[32] Australian Journal of Chemistry, 1993, vol. 46, # 6, p. 953 - 958
[33] Journal of Organic Chemistry, 1998, vol. 63, # 5, p. 1737 - 1739
[34] Bioorganic and Medicinal Chemistry, 2000, vol. 8, # 3, p. 647 - 652
[35] Tetrahedron Letters, 2003, vol. 44, # 2, p. 241 - 245
[36] Carbohydrate Research, 2014, vol. 400, p. 19 - 25
[37] Beilstein Journal of Organic Chemistry, 2014, vol. 10, p. 2428 - 2440
[38] Chemical Communications, 2017, vol. 53, # 16, p. 2463 - 2466
[39] Reactive and Functional Polymers, 2018, vol. 131, p. 12 - 21
[40] Chemical Communications, 2018, vol. 54, # 70, p. 9769 - 9772
[41] RSC Advances, 2015, vol. 5, # 20, p. 15547 - 15558
[42] Angewandte Chemie - International Edition, 2015, vol. 54, # 41, p. 12127 - 12133[43] Angew. Chem., 2015, vol. 127, p. 12295 - 12301,7
[44] Tetrahedron Letters, 1984, vol. 25, # 31, p. 3331 - 3334
[45] Tetrahedron Letters, 1984, vol. 25, # 31, p. 3331 - 3334
[46] Carbohydrate Research, 1993, vol. 238, # 1, p. 193 - 214
[47] Journal of Organic Chemistry, 1982, vol. 47, # 20, p. 3890 - 3893
[48] Angewandte Chemie - International Edition, 2008, vol. 47, # 18, p. 3435 - 3437
[49] Carbohydrate Research, 1989, vol. 192, p. 251 - 258
[50] Patent: US2008/275139, 2008, A1, . Location in patent: Page/Page column 11
[51] Organic Syntheses, 2000, vol. 77, p. 220 - 220
[52] European Journal of Organic Chemistry, 2018, vol. 2018, # 17, p. 1964 - 1974
[53] Green Chemistry, 2013, vol. 15, # 8, p. 2081 - 2085
[54] RSC Advances, 2013, vol. 3, # 42, p. 19255 - 19258
[55] Biomacromolecules, 2013, vol. 14, # 11, p. 4125 - 4134
[56] Carbohydrate Polymers, 2014, vol. 102, # 1, p. 278 - 287
[57] RSC Advances, 2014, vol. 4, # 34, p. 17768 - 17779
[58] RSC Advances, 2014, vol. 4, # 97, p. 54268 - 54281
[59] Chemistry - A European Journal, 2010, vol. 16, # 33, p. 10195 - 10201
[60] RSC Advances, 2016, vol. 6, # 98, p. 96006 - 96014
[61] Journal of Materials Chemistry B, 2015, vol. 3, # 37, p. 7417 - 7426
[62] Journal of the American Chemical Society, 2018,
[63] Chemical Communications, 2016, vol. 52, # 53, p. 8223 - 8226
[64] Carbohydrate Polymers, 2018, vol. 200, p. 200 - 210
[65] Journal of the American Chemical Society, [66] Journal of the American Chemical Society, 2009, vol. 131, p. 1386 - 1387
[67] Chemical Communications, 2011, vol. 47, # 45, p. 12388 - 12390
[68] Chemistry - A European Journal, 2013, vol. 19, # 32, p. 10526 - 10535
[69] Tetrahedron, 2013, vol. 69, # 39, p. 8360 - 8367
[70] Chemical Communications, 2013, vol. 49, # 83, p. 9678 - 9680
[71] RSC Advances, 2015, vol. 5, # 103, p. 84410 - 84422
[72] Journal of the American Chemical Society, 1990, vol. 112, # 10, p. 3860 - 3868
[73] Angewandte Chemie - International Edition, 2006, vol. 45, # 48, p. 8210 - 8214
[74] Journal of Organic Chemistry, 2007, vol. 72, # 22, p. 8227 - 8234
[75] Journal of the American Chemical Society, [76] Journal of the American Chemical Society, 2009, vol. 131, p. 1686 - 1688
[77] New Journal of Chemistry, 2014, vol. 38, # 8, p. 3630 - 3636
[78] Patent: WO2008/17029, 2008, A2, . Location in patent: Page/Page column 23
[79] ChemMedChem, 2014, vol. 9, # 5, p. 1060 - 1070
[80] Chemistry - A European Journal, 2014, vol. 20, # 30, p. 9372 - 9380
[81] Chinese Chemical Letters, 2018,
[82] RSC Advances, 2015, vol. 5, # 98, p. 80434 - 80440
[83] RSC Advances, 2016, vol. 6, # 26, p. 22034 - 22042
[84] Catalysis Science and Technology, 2016, vol. 6, # 12, p. 4283 - 4293
[85] Patent: CN104826123, 2017, B, . Location in patent: Paragraph 0030
[86] Journal of Organic Chemistry, 1997, vol. 62, # 25, p. 8760 - 8766
[87] Angewandte Chemie - International Edition, 2012, vol. 51, # 2, p. 450 - 454
[88] Carbohydrate Research, 1989, vol. 192, p. 111 - 118
[89] Carbohydrate research, 1985, vol. 143, p. 107 - 116
[90] Heterocycles, 1981, vol. 15, # 2, p. 815 - 818
[91] Bulletin of the Chemical Society of Japan, 1993, vol. 66, # 5, p. 1356 - 1360
[92] Journal of medicinal chemistry, 1997, vol. 40, # 17, p. 2755 - 2761
[93] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1989, p. 1409 - 1416
[94] Chemical Communications, 2005, # 33, p. 4208 - 4210
[95] Journal of the American Chemical Society, 1993, vol. 115, # 12, p. 5035 - 5040
[96] Bulletin of the Chemical Society of Japan, 1994, vol. 67, # 2, p. 495 - 499
[97] Journal of the Chemical Society. Perkin Transactions 2, 1996, vol. 5, p. 965 - 972
[98] Journal of Organic Chemistry, 1998, vol. 63, # 5, p. 1444 - 1454
[99] Journal of Organic Chemistry, 1999, vol. 64, # 5, p. 1487 - 1493
[100] European Journal of Organic Chemistry, 2002, # 4, p. 607 - 613
[101] Organic and Biomolecular Chemistry, 2004, vol. 2, # 10, p. 1542 - 1548
[102] Journal of the Chemical Society. Perkin Transactions 2, 2002, # 3, p. 463 - 469
[103] Journal of Organic Chemistry, 2003, vol. 68, # 18, p. 7071 - 7076
[104] Journal of Physical Chemistry B, 2004, vol. 108, # 26, p. 8836 - 8843
[105] Journal of the American Chemical Society, 2005, vol. 127, # 2, p. 657 - 666
[106] Organic and Biomolecular Chemistry, 2004, vol. 2, # 16, p. 2359 - 2364
[107] Tetrahedron Letters, 2005, vol. 46, # 14, p. 2507 - 2511
[108] Organic and Biomolecular Chemistry, 2005, vol. 3, # 6, p. 1129 - 1133
[109] Bioorganic and Medicinal Chemistry, 2006, vol. 14, # 1, p. 131 - 137
[110] Tetrahedron, 2004, vol. 60, # 7, p. 1557 - 1562
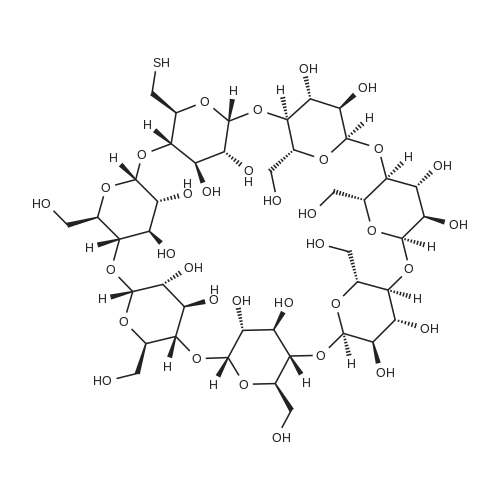
9
[ 4124-41-8 ]
[ 7585-39-9 ]
[ 67217-55-4 ]
Reference:
[1] Beilstein Journal of Organic Chemistry, 2014, vol. 10, p. 1390 - 1396
[2] Journal of Physical Chemistry B, 2012, vol. 116, # 6, p. 1765 - 1771
[3] Journal of Inclusion Phenomena and Macrocyclic Chemistry, 2016, vol. 85, # 3-4, p. 303 - 315
[4] Arkivoc, 2016, vol. 2016, # 5, p. 249 - 267
[5] Journal of Drug Delivery Science and Technology, 2018, vol. 45, p. 81 - 92
[6] Food Chemistry, 2019, vol. 278, p. 322 - 332
10
[ 104-15-4 ]
[ 7585-39-9 ]
[ 67217-55-4 ]
Reference:
[1] Journal of Materials Chemistry A, 2014, vol. 2, # 25, p. 9587 - 9593
11
[ 7585-39-9 ]
[ 67217-55-4 ]
Reference:
[1] Chemical Communications, 2015, vol. 51, # 12, p. 2421 - 2424
12
[ 67217-55-4 ]
[ 29390-67-8 ]
[ 7585-39-9 ]
Yield
Reaction Conditions
Operation in experiment
70%
With ammonia In water at 60℃; for 5 h;
Compound 2 (3.0 g, 2.33 mmol) was dissolved in 50 ml concentrated NH4OH solution. The reaction mixture was stirred at 60° C. for 5 h, cooled to room temperature and poured into 500 ml acetone to give a white precipitate. TLC analysis performed on silica plates (EtOAc:2-propanol:conc. NH4OH:water-7:7:5:4) showed a mixture of product 4 (70percent) and β-CD 1 (30percent). The product 4 was recrystallized from ethanol-water (3:7). The 1H NMR data are as in Example 1.
Synthesis was carried out as described in.1 In a 1L- double-necked, round-bottomed flask, beta-CD (40.0 g, 35.2 mmol) was dissolved in deionized water (450 mL) by heating at 60 C with vigorous stirring. To the resulting milky suspension, 1-(p-toluenesulfonyl)-imidazole (12 g, 54 mmol) was added. After 2 hrs stirring, a solution of sodium hydroxide (9 g, 225 mmol) in 25 mL of water was added slowly (in about 20 minutes). After further 20 minutes stirring, unreacted 1-(p-toluenesulfonyl)imidazole was separated by filtration through a sintered glass funnel. The reaction was quenched by the addition of ammonium chloride (24.1 g, 900 mmol) with swirling to dissolve all the solids. The resulting mixture was concentrated to about half of its originalvolume by blowing a stream of air over its surface. The resulting suspension was filtered througha large sintered-glass funnel and collected solid was washed with ice water (2x100 mL) andacetone (1x200 mL) and dried over CaCl2 in vacuum desiccators until constant weight. 20.63 g,16 mmol, 45.39%.
45.39%
Synthesis was carried out as described in [1]. In a 1L- double-necked, round-bottomed flask, beta-CD (40.0 g, 35.2 mmol) was dissolved in deionized water (450 mL) by heating at 60 C with vigorous stirring. To the resulting milky suspension, 1-(p-toluenesulfonyl)-imidazole (12 g, 54 mmol) was added. After 2 hrs stirring, a solution of sodium hydroxide (9 g, 225 mmol) in 25 mL of water was added slowly (in about 20 minutes). After further 20 minutes stirring, unreacted 1-(p-toluenesulfonyl)imidazole was separated by filtration through a sintered glass funnel. The reaction was quenched by the addition of ammonium chloride (24.1 g, 900 mmol) with swirling to dissolve all the solids. The resulting mixture was concentrated to about half of its original volume by blowing a stream of air over its surface. The resulting suspension was filtered through a large sintered-glass funnel and collected solid was washed with ice water (2x100 mL) and acetone (1x200 mL) and dried over CaCl2 in vacuum desiccators until constant weight. 20.63 g, 16 mmol, 45.39%.TLC. Rf: 0.88 (i-Pr-OH:H2O:EtOAc:NH4OH/5:5:1:1), 1-H NMR (500 MHz, DMSO-d6) delta: 2.42 (s, 3 H), 3.20-3.65 (overlap with HDO, m, 40 H), 4.15-4.23 (m, 1 H), 4.30-4.38 (m, 2 H), 4.40-4.54 (m, 2 H), 4.50 (br s, 3 H), 4.76 (br s, 2 H), 4.84 (br s, 4 H), 5.62-5.84 (m, 14 H), 7.43 (d, 2 H, J = 7.75), 7.73 (d, 2 H, J = 7.75).
40.26%
The synthesis of mono-6-(p-toluenesulphonyl)-6-deoxy-beta-cyclodextrin was carried out using a previously reportedprocedure.43,44 In a 1 L double necked round-bottomed flask,beta-cyclodextrin (40.0 g, 35.2 mmol) was dissolved in deionized water(450 mL) at 60 C with vigorous stirring. 1-(p-Toluenesulphonyl)-imidazole (12 g, 54 mmol) was added to the resulting milkysuspension at room temperature and stirred for 2 h. After that,a solution of sodium hydroxide 9 g (225 mmol, in 25 mL water)was added dropwise over 20 min. After addition of thesodium hydroxide solution, the separation of unreacted 1-(ptoluenesulphonyl)imidazole was carried out by simple filtrationusing a sintered glass funnel. Finally the reaction was quenchedby the addition of ammonium chloride (24.1 g, 900 mmol), withagitation to dissolve the added ammonium chloride. The resultingmixture was concentrated to about half of its original volume byblowing a stream of air over its surface. The resulting suspensionwas filtered using a sintered-glass funnel and the collected solidwas washed twice using 200 mL ice-cold water and 100 mL ofacetone in one portion. The collected solid was well dried overnightin a vacuum at 60 C and kept over CaCl2 in vacuum desiccatorsyielding 18.30 g, or 40.26%.
35.1%
beta-Cyclodextrin (35.0 g, 30.8 mmol) was dissolved in 350 mL H2O with 1-(p-toluenesulfonyl)imidazole (8.9 g, 40.0 mmol) and stirred at 20 C. for 4 h. NaOH (50 mL, 20% w/v) was then added to the mixture and stirred for 10 min, inducing a precipitate. The precipitate was filtered off and the filtrate was collected, then neutralized to pH 7 with 25 g NH4Cl to form another precipitate. This precipitate was collected by filtration and washed with 100 mL of H2O three times and 100 mL of acetone twice and dried overnight under vacuum. Since both mono- and ditosylate forms existed along with unreacted beta-CD, an HP20 (C 18) column was run. The mixture was loaded in bulk water and eluted with water until no more beta-CD-OH emerged, at which point methanol was used as eluent and fractions were collected. Fractions were confirmed via TLC using isopropanol:H2O:EtOAc:30% NH4OH (3:2:1:1) as solvent and acid stain (20% H2SO4) visualization. Yield: 12.3 g (35.1%). 1H NMR (300 MHz, DMSO-D6, delta): 7.80-7.66 (d, 2H, S-Benzene), 7.50-7.33 (d, 2H, Benz-CH3), 5.93-5.48 (b, 14H, OH on C2, C3 of CD), 4.87-4.70 (s, 7H, C1H of CD), 4.63-4.08 (b, 6H, OH on C6 of CD), 3.75-3.43 (m, 28H, C2H, C3H, C4H, and C5H of CD, overlap with HDO), 3.43-3.11 (m, 14H, C6H of CD), 2.43-2.34 (s, 3H, CH3 on OTs).
28.2%
In water; at 20 - 60℃; for 6h;
In a 250-mL three-necked round-bottomed flask, bCD (5.0 g,4.4 mmol) was dissolved in 112.5 mL of water by heating to60 C under vigorous stirring.28 After cooling to room temperature,finely powdered 1-(p-toluenesulfonyl)imidazole (3.9 g, 17.7 mmol)was added to the suspension. After 6 h, a solution of sodium hydroxide (2.3 g, 56.3 mmol) in 6.3 mL of water was added over20 min. After 10 min, unreacted 1-(p-toluenesulfonyl)imidazolewas removed by filtration. To the filtrate was added ammoniumchloride (6.1 g, 112.5 mmol) to quench the reaction. The resultingmixture was concentrated by blowing a stream of air across themixture and the product began to precipitate out of the solution.The suspension was filtered, and the solid was washed with icewater and acetone. The vacuum-dried tosyl bCD was obtained in28.2% yield. The mono-tosylated bCD was treated with an equivalentamount of sodium azide in 16 mL of water at 80 C for 5 h.29After cooling, the solution was precipitated with acetone and thenlyophilized. Lyophilized azido bCD and triphenylphosphine (PPh3)(224 mg, 848 lmol) were dissolved in DMF (8 mL) and stirred for2 h at room temperature. After adding 1.6 mL of water, the solutionwas stirred for 3 h at 90 C, and the resulting product was precipitatedwith acetone. Amino bCD was purified by cation-exchangechromatography (CM-Sephadex C25) using 0.5-M ammoniumbicarbonate as a solvent and desalted with Bio-gel P2. The productwas confirmed using 1H NMR spectra.
28.2%
In water; at 20℃; for 6h;
Tosyl -CD was obtained from the Microbial CarbohydrateResource Bank (MCRB) at Konkuk University, Korea. Tosyl -CDwas prepared according to the method in Byun (Byun, Zhong, &Bittman, 2000). In a 1 L three-necked round-bottom flask, -CD(10.0 g, 8.8 mmol) was dissolved in 250 mL of water by heating it to60C under vigorous stirring. After cooling to room temperature,finely powdered 1-(p-toluenesulfonyl)imidazole (6 g, 27 mmol)was added to the suspension. After 6 h, a solution of sodium hydrox-ide (4.5 g, 112.5 mmol) in 12.5 mL of water was added over 20 min.After 10 min, the unreacted 1-(p-toluenesulfonyl)imidazole wasseparated by filtration. Ammonium chloride (12.05 g, 225 mmol)was then added to the reaction mixture to quench the reaction.The resulting mixture was concentrated by blowing a stream of aironto the surface overnight, after which the product precipitatedfrom the solution. The suspension was filtered, and the solid waswashed with ice water and acetone twice. The vacuum-dried tosyl-CD was obtained at a 28.2% yield.
28.2%
In a 1L three-necked round-bottom flask, beta-CD (10.0g, 8.8mmol) was dissolved in 250mL of water by heating it to 60C under vigorous stirring. After cooling to room temperature, finely powdered 1-(p-toluenesulfonyl)imidazole (6g, 27mmol) was added to the suspension. After 6h, a solution of sodiumhydroxide (4.5g, 112.5mmol) in 12.5mL of water was added over 20min. After 10min, the unreacted 1-(p-toluenesulfonyl)imidazole was separated by filtration. Ammonium chloride (12.05g, 225mmol) was then added to the reaction mixture to quench the reaction. The resulting mixture was concentrated by blowing a stream of air onto the surface overnight, after which the product precipitated from the solution. The suspension was filtered, and the solid was washed with ice water and acetone twice. The vacuum-dried tosyl beta-CD was obtained at a 28.2% yield.
28%
General procedure: Biotin tethering to cyclooligosaccharides starts from monotosylation on the primary hydroxylgroup, but the monotosylation method of both cyclooligosaccharides slightly differed from each other.For monotosylation of beta-CD, beta-CD (10.0 g, 8.8 mmol) was dissolved in water (250 mL) and heated to60 C. After cooling to room temperature, 1-(p-toluenesulfonyl) imidazole (6 g, 27 mmol) was addedand mixed for 6 h. Sodium hydroxide (4.5 g, 112.5 mmol) solution in water (12.5 mL) was addeddropwise for 20 min. After filtration, the filtrate was quenched using ammonium chloride (12.05 g,225 mmol), and the resulting mixture was subjected to air flow for drying. The precipitated productwas washed with water and acetone. The yield of monotosylated beta-CD was 28%. For monotosylatedCyS, CyS (1 g, 0.28 mmol) was dissolved in water (50 mL), and copper sulfate (1 g, 4 mmol) in water(100 mL), was added. NaOH (1 g, 25 mmol) solution in water (50 mL) was mixed and stirredfor 10 min. The suspension became dark blue, and p-toluenesulfonyl chloride (2.5 g, 13.2 mmol)dissolved in 0.1 mL of acetonitrile was added in a dropwise manner for 1 h. The suspension wasstirred at room temperature for 4 h then neutralized with ammonium chloride. After removing the precipitate, the filtrate was concentrated and desalted on a Bio-Gel P2 column. The sample wassubjected to semi-preparative HPLC (Agilent Technologies 1200 series, Santa Clara, CA, USA) on areverse-phase column (Eclipse XDB-C18, 9.4 x 250 mm, 5 mum) at room temperature. The yieldof monotosylated CyS was 25%. Further azidation and amination were performed, as describedin our previous report [43,44]. The mono-6-amino-beta-CD or mono-6-amino-CyS were dissolved inDMF and reacted with biotinamidohexanoic acid N-hydroxysuccinimide ester for 24 h at 40 C.After acetone precipitation, the samples were desalted and lyophilized. The overall synthetic yield fromoriginal cyclooligosaccharide to biotinylated one is about 8%. The structures of the resulting biotinylcyclooligosaccharides were analyzed by NMR spectroscopy and MALDI-TOF mass spectrometry.
28.1%
This compound was prepared following a previoulsy reported procedure.[2] In a 1 L, three-necked, round-bottomed flask equipped with a thermometer, a pressure-equalized dropping funnel, and a large magnetic stir bar, beta-cyclodextrin hydrate (11.23 g, 9.89 mmol) was dissolved in 250 mL of distilled water by heating to 50 C with vigorous stirring. Vigourous stirring was continued as the solution was allowed to cool to room temperature, and to the resulting milky suspension was added finely powdered 22 (8.79 g, 39.57 mmol), in one portion. After 2 hours, a solution of sodium hydroxide (5.05 g, 0.126 mol) in 14 mL of water was added dropwise over 20 minutes. Stirring was then stopped for 10 minutes, and unreacted 22 was separated by filtration through sintered glass. The reaction was quenched by the addition of ammonium chloride (13.53 g, 0.253 mol) with swirling to dissolve all the solids. The resulting mixture was concentrated (to about half of its original volume) by passing a stream of air over its surface overnight. The product begins to precipitate almost immediately as the mixture becomes more concentrated. The resulting suspension was filtered through a large sintered-glass funnel and the collected solid was washed with ice water (2 × 25 mL) and acetone (40 mL) and then dried to constant weight over calcium chloride in a vacuum desiccator to give the title compound 23 (3.61 g, 28.3 % yield) as a white solid. All the spectroscopic data matched with those reported in the literature.[2] 1H NMR (400 MHz, DMSO-d6): delta 7.73 (d, 3J(H-H) = 8.1 Hz, 2H), 7.42 (d, 3J(H-H) = 8.1 Hz, 2H), 5.62-5.83 (m, 14H), 4.83 (br s, 4H), 4.76 (br s, 2H), 4.51 (br s, 3H), 4.44-4.57 (m, 2H), 4.30-4.38 (m, 2H), 4.15-4.20 (m, 1H), 3.20-3.65 (overlap with HDO, m, 40H), 2.49 ppm (s, 3H, CH3).
21%
With sodium hydroxide; In water; for 1h;
25 g (0.022 mol; 1 eq.) of beta-cyclodextrin (Roquette Freres SA) are suspended in 200 ml of distilled water, in a 500 ml Erlenmeyer flask, and then sodium hydroxide chips (8.8 g) are added in a single fraction. The reaction medium becomes clear. Compound 1 (5 g; 0.022 mol; 1 eq.) is rapidly added to the reaction medium (the tosylimidazole remains in suspension). After one hour, the pH is acidified to pH 6 with concentrated HCl. The white precipitate formed is filtered off and then washed with hot distilled water (2*100 ml) and with acetone (3*100 ml), and then recrystallized from water. After filtration and washing with acetone, compound 2 is obtained with a 21% yield. TLC: Rf=0.4 eluent: 6% NH4OH/EtOH/BuOH 5/5/4 (v/v/v) M.p.: 180 C. (decomposition point between 175 C. and 210 C.) 1H NMR DMSO-d6 delta (ppm): 7.75 (d, 2H, Hb/b', 3Ja-b=9 Hz); 7.4 (d, 2H, Hc/c', 3Jb-a=9 Hz); 5.8-5.5 (m, OH); 4.8 (m, 7H, H1-CD); 4.5-4.3 (m, 2H, H6I-CD/H6'I-CD); 3.8-3.5 (m, 20H, H5-CD/H6II-VII-CD/H6II-VII-CD/H3-CD); 3.3 (m, 14H, H2-CD/H4-CD); 2.4 (s, 3H, CH3) ESI-MS+: m/z measured at 1290.2 for [M+H]+, calculated at 1290.2 for C49H77O37S
The cysteinyl beta-CD was synthesized from mono-6-O-p-toluenesulfonyl-beta-CD (tosyl beta-CD), themost important intermediate for mono-functionalization on the primary side of beta-CD [28]. The tosyl beta-CD was synthesized as described in our previous report [29]. The beta-CD (5.0 g, 4.4 mmol) wasdissolved in water by heating it to 60 C, and the cooled solution was mixed with 1-(p-toluenesulfonyl)imidazole (3.9 g, 17.7 mmol). After 6 h, a NaOH aqueous solution was added over a time of 20 min,and the unreacted precipitates were removed. Ammonium chloride (6.1 g, 115 mmol) was added tostop the reaction, and the mixture was concentrated to half of the original volume by blowing air. Tosyl beta-CD began to precipitate, and the precipitates were washed and dried. Tosyl beta-CD (921 mg, 0.7 mmol)and L-cysteine (251 mg, 2.1 mmol) were dissolved in a 40% (v/v) TEA aqueous solution (10 mL) at85 C while stirring for two days under N2. After 48 h, excess solvent was removed by evaporationunder reduced pressure, and the concentrated sample was precipitated using ethanol (100 mL). Theprecipitate was collected, and the cysteinyl beta-CD was purified using DEAE-Sephadex A-25 and aBio-gel P-2 column. The lyophilized product was obtained at a 30% yield starting with beta-CD.
With sodium hydroxide; In water; at 20℃; for 2h;
beta-cyclodextrin (1.0 g, 0.88 mmol, 1 eq) was added into water (11 mL) and then stirred at 60 C. until fully dissolved. The solution was then cooled to 20 C., and tosylimidizole (0.35 mg) was added. The reaction was then stirred at room temperature for 2 hours, and NaOH (0.23 g) was added to the mixture. The mixture was then filtered, and NH4Cl (0.64 g) was added to the filtrate. The mixture was then concentrated and again filtered. The resulting solid was washed with water (2*0.5 mL) and acetone (3 mL) to afford mono-tosylated beta-cyclodextrin.
In water; at 20℃; for 6h;
Cysteinyl beta-cyclodextrin (cysteinyl beta -CD) can be prepared from an intermediate tosyl beta-cyclodextrin (tosyl beta -CD) (see FIG. 12). beta-CD (about 5.0 g, about 4.4 mmol) was dissolved in water at about 60 C.,The cooled solution is mixed with 1- (p-toluenesulfonyl) imidazole (3.9 g, 17.7 mmol).After about 6 hours,Aqueous NaOH solution was added over about 20 minutes,Unreacted precipitate is removed.Ammonium chloride (about 6.1 g, about 115 mmol) was added to stop the reaction,Air is blown into the mixture and condensed to half volume.tosyl [beta] -CD begins to precipitate,The precipitate is extracted, washed and dried.tosyl [beta] -CD (921 mg, 0.7 mmol)And L-cysteine (251 mg, 2.1 mmol) were dissolved in about 40% (v / v) TEA (Triethylamine) aqueous solution (10 mL) under nitrogen atmosphere at about 85 C for about 2 days.After about 48 hours,Excess solvent was removed by evaporation under reduced pressure,The concentrated sample is precipitated using ethanol (100 mL).The precipitate was collected,Cysteinyl beta-CD is prepared by using DEAE-Sephadex A-25 and Bio-gel P-2 column.here,The lyophilized product is produced in about 30% yield relative to the starting beta-CD.
In water; at 20℃; for 2h;
[00125] beta-cyclodextrin (1.0 g, 0.88 mmol, 1 eq) was added into water (11 mL) and then stirred at 60 C until fully dissolved. The solution was then cooled to 20 C, and tosylimidizole (0.35 mg) was added. The reaction was then stirred at room temperature for 2 hours, and NaOH (0.23 g) was added to the mixture. The mixture was then filtered, and NH4CI (0.64 g) was added to the filtrate. The mixture was then concentrated and again filtered. The resulting solid was washed with water (2 chi 0.5 mL and acetone (3 mL) to afford mono-tosylated beta-cyclodextrin.
beta-cyclodextrin (5.0 g) and water (1.5 mL) were ground to a paste in a mortar. 5-[4-[2-(N-Methyl-N-(2-pyridyl)arnrno)ethoxy]benzyl]thiazolidine-2,4-dione (1.57 g) was then added to the paste along with water (1.5 mL). The mixture was ground for a further 10 minutes and then dried for 1 hour at 21C and for 2 hours at 50C under vacuum to afford a 1:1 5-[4-[2-(N-methyl-N-(2-pyridyl)amino)ethoxy]benzyl]thiazolidine-2,4-dione beta cyclodextrin complex. The solid was recrystallised from a mixture of ethanol (25 ml) and water (20 ml), and then dried under vacuum for 19 hours at 50C to afford a 1:1 5-[4-[2-(N-methyl-N-(2-pyridyl)amino)ethoxy]benzyl]thiazolidine-2,4-dione beta-cyclodextriacomplex. 1H-NMR (d6-DMSO): consistent with approximately 1:1 5-[4-[2-(N-methyl-N-(2-pyridyl)amino)ethoxy] benzyl]thiazolidine-2,4-dione beta-cyclodextrin complex. XRPD: See Figure 1.
5-[4-[2-(N-methyl-N-(2-pyridyl)amino)ethoxy]benzyl]thiazolidine-2,4-dione β-cyclodextrin complex 1:2[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In ethanol; water;Heating / reflux;
A stirred mixture of 5-[4-[2-(N-methyl-N-(2-pyridyl)arnino)ethoxy]benzyl]thiazolidine-2,4-dione (0.79 g) and beta-cyclodextrin (5.0 g) in water (40 mL) and ethanol (40 mL) was heated to reflux and maintained at that temperature until a clear solution was formed. The solution was cooled to 21C over approximately 1 hour. The mixture was maintained at 21 C until crystallisation was observed to be complete before the solid was recovered by filtration and washed with ethanol. The solid was dried at 50C under vacuum to afford 1:2 5-[4-[2-(N-methyl-N-(2-pyridyl)amino)ethoxy]benzyl]thiazolidrne-2,4-dione beta-cyclodextrin complex (4.61 g). 1H-MNR (d6-DMSO): consistent with approximately 1:2 5-[4-[2-(N-methyl-N-(2-pyridyl)amino)ethoxy]benzyl]thiazolidine-2,4-dione beta-cyclodextrincomplex. XRPD: See Figure 4. A 1.0 g sample of the product of Example 4 was recrystallised from a mixture of ethanol (25 ml) and water (5 ml), where the initial solution formed was concentrated to approximately half volume, to afford 1:2 5-[4-[2-(N-methyl-N-(2-pyridyl)amino)ethoxy]benzyl]thiazolidine-2,4-dione beta-cyclodextrin complex (0.9 g). 1H-NMR (d6-DMSO): consistent with approximately 1:2 5-[4-[2-(N-methyl-N-(2-pyridyl)amino)ethoxy]benzyl]thiazolidine-2,4-dione beta-cyclodextrincomplex., XRPD: See Figure 5.
With ammonia; In water; at 60℃; for 5h;Product distribution / selectivity;
Compound 2 (3.0 g, 2.33 mmol) was dissolved in 50 ml concentrated NH4OH solution. The reaction mixture was stirred at 60° C. for 5 h, cooled to room temperature and poured into 500 ml acetone to give a white precipitate. TLC analysis performed on silica plates (EtOAc:2-propanol:conc. NH4OH:water-7:7:5:4) showed a mixture of product 4 (70percent) and beta-CD 1 (30percent). The product 4 was recrystallized from ethanol-water (3:7). The 1H NMR data are as in Example 1.
a) A separate solution is prepared by addition of 20 g of cyanuric chloride to a mixture of 100 g of water, 90 g of ice and a small amount of a defoaming agent at a temperature of 0 to 5 C. For about 5 hours the mixture is held at a temperature of 0 to 5 C. and a pH of 9 to 12 (by addition of a sodium hydroxide solution, 36%). b) A further solution is prepared by addition of 30.8 g of beta-cyclodextrin to a mixture of 40 g of water and 15.6 g of a sodium hydroxide solution. c) The solution prepared according to b) is added to the solution prepared according to a) over a period of 3 hours keeping the pH at a value of 10 to 12. Stirring is continued for 3 hours at a pH value of 8 to 10. The mixture is filtered and 14 g of the antimicrobial agent of formula (103) as well as 4.5 g of disodium hydrogen phosphate are added to the clear solution. Stirring is continued for 16 hours at a temperature of 40 C. The resulting solution having a turbid appearance is filtered by suction and the resulting clear solution is dried at a pressure of 50 mbar and a temperature of about 40 C. in a paddel drier (Venulett). Obtained are 60 g of a white product which can be applied to cotton fabric according to the procedure given in Example 2.
raloxifene hydrochloride - β-cyclodextrin[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In methanol; water;Reflux;
20.0 g of <strong>[82640-04-8]raloxifene hydrochloride</strong> (0.0392 mol), 180 g of methanol and 360 g of water were mixed together. The mass was heated at 50-60C until complete dissolution. 45.6 g of beta-cyclodextrin (mol 0,040) were then added. The mass was heated to reflux until complete dissolution, 190 g of methanol-water mixture are then distilled under atmospheric pressure. The mass was cooled to 0-30C and kept at such a temperature to assist the precipitation. The obtained product was then filtered by washing the panel with 40 g of water, and dried at 85-95C. About 58 g of <strong>[82640-04-8]raloxifene hydrochloride</strong> - beta-cyclodextrin were obtained.
b-CD (1.42 g, 1.25 mmol) or HP-b-CD (1.96 g,1.25 mmol) was dissolved in 200 mL distilled water at30 C. The HFlu solution (0.38 g, 1.25 mmol) was addeddropwise to the solutions. The resulting mixture was stirredfor an additional 24 h at room temperature and filtered. Thefiltrate was concentrated by evaporating, and the resultingprecipitate was collected and dried at 50 C for 12 h. Thewhite inclusion complexes (abbr. HFlu-b-CD and HFlu-HP-b-CD, respectively) were used to characterize by differentphysical methods thereinafter.
General procedure: A solution containing 150 mg (0.83 mmol) of inositol (d-chiro-inositol or l-chiro-inositol) or d-pinitol (150 mg, 0.77 mmol) and 150 mg of beta-CD (0.13 mmol) was incubated with 8.6 units/mL of CGTase from Thermoanaerobacter sp. in 1 mL of phosphate buffer (pH 6.0, 50 mM Na2HPO4) at 50 C for 24 h. muco- and allo-Inositols (84 mg each, 0.46 mmol) were incubated in the same conditions using 84 mg (0.074 mmol) of beta-cyclodextrin. The reaction mixtures were immersed in boiling water for 10 min to inactivate the enzyme and were stored at -18 C until subsequent analyses by TLC and HPLC and compared to blank experiments obtained with the enzyme in the absence of inositol. The products obtained after transglucosylation reactions from inositols were subjected to the action of A. niger glucoamylase. Reactions were incubated at 50 C for 24 h with 258 U/mL of the enzyme, which was directly added to the CGTase reaction medium. Samples were taken and analyzed by TLC and HPLC. The reaction was stopped by boiling for 10 min and was stored at -18 C until subsequent high-performance chromatographic separation.
In methanol; aq. phosphate buffer; at 24.99℃;pH 7.2;
General procedure: The inclusion complex formation phenomenon of CDs with guest DIMs in aqueous phosphate buffer solutions was examined at pH 7.2 by means of UV?visible spectral titration in a Shimadzu 1700 Spectrophotometer (Shimadzu Corporation, Kyoto, Japan). A 1.0 mM stock solution of DIMs (5.0 ml) was prepared in methanol, and 6.0 ll of this stock solution was added to the phosphate buffer solution to maintain the final concentration of molecules as 2.5 10 7 mol dm 3 in the cuvette. Then gradually a-, b-, c-CD were added in the cuvette so that the concentrations of CDs ranged from 0 to 12.5 10 3 mol dm 3. The absorption spectra were measured against an appropriate reagent blank.
General procedure: <strong>[480-41-1]Naringenin</strong> (0.03mM, 8.2mg) and CD (0.01mM) were completely dissolved in a mixed solution of ethanol and water (ca. 10mL, v:v=1:4, given the poor water solubility of <strong>[480-41-1]naringenin</strong>, ethanol was used), and the mixture was stirred for 5 days at room temperature. After evaporating the ethanol from the reaction mixture, the uncomplexed <strong>[480-41-1]naringenin</strong> was removed by filtration. The filtrate was evaporated under reduced pressure to remove the solvent and dried in vacuum to produce the <strong>[480-41-1]naringenin</strong>/CD complexes.
The required amounts of the DIOS:beta-CD in themolar ratio of 1:1 and1% w/w PEG 6000 were weighed accurately following the abovedescribed phase solubility study procedure. The homogenous pastes ofbeta-CD and beta-CD-/PEG were prepared in a mortar by adding water:ethanol mixture (1:1) in small quantities for binary and ternarysystems, respectively. The required amount of DIOS powder was thenadded to these pastes with continuous kneading for 2 h. An appropriateamount of water:ethanol mixture (1:1) was added further to maintainthe suitable consistency of the paste. These were dried in a hot airoven at 40-50 C for 4 h. The dried complexes were then powderedand passed through sieve No. 44 and stored in airtight containers tillfurther use.
Example 1: Preparation of amorphous <strong>[71125-38-7]meloxicam</strong>-BCD inclusion complex (or intermediate) Table 1 lists a series of experiments designed to optimize the spray drying process. Experiments 1-3 were designed to evaluate the molar ratio of <strong>[71125-38-7]meloxicam</strong> and betaCD in the spray solution. The molar ratio (<strong>[71125-38-7]meloxicam</strong> : betaCD) ranges from 1: 1.5, 1:2 to 1:2.5. The key measurement is the physical property evaluation by using DSC and XRPD, related substance (by HPLC), and processability. After the molar ratio was finalized as 1: 2 (<strong>[71125-38-7]meloxicam</strong> : betaCD), Experiments 4-8 were conducted to evaluate the effect of inlet temperature, the single most important parameter in the spray drying process. Inlet temperature testing ranges from 110C to 160C. Table 1 presents all the major evaluation data. It is of note that the dissolution of the intermediate all show significantly improvement at a number of experimental conditions (e.g.: pH 6.1 and pH 7.4), and are not substantially different from each other. Hence, the data are not listed here, though a representative set of dissolution data for the intermediate is listed in the Table 3 along with dissolution data of other testing samples. Table 1 Preparation of various intermediates through varying intermediate composition (molar ratio) and experimental conditions (inlet temperature of the spray dryer) 8 160 None None 0.64% Good: no powder stickiness *: detailed preparation see below text description For illustration purpose, the following lists a detailed description of the preparation procedure for the intermediate (Exp. 6, from Table 1), and its extensive evaluation using various instrumentation techniques: 1. Heated a solution of 500 ml water to 75C and maintained the temperature all through; then added 68.8 g betaCD to the solution (approx. 13.8% w/v), stirred till it was dissolved. Added 10ml ammonium hydroxide (cone. 28%); then added 10.64 g <strong>[71125-38-7]meloxicam</strong> (approx. 2.1% w/v), stirred till all drug particles were dissolved. This made a solid concentration approx. 16%; the molar ratio of <strong>[71125-38-7]meloxicam</strong> : betaCD was 1: 2. 2. Fed the above spray solution into the drying chamber of a spray-dryer (equipment model: SprayDryer YC-015, Shanghai Yacheng Company) through a pump and an atomizing device to form droplets. The feed flow rate was at 5ml/min; the drying gas (compressed air) was introduced into the drying chamber to dry the droplets to form powder particles with the gas flow at 2.5M3/min; the inlet temperature was set at 130C; the outlet temperature was not set, but was usually around 65- 75C; the atomizing device was pressure regulated; the separation of powder particles was carried out in a cyclone by centrifugal action. The yield was approx. 70%. 3. The material obtained above is referred to as "intermediate", i.e., <strong>[71125-38-7]meloxicam</strong>^CD inclusion complex with a molar ratio of 1:2. The intermediate was subject to oven drying at 75C for 1 hour. It was then analyzed for physical and chemical properties including: DSC, XRPD, Raman spectroscopy, SEM (scanning electron microscopy), assay, related substance, dissolution, water content, etc. 4. Evaluation using different instrumentation on physical property of <strong>[71125-38-7]meloxicam</strong>^CD inclusion complex (intermediate) as well as <strong>[71125-38-7]meloxicam</strong>, betaCD, and physical mixture (PM) reveals that the drug <strong>[71125-38-7]meloxicam</strong> is indeed entirely complexed and is in a total amorphous state: from DSC, we observed that the <strong>[71125-38-7]meloxicam</strong>'s characteristic endothermic peak (254C) was absent (see Figure 1). From XRPD, we observed that all diffraction peaks of <strong>[71125-38-7]meloxicam</strong> crystalline characteristics were absent including peaks at 13.1, 14.9, 18.6, 25.9 at 2Theta scale (see Figure 2). The Raman spectroscopy shows that the peaks are also different between the <strong>[71125-38-7]meloxicam</strong> and the intermediate (see Figure 3). The SEM pictures in Figure 4 also shows that the morphology of <strong>[71125-38-7]meloxicam</strong> and beta-CD are large crystalline particles with diameters in the range of 10-20muiotaeta, while the intermediate are showing smooth porous ball-like particles with diameters in the range of l-5um. All of the above indicate that <strong>[71125-38-7]meloxicam</strong>, once complexed with betaCD to the full extent, shows the properties of a total amorphous, which is very different from its original state as a crystalline material. The solubility testing results are also listed (see Table 2).
General procedure: The CDs were prepared using a reported method[32] the required CD (0.01 mM) and SCU (0.03 mM, 13.9 mg) were added to ultra pure water (10 mL), and the mixture was stirred for 4 days at room temperature in the dark. Then, the precipitate was removed by filtration, and the filtrate was evaporated under reduced pressure to remove the solvent and dried under vacuum to obtain the SCU/CDs complexes.
Based on the dimensions of the guest <strong>[95737-68-1]pyriproxyfen</strong> andhost CDs, given in Figs. 1 and 2, two b- or c-CD moleculescan encapsulate one <strong>[95737-68-1]pyriproxyfen</strong> molecule. In order toform, a stoichiometric IC with 1 g of <strong>[95737-68-1]pyriproxyfen</strong> at least6.05 g of a-CD, 7.06 g of b-CD, or 8.06 g of c-CD arerequired. 0.5 g of <strong>[95737-68-1]pyriproxyfen</strong> were partially dissolved in100 ml of DI water at 50 C, while an excess amount ofCDs (more than a 2:1 CD:P molar ratio) were dissolved inwater in a different flask. Finally the <strong>[95737-68-1]pyriproxyfen</strong> suspensionwas added into the CD solution and stirred at roomtemperature for 2 h. 24 h later precipitated inclusioncompound was filtered and vacuum dried at room temperaturefor 24 h before further characterization.
General procedure: To the 0.01 M solution of beta-cyclodextrin (10 mL) the solutionof (+)- or (-)-alpha-terpineol (15.4 mg, 0.01 mM) in methanol (0.5 mL)was added and the obtained solution was left at RT. Monocrystals appropriate for X-ray measurement were obtained after one week.
General procedure: <strong>[3681-99-0]Puerarin</strong> (0.6 mmol, 249 mg) was dissolved in 20 ml ethanol, CD (beta-CD, HP-beta-CD, Me-beta-CD, 0.3 mmol) were dissolved in 80 ml water, and then the CD solutions were added to the <strong>[3681-99-0]puerarin</strong> solution. The mixture was sealed and stirred for 48 h. After evaporating the ethanol from the reaction mixture, the uncomplexed <strong>[3681-99-0]puerarin</strong> was filtered. The filtrate was frozen at -40 °C for 24 h and then lyophilized. The resultant powers collected as the <strong>[3681-99-0]puerarin</strong>/CDcomplexes.
betaCD was wetted in a ceramic mortar with 2:1 (v/v) ethanol/water solution until a paste was obtained. The required amount of PRZ was then added and the slurry was kneaded for about 30 min. The ternary systems were prepared by adding 0.5 % (w/w) HPMC to the PRZ-betaCD slurry. The final product was then dried in an oven with air circulation at 40 C until constant weight.
BCN (0.057 g) was dissolved in 5 mL of methanol; 5 mL of aqueous b-CD (0.227 g) wasprepared separately. A solution of the BCN was added slowly to the b-CD solution at roomtemperature in an Ultra-sonicator and maintained for 30 min to get a homogenous solution. The mixture was warmed to 50 C for 10 min and kept at room temperature for 2 days.The solid obtained was collected and analyzed.
To prepare the IC, 0.5 g of FRAX was dissolved in acetone (4 mL), and then the solution was added to saturated beta-CyD aqueous solution at 55C drop by drop. The mixture was stirred at 50-55C for 1.5 hr, then cooled down at 4C for 12 hr. The solution was filtered and washed with water to remove the residual beta-CyD. This solid IC of beta-CyD-FRAX was further characterized by FT-IR, TGA, XRD, and elemental analyses. FT-IR measurements were recorded at 298 K with a resolution of 4 cm-1 and 64 scans within 4000 and 400 cm-1. TGA was performed using a heating rate of 10C/min under flowing air atmosphere, with in a temperature range of 50- 800C. XRD patterns were obtained over an angular range (2theta) of 4 to 50 with a 2theta 0.01313 and 30 ms/step counting time.
General procedure: Steady-state fluorescence measurements were performed employing a Hitachi F-4500 fluorescencespectrophotometer. Interactions of alpha- and beta-ZOLs with CDs were examined in 0.05 M sodium acetate (pH 5.0), in PBS (pH 7.4), and in 0.05 M sodium borate (pH 10.0) buffers at 25 C, in the presence of air. When testing the effects of CD concentration on the complex formation followed by the enhancement of fluorescence, CDs were applied at concentrations 0, 0.02, 0.05, 0.07, 0.10, 0.20, 0.50, 0.70, 1.0, 1.5 and 2.0 mM together with 2 muM alpha-ZOL or 2 muM beta-ZOL. The fluorescence emission spectra were recorded using the excitation wavelengths of 275 nm or 315 nm (lambdaem = 455 nm). Binding constants (K, with the dimension of dm3/mol) of ZOL-CD complexes were calculated employing the graphical applicationof the Benesi-Hildebrand equation assuming 1:1 stoichiometry: I0/(I-I0)=1/A+1/A x K x [CD]n (1) where I0 and I denote the fluorescence emission intensities of ZOLs in the absence and in the presenceof CDs, respectively; [CD] is the molar concentration of the host molecule, while A is a constant and nis the number of binding sites.
The 1-CD inclusion complex was prepared by reacting equimolar amounts of aqueous solutions of 1 and -CD.Compound 1 (0.162 g, 0.001 mol) in EtOH (10 mL) was treated with -CD (1.135 g, 0.001 mol) in H2O (30 mL) and stirredon a magnetic stirrer at 50C for 5 h. The resulting precipitate was filtered off, rinsed with Me2CO, and dried at roomtemperature to give 1-CD as a white powder, mp 280C (dec.).
beta-Cyclodextrin (3.6 g, 2.7 mmol) was dissolved in DMSO(20 ml) until complete solubility. Succinic dihydrazide(0.2 ml, 1.4 mmol) was added with stirring for 3 h. Afterthat the 1,4,5,6,7,7-Hexachloro-5-norbornene-2,3 dicarboxylicanhydride (1 g, 2.7 mmol) was added. The reactionmixture was stirred for 24 h. White precipitate was formedthen filtered off and wash with distilled water to remove anyunreacted beta-CD. The product was dried to give [2] rotaxane(2) as white crystals (77%)
Solid-state samples of MBZ A and C, in equimolar ratio with beta-CD,were prepared as follows.2.2.1. Freeze drying (FD)The binary complexes of MBZ, A and C (FD A and FD C, respectively),were prepared by mixing the reagents with an ethanol:watersolution (80:20 v/v). The resulting suspension was sonicated at constanttemperature (25.0 ± 0.1 C) until the dissolution was complete.Then, ethanol was removed from the solution by rotary evaporation at65 C (Rotavapor Decalab. Argentina). The aqueous suspension wasfrozen at -40 C for 24 h, and freeze-dried using a Rificor lyophiliser(model L-A-B5).
For the structural characterizations by FTIR, TGA/DTA and NMR, the inclusion complex (CPC/betaCD) was prepared by dissolution equimolar amounts of the guest molecule (CPC) and betaCD in Milli-Q water, followed by stirring for 24 h and freezing and lyophilization [21,22].
Accurately weigh 1.134g (1mmol) of beta-cyclodextrin and 0.482g (1mmol) of <strong>[755037-03-7]regorafenib</strong> nto the eggplant bottle, add 40ml of 70% ethanol to the water bath and stir to a temperature of 85 C. Stirring is continued after mixing, and the stirring time is not less than 24 hours. Then, it is taken out and placed at room temperature (15-30 C), and allowed to stand for crystallizing, and the crystallization time is about 8 to 10 hours until the crystallization is complete, suction filtration, and drying (25 C vacuum drying).
In aq. phosphate buffer; at 24.99℃; for 36h;pH 7.4;Thermodynamic data;
General procedure: Solubility of all the required compounds was specifically checked in triply distilled and de-ionized water. Mettler Toledo AG-285 having uncertainty±0.0003g was used to prepare all the solutions of THC, HSA, alpha-and beta-CD by mass at room temperature. All the stock solutions were prepared by mass dilution and freshly prepared solutions were used during each experiment in phosphate buffer aqueous solution of pH 7.4. Sufficient precautions had been taken during measuring weights, preparing solutions and performing all the respective experiments. Two solid ICs, THC+ alpha-CD and THC+beta-CD had been prepared in 1:1M ratio of THC and CD. 1.0mM of alpha-and beta-CD were each separately mixed with water and stirred for 4h. After that the aqueous solution of 1.0mM of THC was added dropwise to the respective solutions of CD and left for stirring near about 36h at 50-55C to prepare the corresponding two ICs. Just after filtration of the hot solutions, it is allowed to cool down to 5C and kept for 12h without any disturbing. The obtained suspension was then filtered and washed with ethanol and dried in air to get white polycrystalline powder.
A precipitation method as described in literatures [5, 6] with some modifications was adopted to fabricate di<strong>[4437-20-1]furfuryl disulfide</strong>-beta-CD inclusion complex. 10 g of beta-CD was addedin 80 g of deionized water at the temperature of 40 C. The mixture was stirred to form a suspension. Then, 4 g di<strong>[4437-20-1]furfuryl disulfide</strong> was slowly added to the beta-CD suspension. To form di<strong>[4437-20-1]furfuryl disulfide</strong>-beta-CD inclusion complex, the mixture was continuously stirred for 3 h at the temperature of 40C. The suspension was refrigerated overnight at 5 C andthe cold precipitated material was recovered by vacuum filtration.The precipitate was washed with anhydrous ethanol, and was dried in a FD-1A-50 freeze drier (Shanghai Bilang instrument Manufacturing Co., Ltd., Shanghai, China) for 24 h at a temperature of -56 C and pressure of around 24 Pa. The dried sample was stored in a desiccator for further FTIR and TG analysis.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






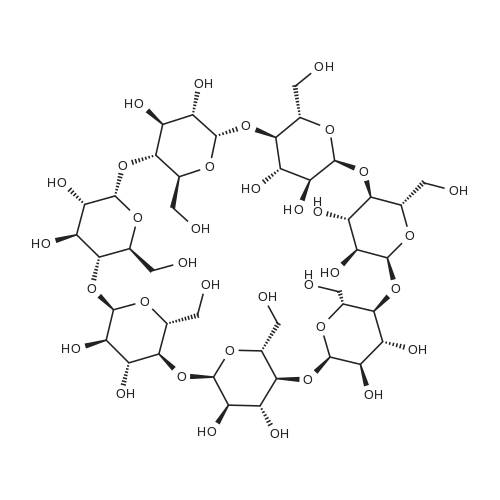

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping