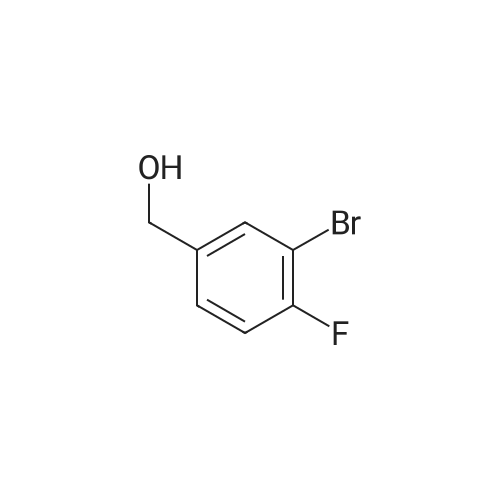
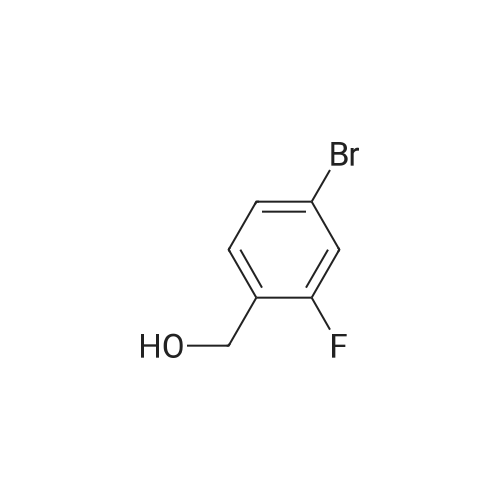
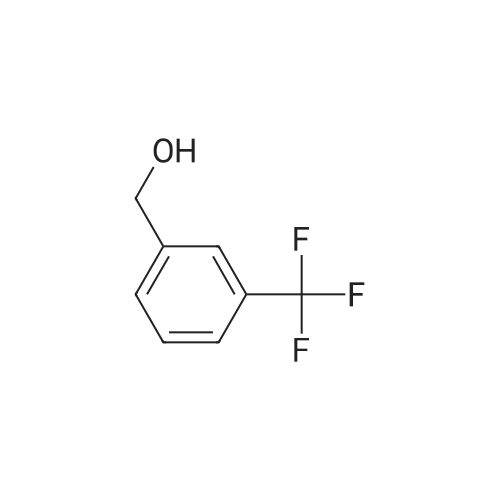
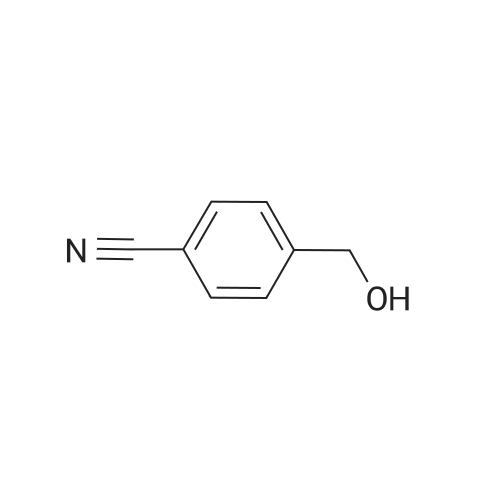
| 65% |
With iron(III) chloride In dichloromethane at 25℃; for 2h; Inert atmosphere; |
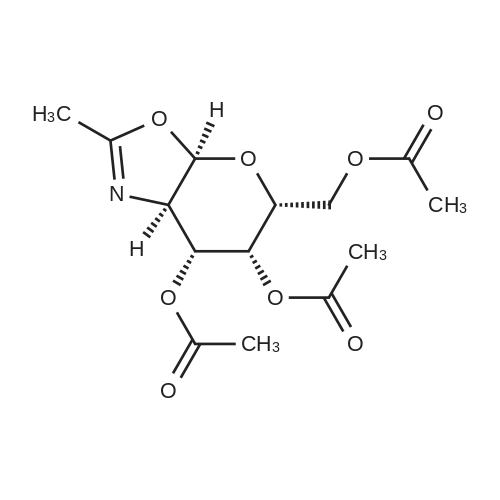
Into a 50 L 3-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed a solution of G7-3 (220 g, 565 mmol, 1.00 equiv.) in dichloromethane (22 L). This was followed by the addition of ferric chloride (275 g), in portions at 25°C. The resulting solution was stirred for 2 h at 25°C. The reaction was then quenched by the addition of 17 L of water/ice. The resulting solution was washed with water and saturated aqueous sodium chloride. The mixture was dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. This resulted in 121 g (65%) of G7-4. |
| 64% |
With trimethylsilyl trifluoromethanesulfonate In 1,2-dichloro-ethane at 50℃; for 16h; |
1
Alternatively, the following procedures were used to provide 4 (eg, 2-methyl-(3,4,6-tri-O-acetyl-1,2-dideoxy-α-D-galactopyrano)[1,2d]-1,3-oxazoline).galactosamine penta-acetate 15 (2Mg, 5.15 rnmol) was dissolved in dichioroethane (DCE) (2() mL), Then trirnethylsilyl trifluoromethanesufonate (TMSOTf (1 ml, 5.53 mmol) was added, and the mixture was stirred at 50°C for 9h. The mixture was then removed from the heat and stirred for 7h. Triethylarnine (2m1) was added to the mixture at room temperature. The mixture was then washed with a saturated so’ution of NaHCO3 and then dried with sodium sulfate. The organic phase was then filtered and the solvent was removed via rotary evaporation and the residue was loaded onto silica gel. The product was purified via column chromatography on silica gel with EtOAc (100) to yield 16 as a yellow viscus solid, (Yield:64%) IH NMR: (400 MHz, CDCI3-d6): ö (ppm), 5.97 (d, J=69 Hz, 1H, H-4); 5.45 (t, J=3,O Hz, 1H, H5); 4.92 (dd, J=7,6 Hz, 14 Hz, 1H, H-4); 4.26 (td, J=63 Hz, 2.8 Hz, 1W); 425- 4J3 (m, IH, H-3); 3.99 (s, IH); 2.13 (s, 3H); 2M7 (s, 6H); 2M5 (s, 3Ff). 13C NMR: (125 MHz, DMSOd6): ö (ppm), 170M; 169.55; [68.11; 165.21; 100.9; 70.66; 68.2; 65.02, 63.00, 61.8, 20.5, 20.44, 20.42, 13.91. MS miz: [M + H]± 330.12. |
| 62% |
With trimethylsilyl trifluoromethanesulfonate In 1,2-dichloro-ethane at 50℃; Inert atmosphere; |
|
| 54% |
With trimethylsilyl trifluoromethanesulfonate In 1,2-dichloro-ethane at 50℃; for 9h; Inert atmosphere; |
|
|
With iron(III) chloride In dichloromethane at 25℃; for 2h; Inert atmosphere; |
1-2 Synthesis of (3aR.5R.6R.7R.7aR)-5-(acetoxymethyl)-2-methyl-5,6.7.7a-tetrahvdro-3aH- pyrano|"3.2-d"|oxazole-6,7-diyl diacetate (Compound 7)
Synthesis of (3aR.5R.6R.7R.7aR)-5-(acetoxymethyl)-2-methyl-5,6.7.7a-tetrahvdro-3aH- pyrano|"3.2-d"|oxazole-6,7-diyl diacetate (Compound 7) Into a 2000-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen was placed a solution of (3R,4R,5R,6R)-3-acetamido-6- (acetoxymethyl)tetrahydro-2H-pyran-2,4,5-triyl triacetate (Compound 6, 30 g, 77.05 mmol, 1.00 equiv) in dichloromethane (1500 mL), then added iron (III) chloride (30 g, 184.95 mmol, 2.40 equiv). The resulting mixture was stirred for 2 h at 25°C. The reaction was then quenched by the addition of 1000 mL of water/ice. The organic layer was washed with 1x1000 mL of sodium aq. bicarbonate and 1x1000 mL of water, dried over anhydrous sodium sulfate and concentrated under vacuum. This resulted in (3aR,5R,6R,7R,7aR)-5- (acetoxymethyl)-2-methyl-5,6,7,7a-tetrahydro-3aH-pyrano[3,2-d]oxazole-6,7-diyl diacetate (Compound 7) as yellow oil. ^MR CDCb, 300MHz, ppm): 2.03(s, 9H), 2.12(s, 3H), 3.97-4.27(m, 4H), 4.90-4.93(m, J = 3.3Hz, 1H), 5.45-5.47(t, J= 3.0Hz, 1H), 5.98-6.00(d, J= 6.6Hz, 1H). |
|
With iron(III) chloride In dichloromethane at 25℃; for 2h; Inert atmosphere; |
A.1-2 Synthesis of (3aR,5R,6R,7R,7aR)-5-(acetoxymethyl)-2-methyl-5,6,7,7a-tetrahvdro-3aH- pyranor3,2-d1oxazole-6,7-diyl diacetate (Compound A7)
Synthesis of (3aR,5R,6R,7R,7aR)-5-(acetoxymethyl)-2-methyl-5,6,7,7a-tetrahvdro-3aH- pyranor3,2-d1oxazole-6,7-diyl diacetate (Compound A7) Into a 2000-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen was placed a solution of (3R,4R,5R,6R)-3-acetamido-6- (acetoxymethyl)tetrahydro-2H-pyran-2,4,5-triyl triacetate (A6, 30 g, 77.05 mmol, 1.00 equiv) in dichloromethane (1500 mL), then added iron (III) chloride (30 g, 184.95 mmol, 2.40 equiv). The resulting mixture was stirred for 2 h at 25°C. The reaction was then quenched by the addition of 1000 mL of water/ice. The organic layer was washed with 1x1000 mL of sodium aq. bicarbonate and 1x1000 mL of water, dried over anhydrous sodium sulfate and concentrated under vacuum. This resulted in (3aR,5R,6R,7R,7aR)-5-(acetoxymethyl)-2- methyl-5,6,7,7a-tetrahydro-3aH-pyrano[3,2-d]oxazole-6,7-diyl diacetate (Compound A7) as yellow oil. 1HNMR(CDC13, 300MHz, ppm): 2.03(s, 9H), 2.12(s, 3H), 3.97-4.27(m, 4H), 4.90-4.93(m, J = 3.3Hz, 1H), 5.45-5.47(t, J= 3.0Hz, 1H), 5.98-6.00(d, J= 6.6Hz, 1H). |
|
With trimethylsilyl trifluoromethanesulfonate In 1,2-dichloro-ethane at 20 - 55℃; Inert atmosphere; |
1.NAG7 Preparation of benzyloxycarbonylbutyl 2-deoxy 2-A/-acetyl -3,4,6-tri-0-acetyl^-D- galactopyranoside (NAG7) - Method A.
Under an inert atmosphere, TMSOTf (8.56 g, 38.4 mmol) was added to a solution of NAG2 (10.0 g, 25.6 mmol) in DCE (100 mL) at ambient temperature. The mixture was stirred at 55 °C for 2 h, removed from heat, and stirred overnight. The reaction mixture was poured onto ice cold sat NaHC03 (aq.) and extracted with CH2CI2. The organic layer was dried over Na2S04 andconcentrated in vacuo to give syrup NAG6. A solution NAG6 in DCE (60 ml_) was charged with alcohol NAG5 (8.00 g, 38.4 mmol) and molecular sieves. The mixture was placed under an inert atmosphere, treated with TMSOTf (2.85 g, 12.8 mmol), and stirred overnight at rt. The mixture was poured over ice cold sat NaHC03 (aq.) and extracted with CH2CI2. The organic layer was dried over Na2S04 and concentrated in vacuo to give syrup. This crude material was purified by Si02 gel chromatography to afford glycoside NAG7 (3.3 g, 24% yield). H NMR (CDCI3, 500 MHz): δ 7.35 (m, 5H), 5.98 (d, 1 H, J 7.0 Hz), 5.57 (m, 1 H), 5.34 (d, 1 H, J 3.0 Hz), 5.25 (dd, 1 H, J 3.0 Hz, 1 1 Hz), 5.10 (s, 2H), 4.63 (d, 1 H, J 8.5 Hz), 4.1 1 (m, 2H), 3.95 (m, 1 H), 3.88 (m, 2H), 3.49 (m, 1 H), 2.37 (m, 2H), 2.13 (s, 3H), 2.03 (s, 3H), 1.99 (s, 3H), 1.90 (s, 3H), 1.70 (m, 2H), 1.61 (m, 2H). |
|
With iron(III) chloride In dichloromethane at 25℃; for 2h; Inert atmosphere; |
Synthesis of (3aR,5R,6R,7R,7aR)-5-(acetoxymethyl)-2-methyl-5,6,7,7a-tetrahydro-3aH- pyrano[3.2-d]oxazole-6.7-diyl diacetate (Compound A7)
Synthesis of (3aR,5R,6R,7R,7aR)-5-(acetoxymethyl)-2-methyl-5,6,7,7a-tetrahydro-3aH- pyrano[3.2-d]oxazole-6.7-diyl diacetate (Compound A7)Into a 2000-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen was placed a solution of (3R,4R,5R,6R)-3-acetamido-6-(acetoxymethyl)tetrahydro-2H-pyran-2,4,5-triyl triacetate (A6, 30 g, 77.05 mmol, 1.00 equiv) in dichloromethane (1500 mL), then added iron (III) chloride (30 g, 184.95 mmol, 2.40 equiv). The resulting mixture was stirred for 2 h at 25°C. The reaction was then quenched by the addition of 1000 mL of water/ice. The organic layer was washed with 1x1000 mL of sodium aq. bicarbonate and 1x1000 mL of water, dried over anhydrous sodium sulfate and concentrated under vacuum. This resulted in Compound A7 as an oil. 1H NMR (CDCl3, 300MHz, ppm): 2.03(s, 9H), 2.12(s, 3H), 3.97-4.27(m, 4H), 4.90-4.93(m, J= 3.3Hz, 1H), 5.45-5.47(t, J= 3.0Hz, 1H), 5.98-6.00(d, J= 6.6Hz, 1H). |
|
With iron(III) chloride In dichloromethane at 25℃; for 2h; Inert atmosphere; |
Synthesis of (3 aR,5R,6R,7R,7aR)-5 -(acetoxymethyl)-2-methyl-5 ,6,7,7a-tetrahydro-3 aH20 pyrano 13 ,2-d] oxazole-6,7-diyl diacetate (Compound AT)
Into a 2000-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen was placed a solution of (3R,4R,5R,6R)-3-acetamido-6- (acetoxymethyl)tetrahydro-2H-pyran-2,4,5-triyl triacetate (A6, 30 g, 77.05 mmol, 1.00 equiv) in dichloromethane (1500 mL), then added iron (III) chloride (30 g, 184.95 mmol, 2.40 equiv). The25 resulting mixture was stirred for 2 h at 25°C. The reaction was then quenched by the addition of1000 mL of water/ice. The organic layer was washed with lx 1000 mL of sodium aq. bicarbonate and lxl000 mL of water, dried over anhydrous sodium sulfate and concentrated under vacuum. This resulted in Compound A7 as an oil.‘HNMR(CDC13, 300MHz, ppm): 2.03(s, 9H), 2.12(s, 3H), 3.97-4.27(m, 4H), 4.90-4.93(m, J= 30 3.3Hz, 1H), 5.45-5.47(t, J= 3.0Hz, 1H), 5.98-6.00(d, J= 6.6Hz, 1H). |
|
With trimethylsilyl trifluoromethanesulfonate In 1,2-dichloro-ethane at 50℃; for 12h; Inert atmosphere; |
|
|
With trimethylsilyl trifluoromethanesulfonate In dichloromethane at 40 - 45℃; for 21h; |
1.2 Step 2
To suspension of peracetylated galactosamine (1 .63g, 4.17mmol) in 30mL DCM. TMSOTf (1 .90mL, 10.4mmol) was added and reaction mixture stirred at 40-45°C for 5 h. An additional portion of TMSOTf (0.30 mL 2.8 mmol) was added and the reaction mixture was stirred for additional 16 h. Then the reaction was quenched with NEt3 (0.90 mL) at 0 °C and diluted with DCM, extracted with cold sat. NaHC03 (2x100 mL), brine (50 mL), dried over Na2S04 and evaporated. Product 1 isolated as a yellow oil, 1 .40 g crude yield. |
| 1.35 g |
With trimethylsilyl trifluoromethanesulfonate In dichloromethane at 40℃; for 24h; |
13 Synthesis of (3aR,5R,6R,7R,7aR)-5-(acetoxym-ethyl)-2-methyl-5,6,7,7a-tetrahydro-3aR-pyrano[3,2-d]oxazole-6,7-diyl diacetate (2)
The crude mixture of compound 1(2.0 g, 5.139 mmol) was dissolved in DCM (25 mE) and TMSOTf (1.85 mE, 10.27 mmol) was added and reaction mixture stirred at 40°C. for 24 hours. The reaction was quenched with Et3N (0.4 mE, pH 7.5), diluted in DCM and extracted with sat. NaRCO3 solution. The organic layer was washed with water, brine and dried over Na2504 before evaporation of solvents under reduced pressure and the residue purified by silica gel column chromatography using DCM:EA:MeOR (7.5:2.0:0.5) as an eluent to obtain compound 2 (1.35 g, 82% yield). ‘R-NMR (500 MHz, CDC13): ö 2.03 (s, 3R, H7), 2.064 (s, 3R, CR3, OAc), 2.067 (s, 3R, CR3, OAc), 2.08 (s, 3R, CR3, OAc), 4.12 (dd, 1R, J=6.5 & 12.0 Hz, H5), 4.18 (d, 1R, J=9.0 Hz, H3), 4.32 (dd, 1R, J=3.5 & 12.0Hz, H5), 4.52 (dd, 1R, J=1.5 & 5.5 Hz, H2), 5.00 (m, 1R, H5), 5.14 (s, 1R, H4), 6.12 (d, 1R, J=6.5 Hz, H1); ‘3C-NMR (125 MHz, CDC13): ö 14.30 (C7), 20.6, 20.7,20.75 (OAc, 3CR3), 62.8 (C5), 69.6 (C2), 76.4 (C3), 77.6 (C5), 84.3 (C4), 107.3 (C1), 167.05 (C8), 169.6, 169.8, 170.3 (OAc, CO). EI-MS: [M+R] C,4R2QN08 calcd 330.12, obsd 330.11, [M+Na] C,4R,9NNaO8 calcd 352.10, obsd 352.11. |
|
With trimethylsilyl trifluoromethanesulfonate In dichloromethane for 24h; Reflux; |
|
| 26.9 g |
With trimethylsilyl trifluoromethanesulfonate In 1,2-dichloro-ethane at 20℃; Inert atmosphere; Cooling with ice; |
1.1-1b (1-1b) Synthesis of GAL-3
Dissolve the GAL-2 (35.1g, 90.0mmol) obtained in step (1-1a) in 213ml of anhydrous 1,2-dichloroethane,Under ice-water bath and nitrogen protection, 24.0 g of TMSOTf (CAS No.: 27607-77-8, purchased from Macleans Company, 108.0 mmol) was added and reacted at room temperature overnight.Add 400ml of dichloromethane to the reaction solution to dilute, filter with diatomaceous earth,Then add 1L saturated sodium bicarbonate aqueous solution, stir evenly, separate the organic phase, and extract the aqueous phase with dichloroethane twice, 300ml each time,The organic phases were combined and washed with 300 ml of saturated aqueous sodium bicarbonate solution and 300 ml of saturated brine respectively. The organic phase was separated, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure to obtain 26.9 g of light yellow viscous sugar thin product GAL-3. |
| 26.9 g |
With trimethylsilyl trifluoromethanesulfonate In 1,2-dichloro-ethane at 20℃; |
1.1-1b (1-1b) Synthesis of GAL-3
The GAL-2 (35.1g, 90.0mmol) obtained in step (1-1a) was dissolved in 213ml of anhydrous 1,2-dichloroethane, and 24.0g of TMSOTf(CAS No.: 27607-77-8, purchased from Macleans Company, 108.0 mmol), reacted at room temperature overnight.Add 400ml of dichloromethane to the reaction solution to dilute, filter with diatomaceous earth, then add 1L of saturated sodium bicarbonate aqueous solution, stir evenly, separate the organic phase, and extract the aqueous phase with dichloroethane twice, 300ml each time, and combine The organic phase was washed with 300 ml of saturated sodium bicarbonate aqueous solution and 300 ml of saturated brine respectively, the organic phase was separated, dried with anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure to obtain 26.9 g of light yellow viscous syrupy product GAL-3. |
| 26.9 g |
With trimethylsilyl trifluoromethanesulfonate In 1,2-dichloro-ethane at 20℃; Cooling with ice; Inert atmosphere; |
1.1-1-1; 1.1-1-1b (1-1-1b) Synthesis of GAL-3
Dissolve the GAL-2 (35.1g, 90.0mmol) obtained in step (1-1-1a) in 213ml of anhydrous 1,2-dichloroethane, Under ice-water bath and nitrogen protection, 24.0g TMSOTf (CAS number: 27607-77-8, purchased from Macleans Company, 108.0 mmol) was added, and reacted at room temperature overnight. Add 400ml of dichloromethane to the reaction solution to dilute, filter with diatomaceous earth, then add 1L of saturated sodium bicarbonate aqueous solution, stir well, separate the organic phase, The aqueous phase was extracted twice with 300ml of dichloroethane each time, and the organic phases were combined and washed with 300ml of saturated aqueous sodium bicarbonate solution and 300ml of saturated brine, respectively. Separate the organic phase, dry with anhydrous sodium sulfate, and evaporate the solvent under reduced pressure to obtain 26.9 g of light yellow viscous sugar thin product GAL-3. |
|
With trimethylsilyl trifluoromethanesulfonate In 1,2-dichloro-ethane at 0 - 50℃; for 3.16667h; |
3.2
Step 2: To a solution of (3R,4R,5R,6R)-3-acetamido-6-(acetoxymethyl)tetrahydro-2H-pyran- 2,4,5-triyl triacetate (100 g, 0.257 mol) in 1 ,2-dichloroethane (500 mL) cooled to 0 °C was added TMSOTF (85.5 g, 0.385 mol), the mixture was stirred for 10 min, then heated to 50 °C and stirred for 3 hours. TLC showed the start material was completely consumed. After cooling, the resultant mixture was treated with sat. aqueous NaHCO3 (1000 mL) at 0 °C, extracted with DCM (500 mL x 2). The combined organic layers were dried over Na2SO4 and concentrated. The residue was dried under high vacuum overnight to give (3aR,5R,6R,7R,7aR)-5-(acetoxymethyl)-2-methyl-3a,6,7,7a-tetrahydro-5H-pyrano[3,2- d]oxazole-6,7-diyl diacetatel. 1H NMR (400 MHz, CDCIa) δ ppm 6.00 (d, 1 H, J = 2.8Hz), 5.47- 5.46 (m, 1 H), 4.93-4.90 (m, 1 H), 4.27-4.18 (m, 2H), 4.13-4.09 (m, 1 H), 4.02-3.98 (m, 1 H), 2.13 (s, 3H) , 2.07 (s, 6H), 2.06 (s, 3H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping