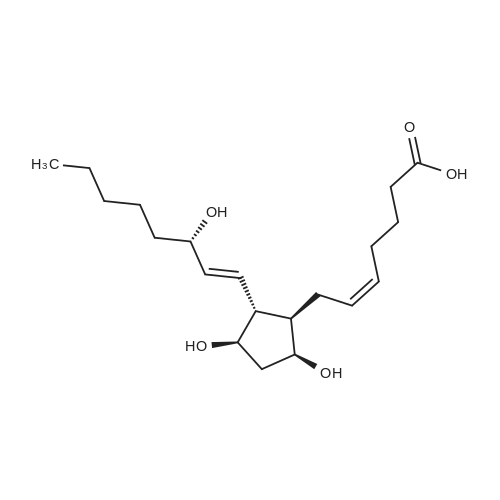
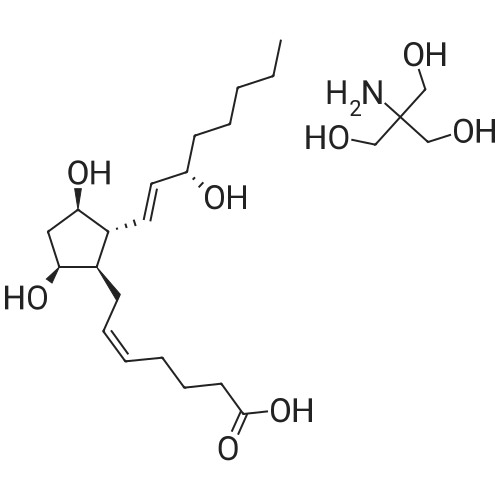
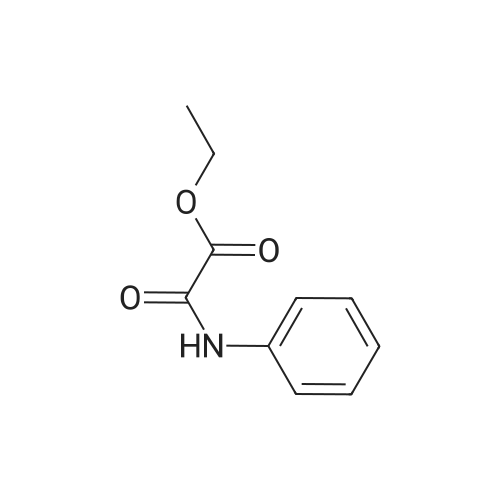
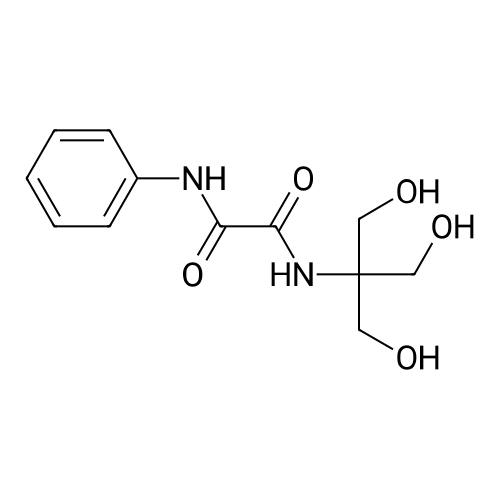
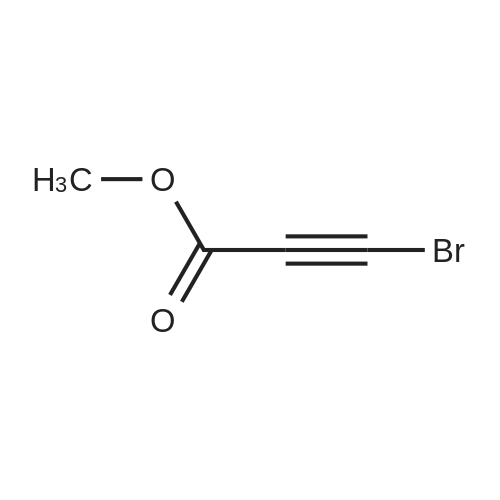
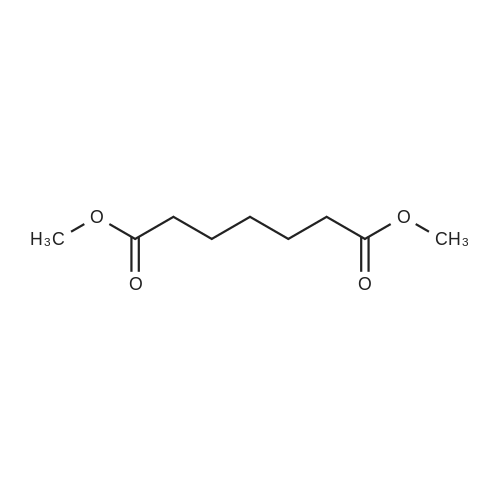
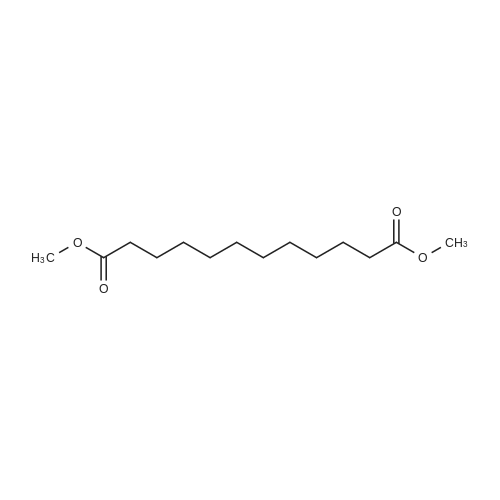
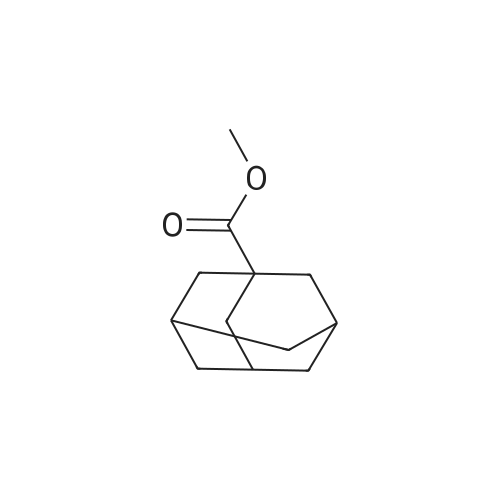
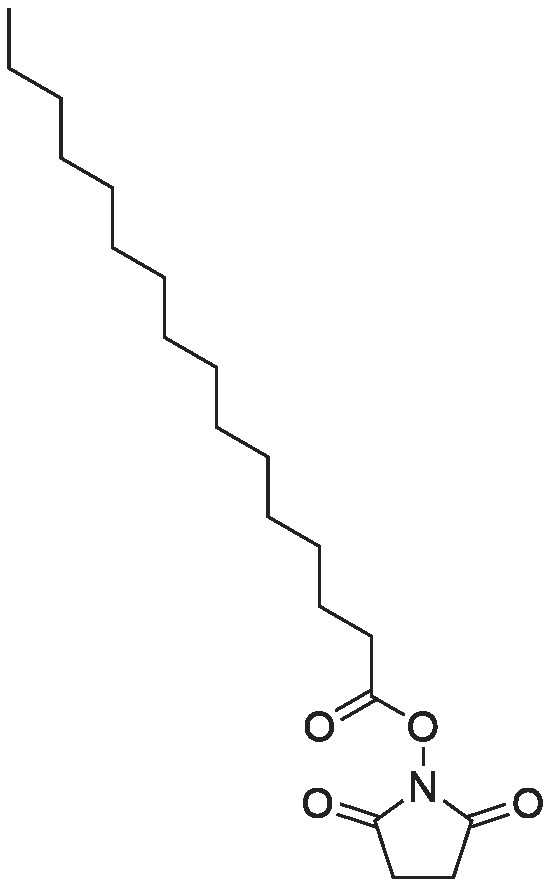
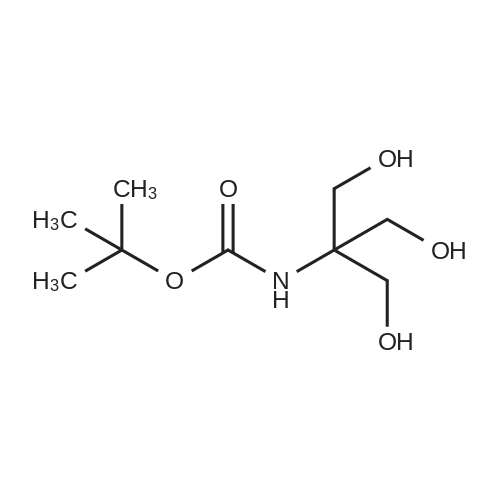
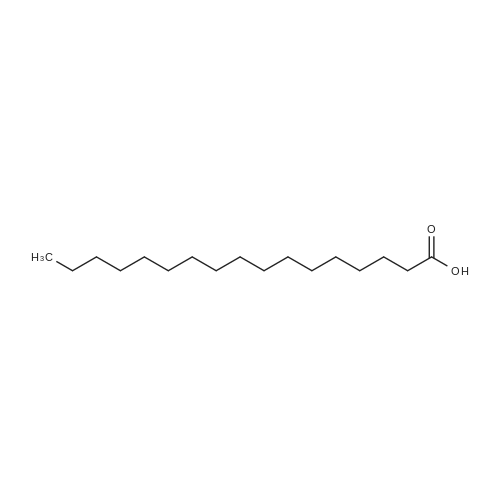
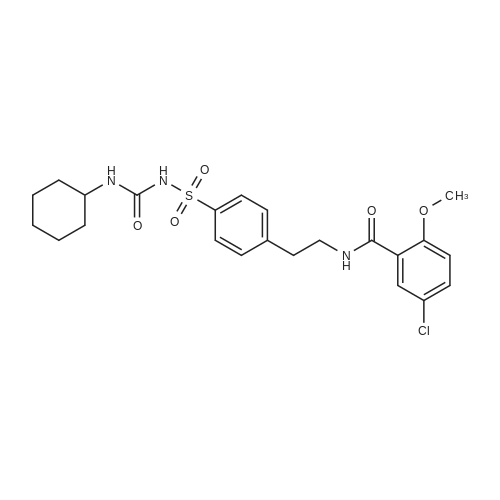
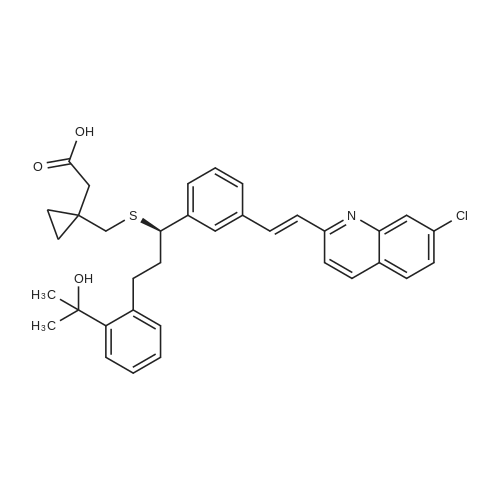
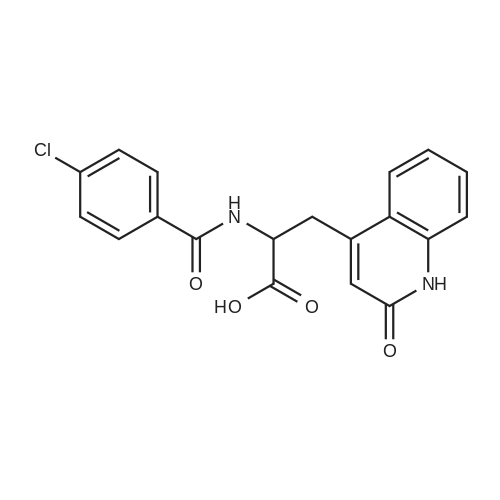
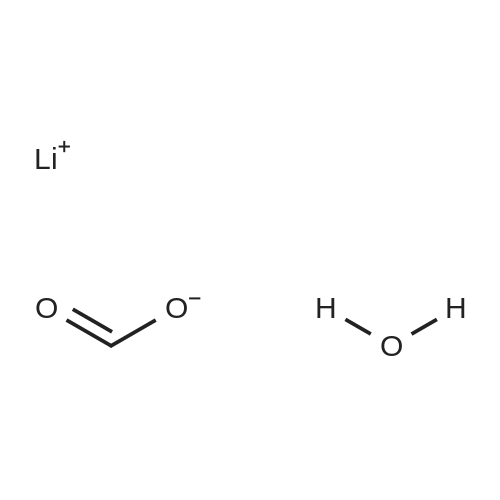
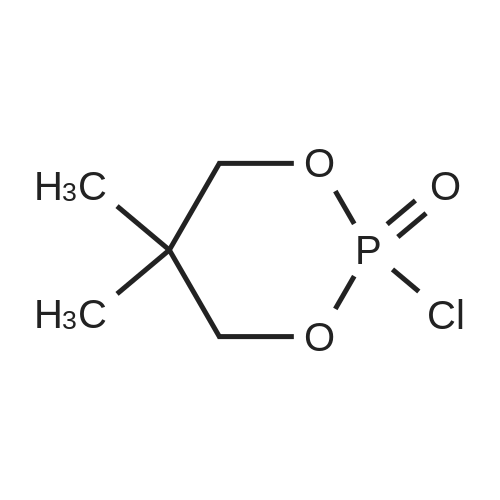
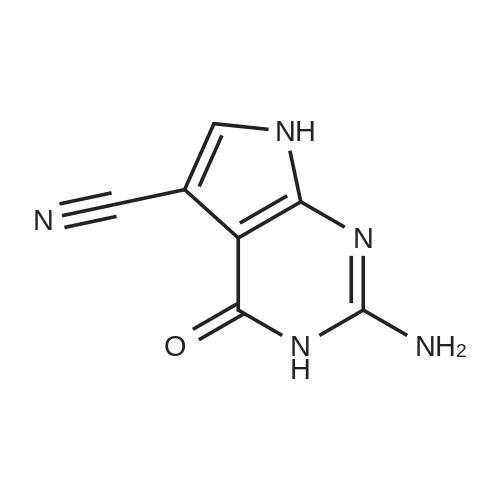
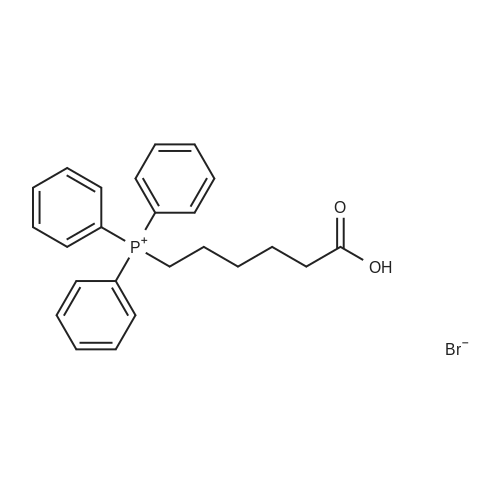
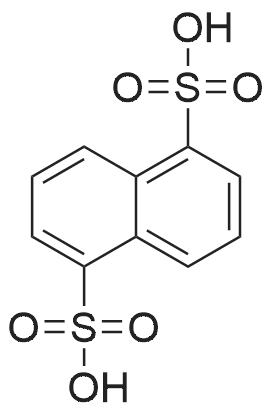
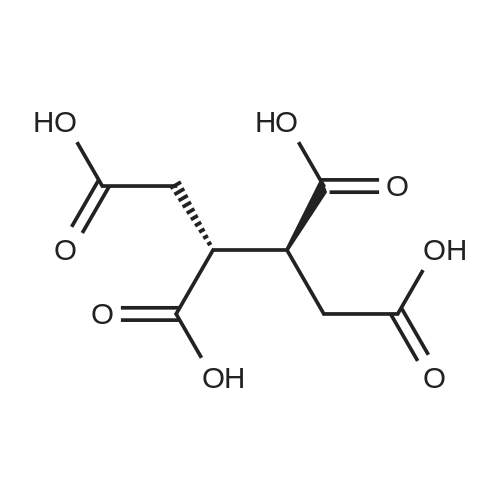
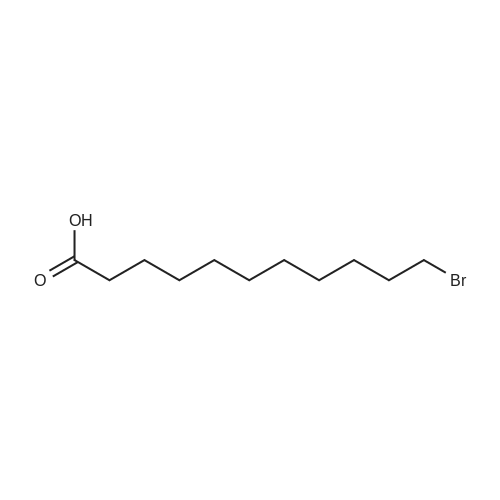
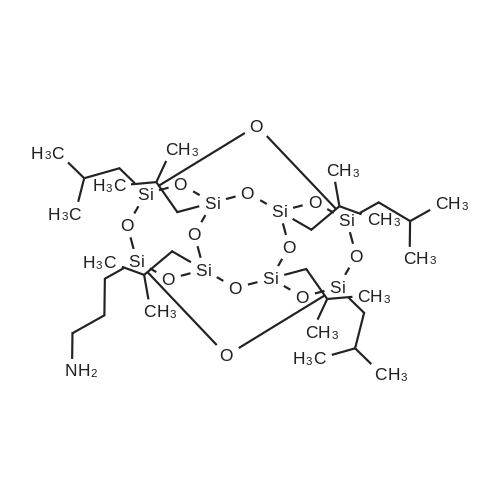
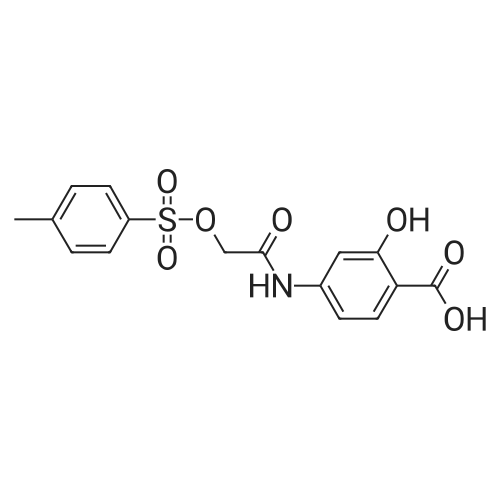
| 99% |
With trimethylamine; In dichloromethane; for 4.0h; |
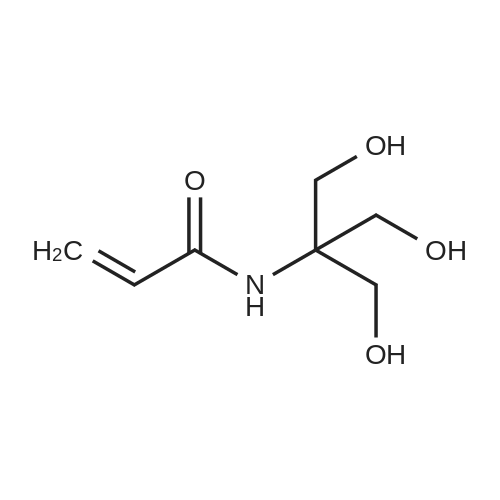
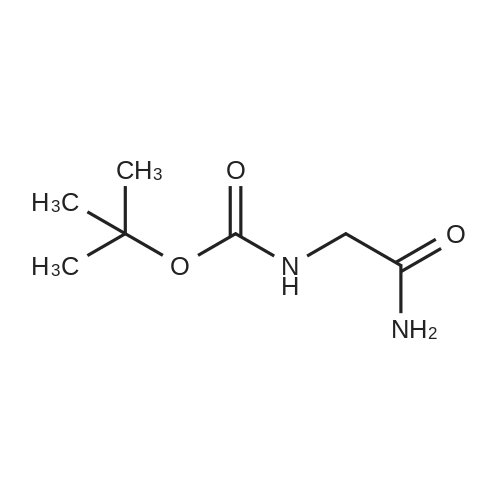
Tris base (5,00 g 41,3 mmol) was dissolved in diehloromethane (80 mL) and trimcthylamine (20 mL). Di-tert-butyl diearbonate (10.81 g, 49.6 mmol, f .2 eq) was then added, and the reaction stirred for 4 hours. The mixture was evaporated arid the residue portioned between ethyl acetate and water. The organic fraction was washed with wate { lx), M BC1 (2x), saturated sodium bicarbonate (lx), and brine (lx) before drying over sodium sulfate and evaporation to give compound 23 (9.04g, 40.9 mmol) in 99% yield, which was used without purification in further steps. |
| 94% |
In water; tert-butyl alcohol; at 20℃; for 24.0h; |
A solution of Boc2O (2.39 g, 10.7 mmol) in tBuOH (10 mL) was added to a suspension of TRIS (1.01 g, 8.30 mmol) in tBuOH:H2O (1:1, 15 mL) and the reaction mixture was stirred at r.t. for 1 d. The solvent was removed under vacuum and the product was purified by precipitation with cold EtOAc. Compound 17 (1.72 g, 7.77 mmol, 94%) was obtained as a white solid. |
| 93% |
In methanol; tert-butyl alcohol; at 20℃; for 20.0h; |
To a suspension of <strong>[77-86-1]tris(hydroxymethyl)aminomethane</strong> (3) (10.0 g, 63.0 mmol) in t-BuOH (100mL), a mixture of di-tert-butyl dicarbonate (18.0 g, 82.4 mmol) in 1:1 mixture of MeOH : t-BuOH (160mL) was added slowly and the reaction mixture was allowed to stir at room temperature for 20h. After 20h, the solvents are evaporated in vacuo to give a crude white residue which is recrystallized from cold ethyl acetate. Vacuum filtration of the white solid subsequently afforded the pure white product 4 (13.0g,93%). 1H NMR (500 MHz, DMSO-d6) delta: 5.77 (br s, 1H, NH), 4.50 (t, 3H, J 5.5 Hz, 3 × OH), 3.53 (d, 6H,J 6.0 Hz, CH2OH), 1.38 [s, 1H, 3 × C(CH3)3]. 13C NMR (125 MHz, DMSO-d6) delta: 155.5 (COCH2), 78.3,60.9(2), 60.7, 28.7(3). HRMS calcd. for C9H19O5NNa (M+Na)+: 221.1263, found: 221.1267. |
| 93% |
In methanol; tert-butyl alcohol; at 20℃; |
5 g of tris(hydroxymethyl)methylaminomethane was dissolved in a mixed solution of 30 mL of methanol and 30 mL of t-butanol, A solution of BOC anhydride (11.75 g) tert-butanol (50 mL) was added with stirring, Room temperature reaction overnight, Remove the solvent, Add ethyl acetate to cool overnight, Filtration gave a white solid 8 (8.5 g, 93%). |
| 64% |
In methanol; tert-butyl alcohol; at 20℃; for 22.0h; |
To a suspension of <strong>[77-86-1]tris(hydroxymethyl)aminomethane</strong> (2 g, 16.5 mmol) in t-BuOH (20 mL), a mixture of di-tert-butyl dicarbonate (3.6 g, 16.4 mmol) in 1:1 mixture of MeOH:t-BuOH (32 mL) was added slowly and the reaction mixture could stir at room temperature for 22h. Then, the solvents were evaporated in vacuo to give a crude white residue which was recrystallized from cold ethyl acetate. Vacuum filtration of the white solid subsequently afforded pure tert-butyl (1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl)carbamate in 64% yield. To a solution of protected <strong>[77-86-1]tris(hydroxymethyl)aminomethane</strong> (2.32 g, 10.5 mmol) in dry DMF, propargyl bromide (80 wt.% in toluene) (5.7 mL, 364.3 mmol) was added and the reaction mixture was stirred at 0 C for 10 min. It was followed by the addition of finely powdered KOH (3.56 g, 63.4 mmol) in small portions. The reaction mixture was then stirred at room temperature for 40h. To the resulting brown coloured mixture, ethyl acetate was added and stirred for another 10 min. Further, the entire reaction mixture was washed successively with H2O (2 × 10 mL) and brine (10 mL). The organic layer was then collected, dried over anhydrous MgSO4 and concentrated under reduce pressure. The crude material thus obtained was purified by flash chromatography (n-hexane: EtOAc) to afford compound 28 as a yellowish powder (1.56 g, 4.7 mmol, 44%). 1H NMR (300 MHz, CDCl3) delta: 4.93 (1H, br s, NH), 4.15 (6H, d, J 2.3 Hz, 3 × CH2CCH), 3.79 (6H, s, CH2OH), 2.43 (3H, t, J 2.3 Hz, CCH), 1.43 (1H, s, 3 × CH3-Boc). 13C NMR (75 MHz, CDCl3) delta: 154.7 (COCH2), 79.6, 74.5, 68.8, 58.6, 58.0, 28.3. HRMS calcd. for C18H25NNaO5+ 358.1625, found 358.1625 [M + Na+]. |
| 50% |
In methanol; tert-butyl alcohol; at 20℃; for 24.0h; |
A solution of 554 Boc anhydride (81.2 mL, 619.0 mmol) in 555 tert-butanol (150 mL) was charged with a solution of 556 <strong>[77-86-1]2-amino-2-(hydroxymethyl)propane-1,3-diol</strong> A (50 g, 413.00 mmol) in mixture of tert-butanol: 124 methanol (1:1, 250 mL) and stirred at room temperature for 24 h. The reaction mixture was concentrated in vacuo, resulting in the crude residue as white powder which was purified by recrystallisation in 236 ethanol to afford 45.6 g, 50% yield of the title compound as a white solid. 1H NMR (400 MHz, DMSO-d6) delta=5.74 (s, 1H), 4.45-4.51 (m, 3H), 3.52 (d, J=5.87 Hz, 6H), 1.37 (s, 9H). |
|
In N,N-dimethyl-formamide; at 20℃; for 24.0h; |
Tris(hydroxymethyl)aminomethane (3.210g, 26.50mmol, 1eq) and Boc2O (6.652g, 30.48mmol, 1.15eq) were dissolved in dry DMF (24 mL) and stirred for 24h at ambient temperature at which time benzaldehyde dimethyl acetal (5.1g, 5.0 mL, 33mmol, 1.25eq) and catalytic para-toluenesulfonic acid monohydrate (PTSA) (0.192g, 1.01mmol, 0.06eq) were added. Stirring was continued for an additional 24h, at which time the mixture was diluted with Et2O (60 mL) and extracted with sat?d NaHCO3 (60 mL) and additional water (20 mL). The aqueous layer was further diluted with water (30 mL) and washed with Et2O (60 mL × 3), followed by a final dilution with water (20 mL) and final wash with Et2O (100 mL). The organic layers combined, dried over MgSO4 and the solvent removed under reduced pressure. The product was recrystallized from Et2O/hexane which deposited colourless crystals (6.666g, 21.55mmol, 81.3%) as a mixture of two isomers (60:40 ratio). For characterization purposes, the isomers were separated on silica-gel. |
|
In methanol; tert-butyl alcohol; at 20℃; for 24.0h;Inert atmosphere; |
A solution of Boc2O (7.0 mL, 32.5 mmol) in t-BuOH (12.5 mL) was added to a solution of trishydroxymethylaminomethane (Tris) 2 (3.0 g, 25.0 mmol) in a mixture t-BuOH/MeOH (1:1) (50 mL). The reaction mixture was stirred at room temperature for 24 h. The solvent was removed under reduced pressure and the crude tert-butyl-1,3-dihydroxy-2-(hydroxymethyl)propan-2-ylcarbamate was isolated as a white powder.1H NMR (400 MHz, CDCl3): delta=5.44 (s, 1H), 3.61 (d, J=5.9 Hz, 6H), 3.30 (t, J=5.9 Hz, 3H), 1.20 (s, 9H) ppm. 13C NMR (100 MHz, CDCl3): delta=157.0, 80.5, 63.9, 59.6, 28.3 ppm.2,2-Dimethoxypropane (9.4 mL, 75.0 mmol) and PTSA (240 mg, 1.25 mmol) were added to a solution of the previous crude product (25.0 mmol in theory) in CH2Cl2 (125 mL). The reaction mixture was stirred at room temperature for 30 min and then neutralized with Et3N (210 muL, 1.5 mmol). The solvent was removed under reduced pressure and the residue was purified by chromatography on silica gel (cyclohexane/EtOAc 1:1) to give 5.89 g (90% yield over two steps) of the required product 12 as a white powder. |
|
In N,N-dimethyl-formamide; at 20℃; for 2.0h; |
Tris(hydroxymethyl)aminomethane (2.50 g, 20.6 mmol) was suspended in DMF (50 mL) and Boc anhydride (5.00 g, 22.7 mmol, 1.1 eq.) was added. The reaction mixture was stirred for 2 h at r.t.. Then dimethoxypropane (3.0 mL, 24.8 mmol, 1.2 eq.) and para-toluenesulfonic acid monohydrate (200 mg, 1 .04 mmol, 0.1 eq.) were added and the resulting mixture was stirred overnight at r.t.. The reaction was quenched by addition of diethyl ether (50 mL). The organic phase was washed with saturated sodium bicarbonate solution (1 chi 30 mL) and brine (1 x 20 mL) and then dried over Na2S04. The solvent was removed under reduced pressure. The product was obtained as white solid and was used without purification in the next step. Yield: 5.38 g, (95%), purity: 91 % (GC). 36 M.p.: 100 C (lit. 100-102 C) 1H-NMR (300 MHz, CDCI3) delta [ppm]: 1 .38-1.52 (m, 15 H, 6-CH3, 7-CH3, 10-CH3 to 12-CH3), 3.61 -4.06 (m, 6 H, 1 -CH2, 3-CH2, 4-CH2), 4.18 (s, 1 H, 2-NH). 13C-NMR (75 MHz, CDCI3) delta [ppm]: 28.0 (q, C-10, C-1 1 , C-12), 30.6 (q, C-6, C-7), 59.8 (s, C-2), 62.2 (t, C-3, C-4), 62.9 (t, C-1 ), 80.0 (s, C-9), 98.4 (s, C-5), 162.9 (s, C-8). Exact mass (ESI+): Ci2H23N05 + H+: calcd. 262.1649, found: 262.1650; Ci2H23N05 + Na+: calcd. 284.1468, found: 284.1468. Ref.: Synthesis according to H. Ooi, N. Ishibashi, Y. Iwabuchi, J. Ishihara, S. Hatakeyama, J. Org. Chem. 2004, 69, 7765-7768. Spectroscopic data agree with those given in the literature. |
|
In methanol; tert-butyl alcohol; at 20℃; |
Synthesis of Compound (6) Dissolve 5 parts of 2-amino-2-hydroxymethy-propane-1,3-diol in 70 parts of 1:1 methanol/tert-butanol mixed solvent. Dissolve 6.4 parts of Di-tert-butyl carbonate in 50 parts of melting tert-butanol. Add Di-tert-butyl carbonate/tert-butanol solution into 2-amino-2-hydroxymethy-propane-1,3-diol/1:1 methanol/tert-butanol solution slowly at room temperature and keep stirring overnight. Remove solvents. Re-crystallize by ethyl acetate. |
|
In methanol; tert-butyl alcohol; at 20℃; |
2-amino-2-hydroxymethyl-propane-1,3-diol of 5 parts is dissolved inthe 1:1 methanol / tert-butanol mixed solvent of 70 parts. Thedi-tert-butylcarbonate of 6.4 parts is dissolved in melting tert-butanol of 50 parts. It maintainsovernight, adding and stirring slowly di-tert-butylcarbonate / tert-butanolsolution at a room temperature to 2-amino-2-hydroxymethyl-propane-1,3-diol /1:1 methanol / tert-butanol solution. A solvent is removed. It is recrystallized from ethyl acetate |
|
With triethylamine; In dichloromethane; for 4.0h; |
Tris base (5.00 g, 41.3 mmol) was dissolved in dichloromethane (80 mL) and trimethylamine (20 mL). Di-tert-butyl dicarbonate ( 10.81 g, 49.6 mmol, 1.2 eq) was then added, and the reaction stirred for 4 hours. The mixture was evaporated and the residue portioned between ethyl acetate and water. The organic fraction was washed with water (l x), 1 M HC1 (2x), saturated sodium bicarbonate (l x), and brine (lx) before drying over sodium sulfate and evaporation to give compound 23 (9.04g, 40.9 mmol) in 99% yield, which was used without purification in further steps. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping