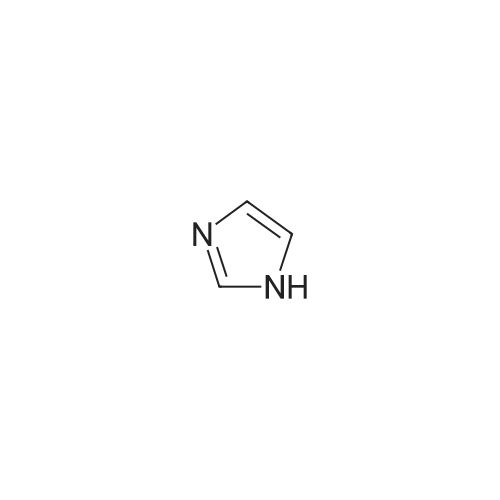
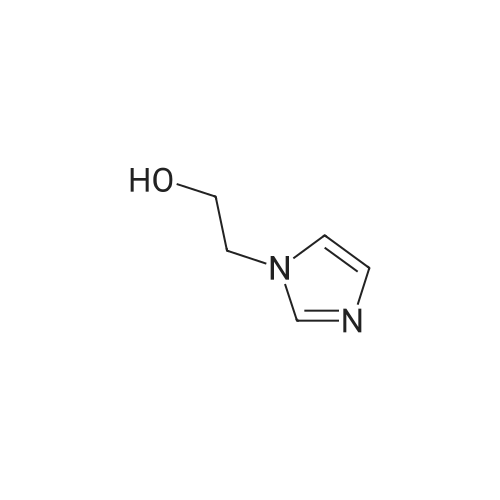
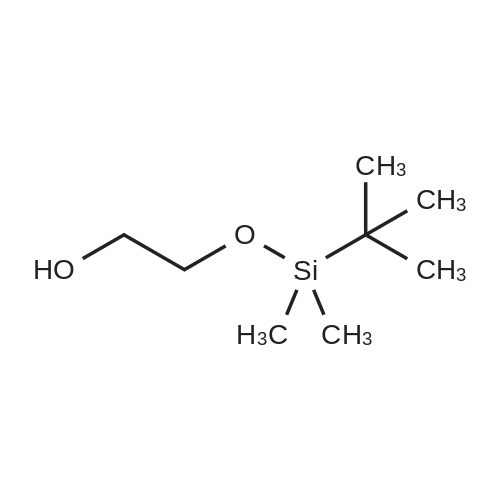
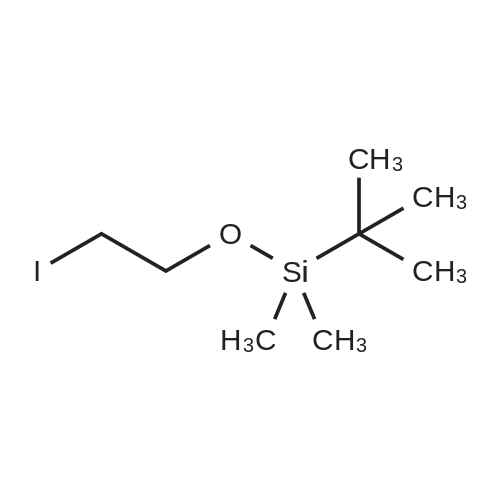
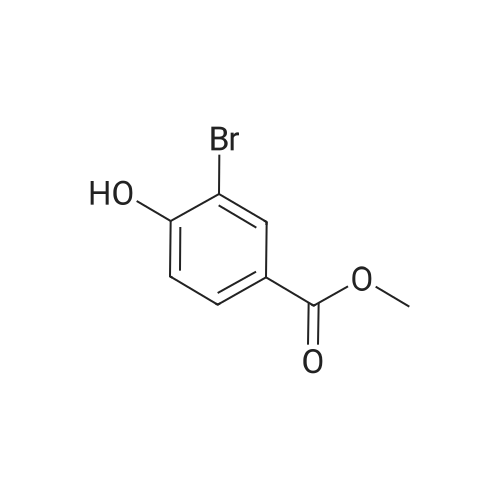
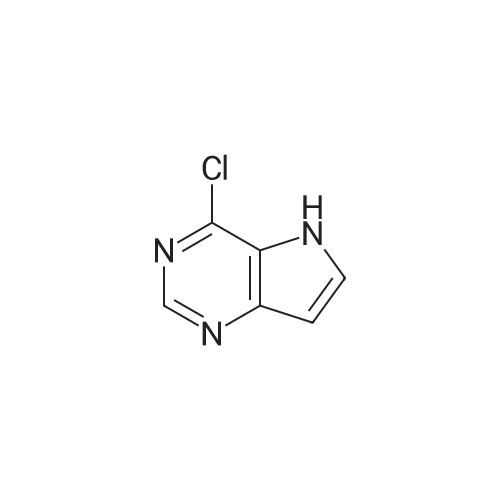
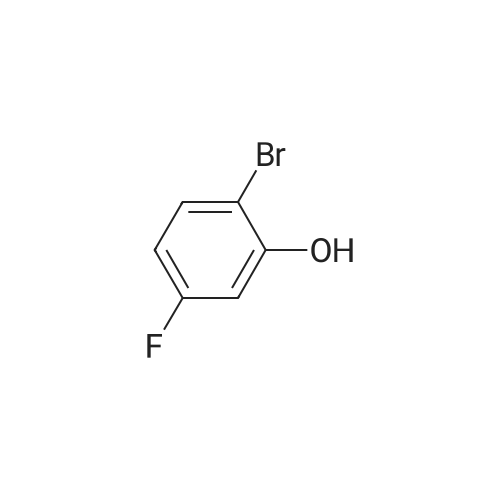
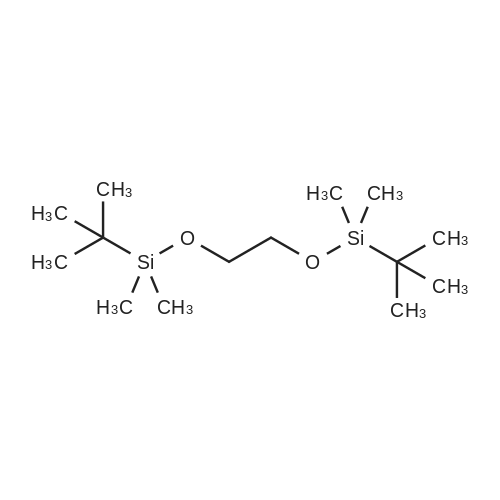
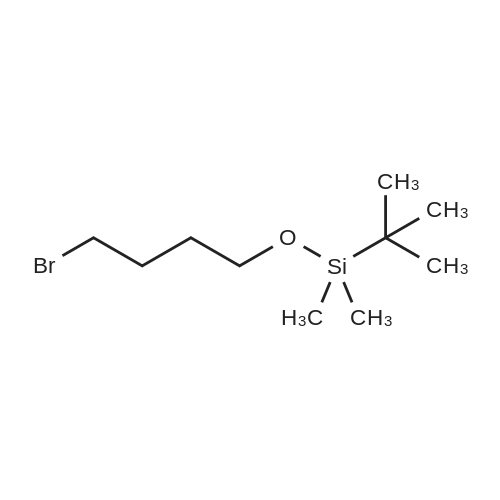
| 98.3% |
With 1H-imidazole In DMF (N,N-dimethyl-formamide) at 20℃; |
[0690] To a solution of P0026-0 (5.0g) and imidazole (3.3g) in DMF (40ml) was added portionwise TBDMSC1 (6.69g) at room temperature. After stirring overnight, water and hexane was added. The aqueous layer was separated and extracted twice with hexane. The combined organic layer was washed with water (twice) and brine, dried over MgSO4, filtered and evaporated under reduced pressure to give 9.49g (98.3percent) of P0026. [0691] IR (film): 2952.5, 2935.1, 1467.6, 1255.4, 1124.3, 1097.3, 838.9, 777.2 cm-1. |
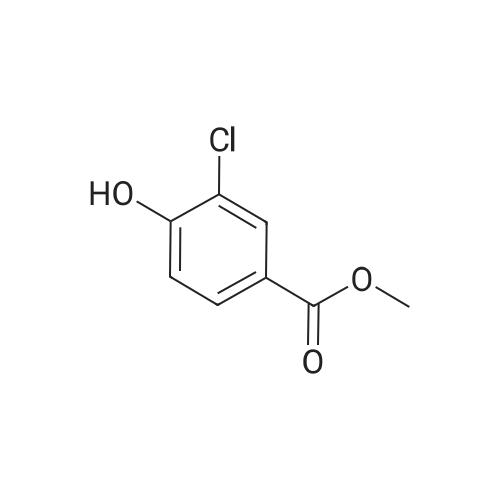
| 98.3% |
With 1H-imidazole In DMF (N,N-dimethyl-formamide) at 20℃; |
To a solution of P0054-0 (5. 0g) and imidazole (3.3g) in DMF (40ML) was added portionwise tert-butyldimethylsilyl chloride (TBDMSC1) (6.69g) at room temperature. After stirring overnight, water and hexane was added. The aqueous layer was separated and extracted twice with hexane. The combined organic layer was washed with water (twice) and brine, dried over MGS04, filtered and evaporated under reduced pressure to give 9.49g (98. 3percent) of P0054. IR (FILM) : 2952.5, 2935.1, 1467.6, 1255. 4,1124. 3,1097. 3,838. 9, 777.2 CM-1. |
| 97% |
With 1H-imidazole In N,N-dimethyl-formamide at 20℃; for 12 h; |
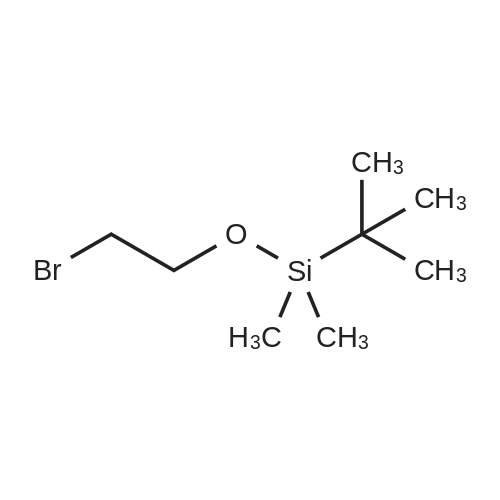
Following a published procedure,24 2-bromoethanol (10.0 mL, 141 mmol) was added to a mixture of imidazole (12.5 g, 184 mmol) and toet-butyldimethylsilyl chloride (21.1 g, 140 mmol) in anhydrous DMF (25 mL). The reaction mixture was stirred at room temperature for 12 h. Water and diethyl ether were added. The phases were separated. The aqueous phase was extracted with diethyl ether. The combined organic phases were washed with water and brine. The solution was dried (Na2SO4). Evaporation of the solvent followed by bulb-to-bulb distillation (40-45 °C/0.05 mmHg) yielded a colorless liquid (32.5 g, 97percent): IR (film, vmax cm"1) 2951, 2859, 1471; 1H NMR δ 0.07 EPO <DP n="87"/>- 85 -(s, 6H), 0.89 (s, 9H), 3.36-3.41 (m, 2H), 3.85-3.90 (m, 2H); 13C NMR δ -5.06, 18.49, 26.04, 33.45, 63.74; EI-MS 137/139, 181/183, calcd 238.0389 (C8H19BrOSi); Anal. Calcd C, 40.17; H, 8.01. Found: C, 40.55; H, 8.25. |
| 96% |
at 20℃; for 3 h; Inert atmosphere |
A modified procedure of Galka et al., J. Lab. Comp. Rad. 2005, 48, 11, 797-809, was used. A mixture of 2-bromoethanol (6.6 mmol; 0.83 g = 0.47 ml), fert.-butyldimethylsilylchloride (6.6 mmol, 1.0 g) and imidazole (7.3 mmol; 0.5 g) was stirred at RT for 3 hours under nitrogen atmosphere. The reaction was quenched with water, extracted with diethylether. The organic phases were dried over Na2SO/i, filtered and concentrated. The purification was achieved by column chromatography (petrolether) to yield 2-bromoethoxy)(ieri.- butyl)dimethylsilane (6.4 mmol, 96 percent). .H NMR (300 MHz, CDC13) 3.91 (t; 3J = 6.5 Hz; 2H; OCH2); 3.41 (t; 3J = 6.5 Hz; 2H; CH2Br); 0.93 (s; 9H; C(CH3)3); 0.11 (s; 6H; 2xCH3). |
| 92% |
With 1H-imidazole In N,N-dimethyl-formamide at 20℃; for 12 h; |
To a solution of tert-butyldimethylsilyl chloride (TBS-Cl) (11.6 g, 77 mmol) in 25 mL of DMF was added imidazole (6.2 g, 91 mmol) followed by the dropwise addition of 2-bromoethanol (8.7 g, 70 mmol). The reaction mixture was stirred for 12 h at rt, extracted with EtOAc and washed with H2O (x3). The combined organic fractions were dried over Na2SO4 and concentrated in vacuo to afford (2-bromoethoxy)(tert-butyl)dimethylsilane as a clear colourless oil (15.3 g, 92percent) The 1H proton and 13C NMR shifts were confirmed in the report by Vader et al. |
| 91% |
With 1H-imidazole In dichloromethane at 20℃; |
(2-Bromoethoxy)(tert-butyl)dimethylsilane (2)TBDMSO. ^„— BrTo a stirring solution of 2-bromoethanol (15.0 mL, 212 mmol) and imidazole (28.9 g, 425 mmol) in dichloromethane (200 mL) was added tert- butylchlorodimethylsilane (32.0 g, 212 mmol). The reaction mixture was stirred at room temperature overnight. The reaction mixture was quenched with water (300 mL) and extracted into dichloromethane (2 x 100mL). The organic layers were combined, washed with brine (100 mL), dried over sodium sulfate, filtered and evaporated to yield (2-bromoethoxy)(tert- butyl)dimethylsilane (46.3 g, 91 percent) as a colourless oil. 1 H NMR (400 MHz CDCI3) ?? ppm 0.00 (s, 6H); 0.82 (s, 9H); 3.27 (t, 2H); 3.80 (t, 2H). |
| 91% |
at 20℃; for 3 h; Inert atmosphere |
A modified procedure of Galka et. al., J. Lab. Comp. Rad. 2005, 48, 11, 797-809 was used to prepare the title compound. A mixture of 2-bromoethanol (40 mmol), tert-butyldimethylsilylchloride (40 mmol) and imidazole (44 mmol)was stirred at rt for 3 hours under inert atmosphere. The reaction was quenched with water and extracted with diethylether.The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The purification was achieved by columnchromatography (PE/EE) to yield the title compound (36.4 mmol; 91 percent). 1H NMR (300 MHz, CDCl3) δ [ppm] = 3.91 (t;3J = 6.5 Hz; 2H; OCH2); 3.41 (t; 3J = 6.5 Hz; 2H; CH2Br); 0.93 (s; 9H; C(CH3)3); 0.11 (s; 6H; 2xCH3). |
| 86% |
With dmap; triethylamine In tetrahydrofuran at 0 - 20℃; for 10 h; |
<Synthesis of Compound>Example 1-1Synthesis of Compound (M-1)In a 1 L reaction flask were charged 40 g of 2-bromoethanol (0.32 mol) and 53 g of t-butyldimethylchlorosilane, and thereto was added 400 mL of tetrahydrofuran (THF). The THF solution thus obtained was cooled to 0° C., and thereto was slowly added dropwise a THF solution prepared by dissolving 36 g of triethylamine (0.35 mol) and 3.9 g of N,N-dimethyl-4-aminopyridine (0.03 mmol) in 100 mL of THF, followed by stirring the mixture at room temperature for 10 hrs. After completing the reaction, precipitates thus generated were removed by suction filtration and then THF in the liquid layer was distilled off by an evaporator, and the residue was extracted with ethyl acetate, followed by washing with water and saturated saline and drying the organic layer over anhydrous magnesium sulphate. Thereafter, a residue obtained by vacuum concentration was purified by vacuum distillation to obtain 66.0 g (yield: 86percent) of a compound represented by the following formula (m-1). |
| 83% |
With 1H-imidazole In N,N-dimethyl-formamide at 20℃; for 16 h; |
To a stirred solution of TBDMSC1 (3.5 g) in DMF (10 ml) was added imidazole (1.77 g). Then 2-bromoethan-l-ol (2.5 g) was added slowly dropwise and the reaction mixture was stirred at room temperature for 16 hours. The reaction was quenched with water and extracted with hexane. The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude material was purified by flash chromatography (silica gel, gradient 0 - 3percent ethyl acetate in hexane). The title compound was obtained as colourless oil (3.96 g, 83percent). NMR complied with literature data. |
| 77.5% |
With 1H-imidazole In dichloromethane at 0 - 20℃; for 24 h; |
Step 4: Preparation of (2-bromoethoxy)(teri-butyl)dimethylsilane To a solution of 2-bromoethanol (3.4 mL, 48.34 mmol) in dry DCM (25 mL) imidazole (9.86 g, 143 mmol) and ieri-butylchlorodimethylsilane (10.9gm, 72.52 mmol) were added at 0 °C. The reaction mixture was stirred at r.t. for 24 h. After completion of reaction, as confirmed by TLC, the reaction mixture was diluted with ethylacetate (350 mL) and washed with water (4 x 20 mL). The organic layer was dried over anhydrous Na2S04 and' volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 0.2: 9.8 Ethyl acetate: Pet. Ether) to afford title compound (204 mg, 77.5 percent) as liquid. ESIMS (m/z): 239.1 (M+l). |
| 77.5% |
With 1H-imidazole In dichloromethane at 0 - 20℃; for 24 h; |
Step 4:
Preparation of (2-bromoethoxy)(tert-butyl)dimethylsilane
To a solution of 2-bromoethanol (3.4 mL, 48.34 mmol) in dry DCM (25 mL) imidazole (9.86 g, 143 mmol) and tert-butylchlorodimethylsilane (10.9gm, 72.52 mmol) were added at 0 °C.
The reaction mixture was stirred at r.t. for 24 h.
After completion of reaction, as confirmed by TLC, the reaction mixture was diluted with ethylacetate (350 mL) and washed with water (4 x 20 mL).
The organic layer was dried over anhydrous Na2SO4 and volatiles were evaporated in vacuo.
The residue obtained was purified by column chromatography (silica gel, 0.2: 9.8 Ethyl acetate: Pet. Ether) to afford title compound (204 mg, 77.5 percent) as liquid.
ESIMS (m/z): 239.1 (M+1). |
| 76% |
With dmap; triethylamine In dichloromethane at 20℃; for 16 h; |
To a solution of 2.0 g (16.0 mmol, 1.0 eq.) of 2-bromoethanol in 10 mL of methylene chloride at 0 °C was added 3.2 g (32.0 mmol, 2.0 eq.) of triethylamine followed by 2.89 g(19.2 mmol, 1.2 eq.) of tert-butyldimethylsilyl chloride and 0.78 g (6.40 mmol, 0.4 eq.) of 4- dimethylamino pyridine and the mixture was stirred at room temperature for 16 h. The mixture was diluted with 50 mL of 1 M HC1 and extracted with 2 x 50 mL of methylene chloride. The combined organic extracts were washed with 30 mL of water, 30 mL of brine, dried (Na2SO4), filtered and the solvent removed in vacuo to provide 2.9 g (12.2 mmol, 76percent)of (2)-bromoethoxy)(tert-butyl)dimethylsilane. ‘H NMR (400 IVIFIz, CDC13): 3.79 (t, 2H),3.29 (t, 2H), 0.81 (s, 9H), -0.01 (s, 6H). |
| 70% |
With 1H-imidazole In dichloromethane at 20℃; for 2 h; |
Tert-butyldimethylchlorosilane (6.4 g, 42 mmol) was added portionwise to a solution of compound 4 (5 g, 40 mmol) and imidazole (5.5 g, 80 mmol) in methylene chloride at room temperature for 2 hours at room temperature. The system was diluted with methylene chloride, washed with saturated aqueous sodium carbonate solution, washed with water,Saturated aqueous sodium chloride solution, the organic phase was dried over anhydrous sodium sulfate, filtered,Rotate to a yellow oil (6.7 g, 70percent). |
| 62.4% |
With dmap; triethylamine In dichloromethane at 20℃; for 15 h; |
Example 123a
(2-Bromoethoxy)(tert-butyl)dimethylsilane 123a
To a solution of 2-bromoethanol (5.0 g, 40.3 mmol) in DCM (20 mL) was added tertbutyldimethylsilyl chloride (9.1 g, 60.5 mmol) followed by the additions of triethylamine (8.14 g, 80.6 mmol) and 4-dimethylaminopyridine (49.2 mg, 0.4 mmol).
The mixture was stirred at room temperature for 15 h and concentrated in vacuo.
The residue was partitioned between 1N HCl and ethyl acetate.
The aqueous portion was extracted with ethyl acetate.
The combined organic portion was washed with brine, dried over sodium sulfate, filtered and concentrated in vacuo to afford yellow oil, which was purified by column chromatography eluting with PE:EA (50:1) to afford 123a as colorless oil (6.0 g, 62.4 percent). LCMS: (M+H)+ 241. |
| 57% |
With 1H-imidazole In acetonitrile at 20℃; for 12 h; |
To a solution of 2-bromoethan-l-ol (6.5 g, 1.0 eq) in ACN (100 mL) at rt, TBDMSCl (5.0 g, 0.65 eq) and imidazole (3.4 g, 1.0 eq) were added and stirred at rt for 12 h. After TLC showed completion of starting material, the mixture was diluted with water (90 mL) and extracted with EtOAc (2 x 80 mL). The organic layer was washed with brine solution (20 mL), dried over anhydrous Na2S04 and concentrated to provide (2-bromoethoxy)(tert-butyl)dimethylsilane (7.5 g, 57percent). 1H NMR (400 MHz, CDC13): δ 3.87 (t, 2H), 3.47 (t, 2H), 0.86 (s, 9H), 0.06 (s, 6H). |
| 55% |
With triethylamine In dichloromethane at 20℃; for 1.33333 h; |
Dry DCM (25ml) in bromoethanol (9.912g, 79.32mmol) to a stirred solution of, in addition tert- butyldimethylsilyl chloride (13.212g, 85.03mmol) at a time, at room temperature the reaction mixture in the mixture was stirred. Then triethylamine in dry DCM (40ml) (8.865g, 12.3ml, 87.61mmol) was added dropwise over a solution for 1 hour and 20 minutes of. The reaction mixture was stirred for 3 days at room temperature, then water was added (30ml). The organic phase was separated, the aqueous phase was extracted with DCM (2 × 20ml). The combined organic extracts were washed with brine (30ml), dried (Na2SO4), the solvent was removed under reduced pressure, to give a pale yellow oil. It was distilled under reduced pressure to give the title compound (10.54g, 55percent) as a colorless oil. |
| 53% |
With 1H-imidazole In N,N-dimethyl-formamide at 20℃; for 20 h; |
Intermediate (69) : Preparation of 2-[2-(tert-butyl-dimethyl-silanyloxy)-ethyl] -methyl-amino}-ethanol; Step 1. Preparation of (2-bromo-ethoxy)-t-butyl-dimethyl-silane; 2-bromoethanol (1.Og, 8.0mmol) was dissolved in DMF (5ml). Therein, tert- butyldimethylsilyl chloride (1.45g, 9.6mmol) and imidazole (1.36g, 20.0mmol) were added, and the reaction mixture was stirred at room temperature for 20 hours. Thereafter, the reaction mixture was poured into diethyl ether, and washed with water and saturated sodium chloride aqueous solution. Combined organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by column chromatography (n-hexane=100percent) to give 1.Og (yield: 53percent, <n="113"/>colorless oil) of the target compound.[1197] 11HH--NNMMIR (CD3OD, 400D) δ 3.89(t/=6.6Hz, 2H), 3.40(t, J=6.6Hz, 2H), 0.90(s, 9H), 0.09(s, 6H) |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping