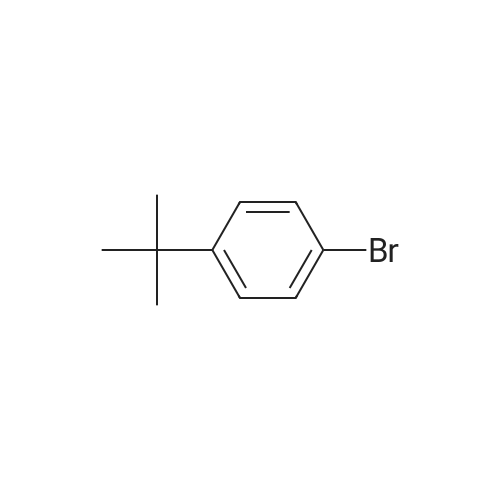
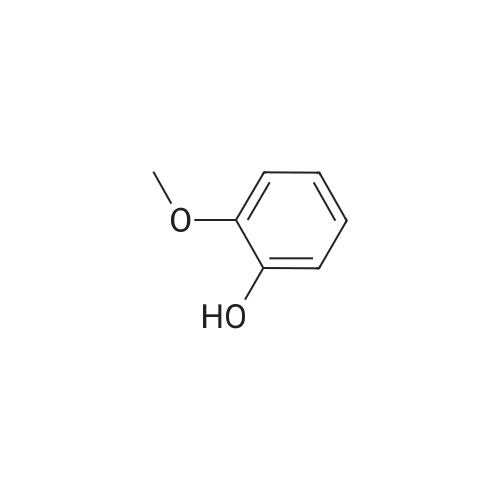
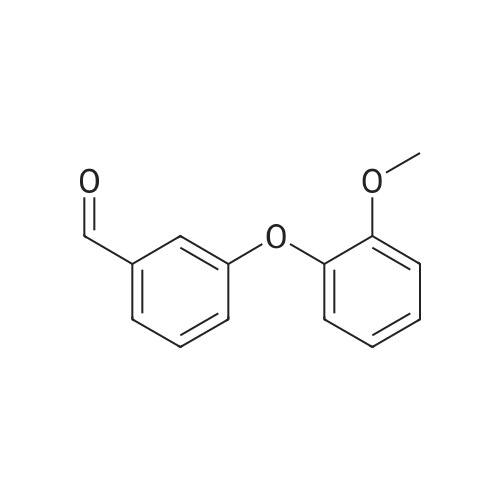
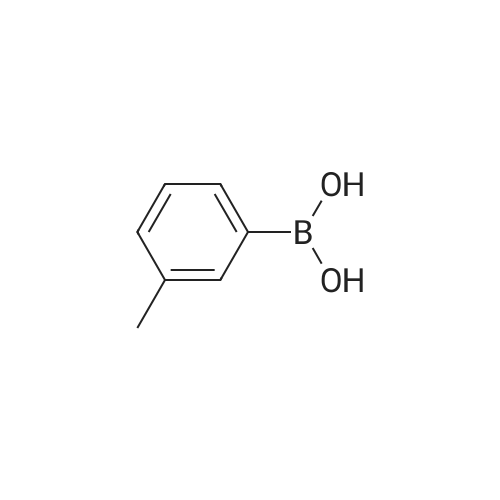
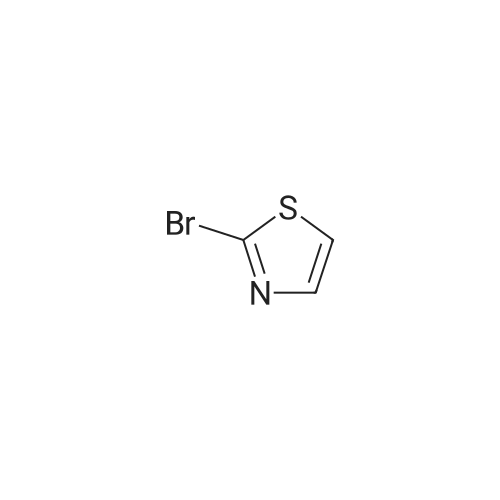
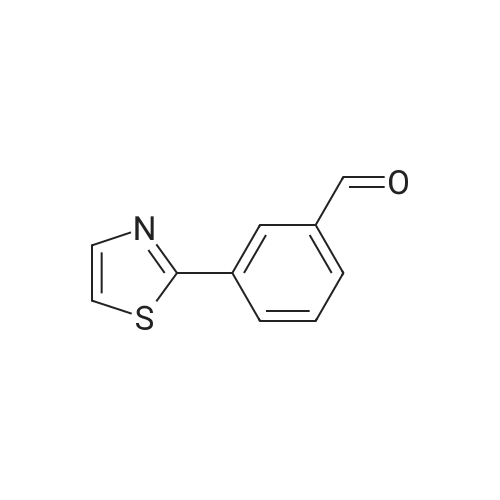
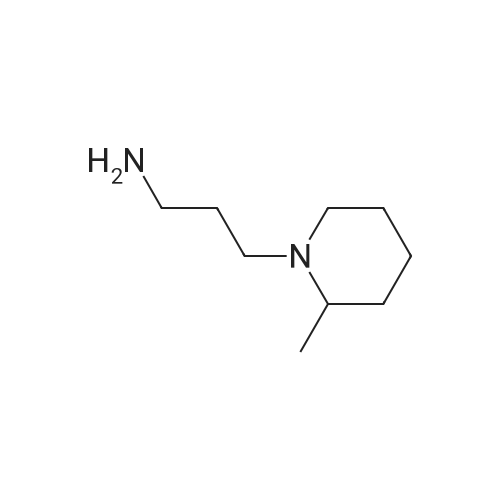
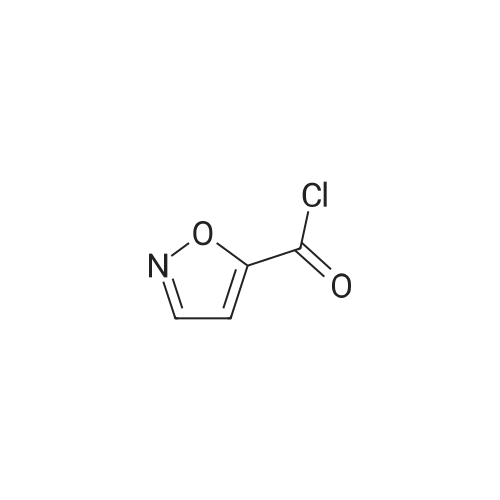
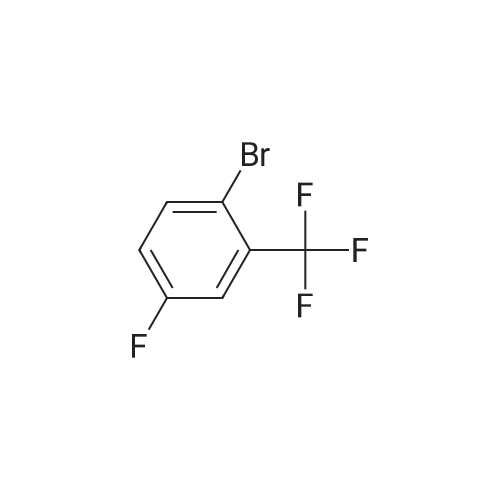
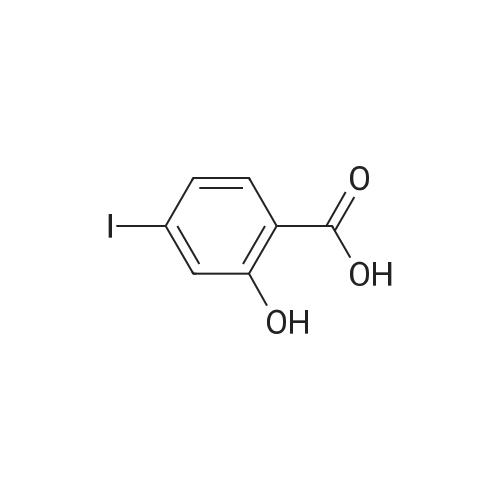
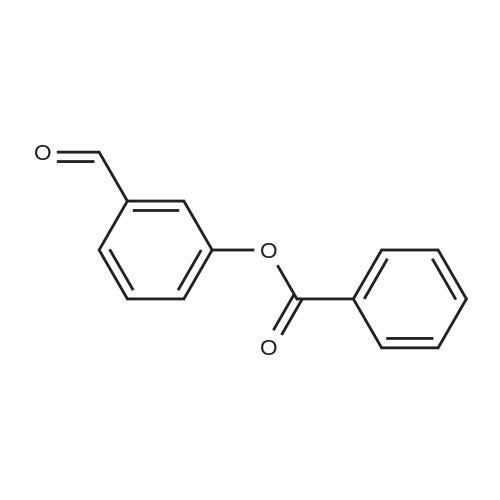
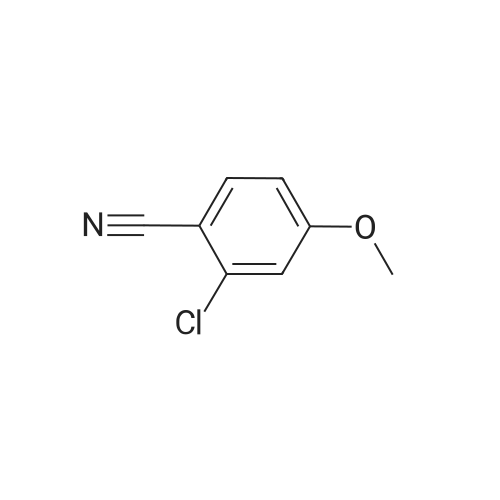
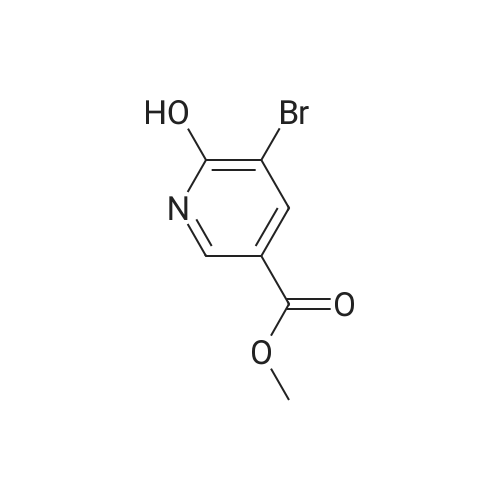
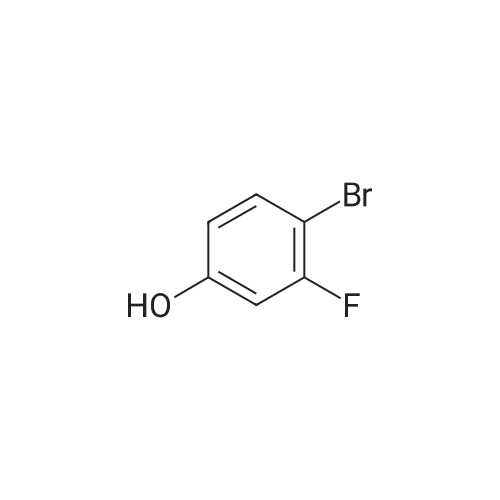
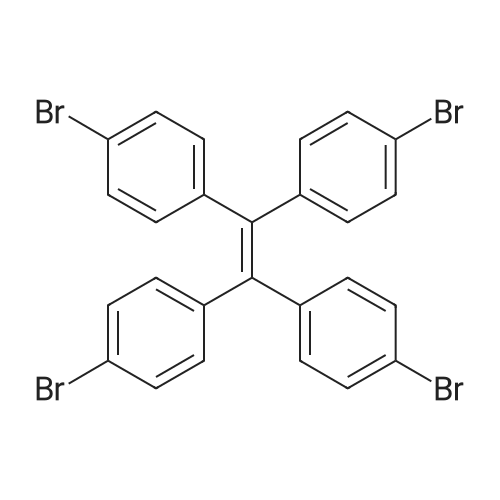
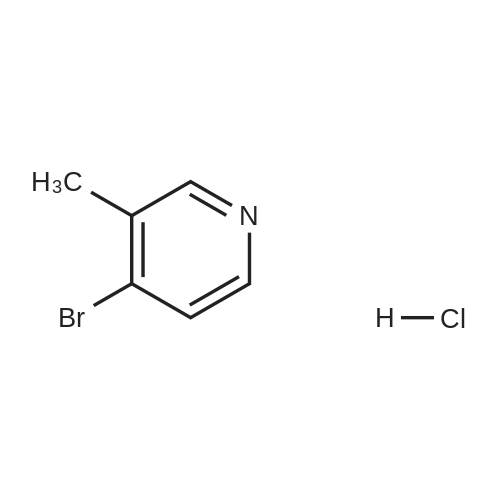
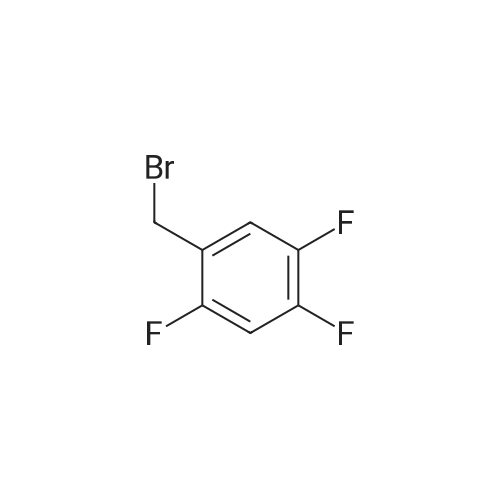
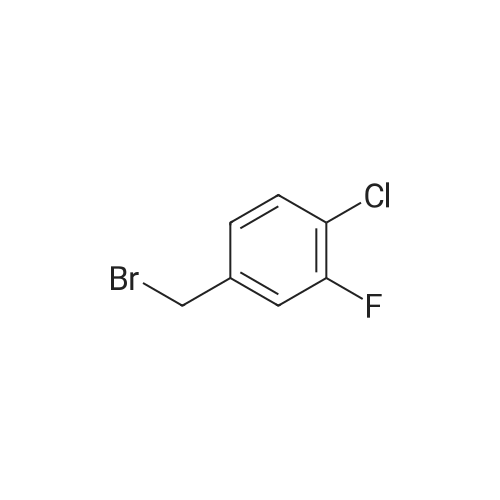
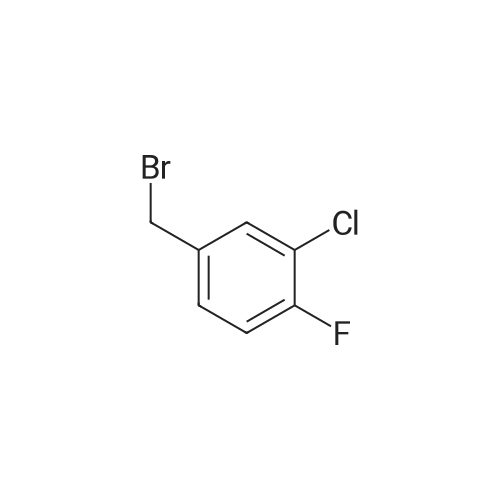
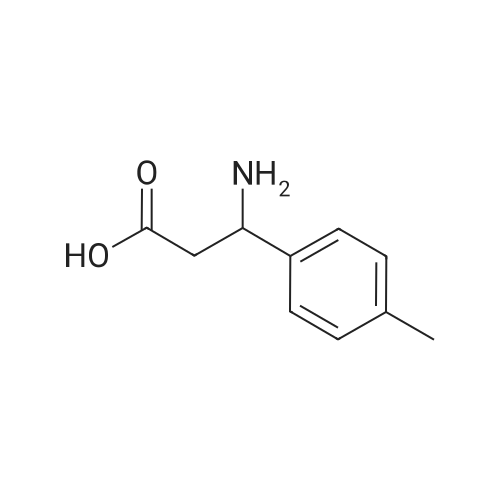
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
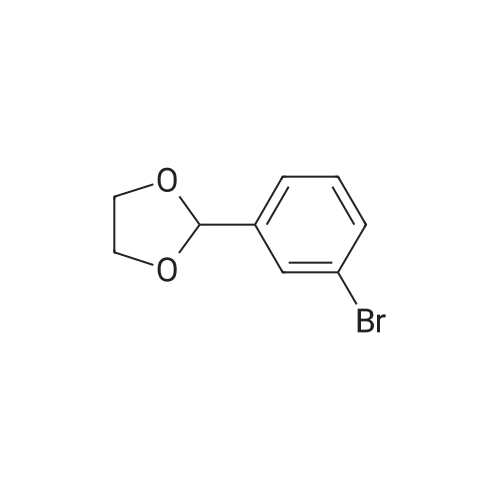
Stage #1: for 1 h; Cooling with ice Stage #2: With sulfuric acid In tetrahydrofuran; water at 20℃; for 14 h;
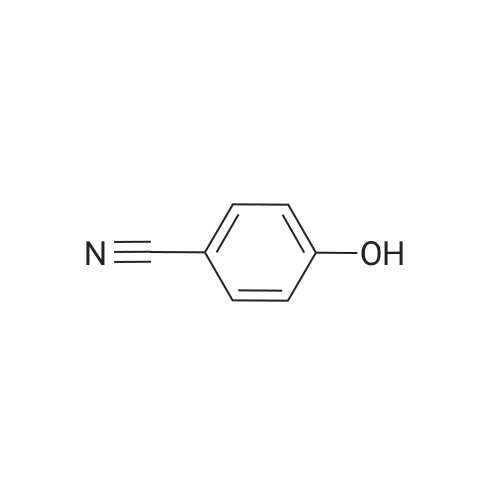
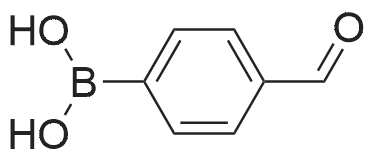
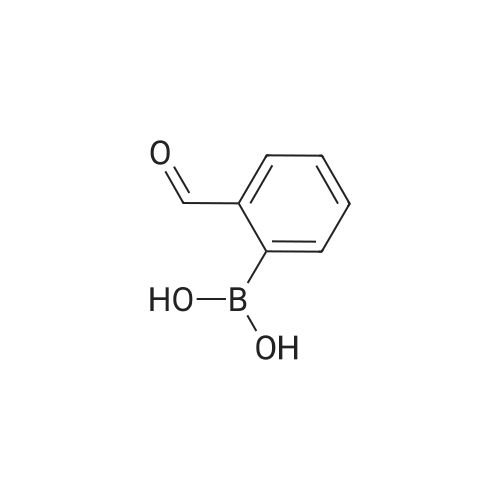
Separately, a three-necked flask having a capacity of 3 liters was charged with 154.8 ml of trimethyl borate and then with 915 ml of THF to prepare a solution of trimethyl borate in THF. The solution was agitated while a nitrogen gas flowed through the flask and the solution was cooled with ice. The ice-cooled THF solution was mixed with the Grignard reagent fed into the 3 liter flask through a stainless steel pipe. The resultant reaction mixture liquid was agitated for one hour while cooling with ice, and then further mixed with an aqueous sulfuric acid solution prepared from 30 ml of concentrated sulfuric acid and 480 ml of water. The temperature of the resultant admixture liquid was raised to room temperature and, then, the admixture liquid was agitated at this temperature for 2 hours. Thereafter, the agitation was stopped and the resultant reaction mixture liquid was left to stand in the ambient atmosphere for one night. The precipitate generated in the reaction mixture liquid was removed by filtration and the resultant filtrate was concentrated. The resultant concentration residue was mixed with water in a volume equal to that of the concentration residue, the resultant mixture liquid was agitated at room temperature for one hour. The resultant solid fraction was collected from the mixture liquid by filtration and dried. The target compound 3-formylphenylboronic acid was obtained in an amount of 123.46g. The yield thereof was 88percent. The results of the1H-NMR measurement of the resultant target compound (200 MHz, δ ppm, CDCl3) were as follows. 7.54 (t, J = 7.5 Hz, 1H), 7.93 (d, J = 6.1 Hz, 1H), 7.9 - 8.1 (br. d, 1H), 8.2 - 8.3 (br. s, 1H), 10.04 (s, 1H)
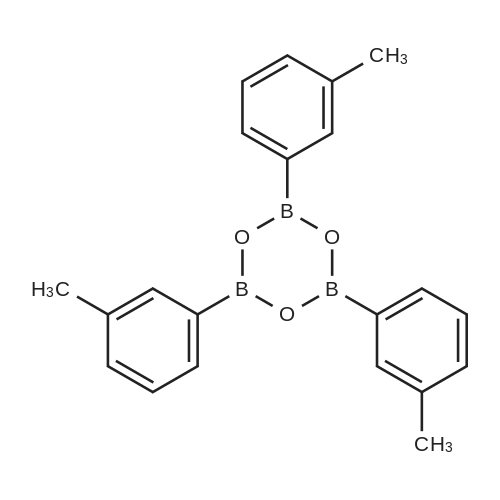
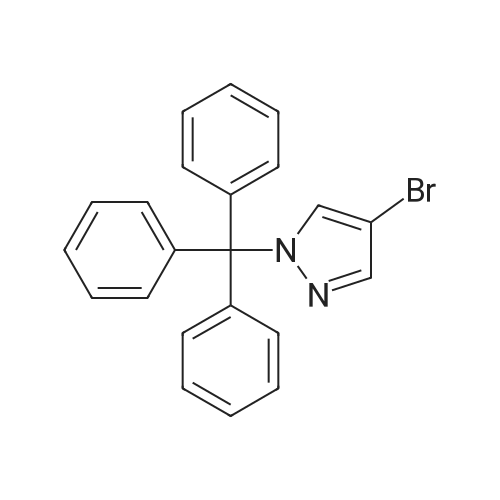
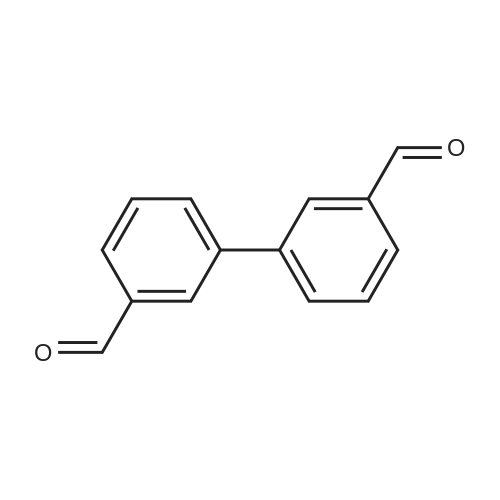
With dipotassium peroxodisulfate; iron(II) acetylacetonate In water; acetonitrile at 80℃; for 12 h;
Ferroacetylacetonate (0.025mmol), polymethylhydrogensiloxane0 75mmol), Potassium persulfate (0.25mmol), lah (0.25mmol), AcetonitrilelmL),waterlmL),in the reaction mixture 80 ° C reaction under 12 h. The reaction is finished adding ammonia water (2 ml) to remove the peripheric, add saturated salt water 10 ml, and extraction with ethyl ether (10 ml × 3), the combined organic phase, pressure reducing evaporate the solvent column chromatography to obtain yield 73percent.
Reference:
[1] Patent: CN107216242, 2017, A, . Location in patent: Paragraph 0092; 0093
5
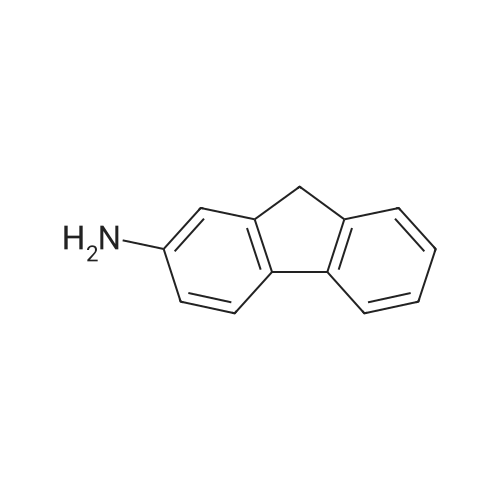
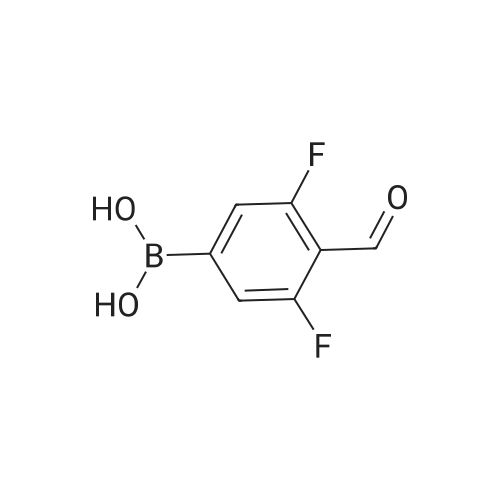
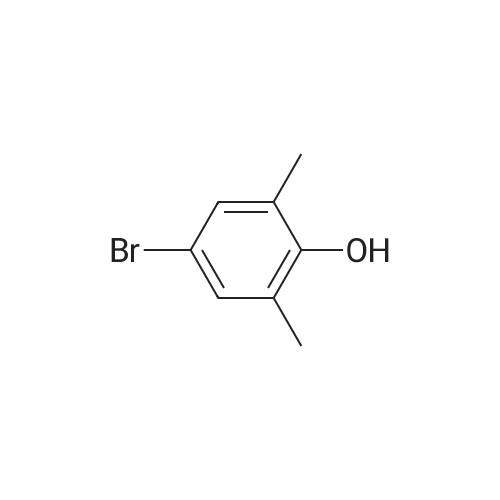
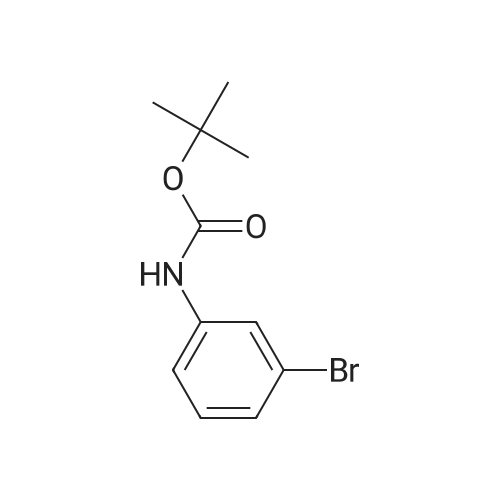
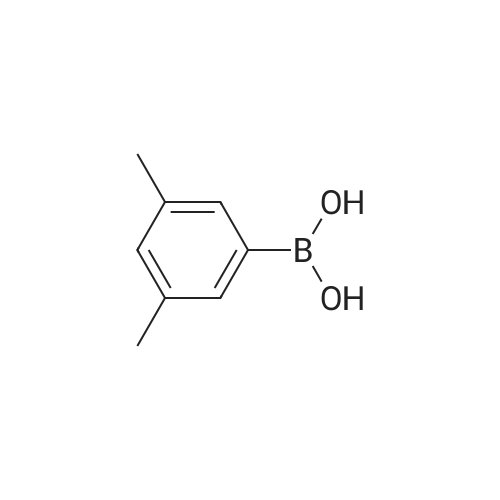
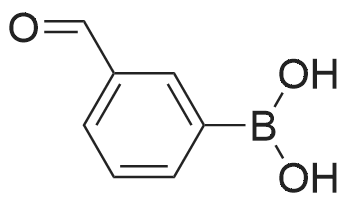
[ 98527-70-9 ]
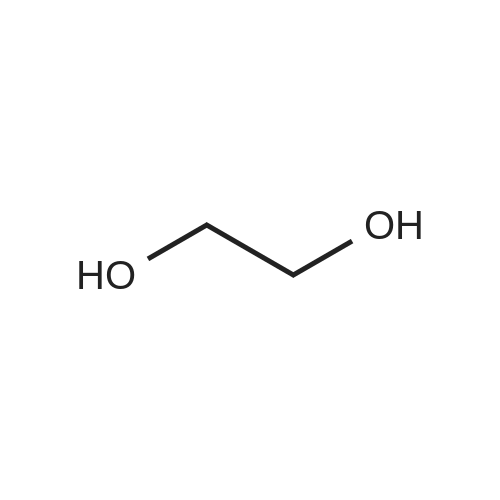
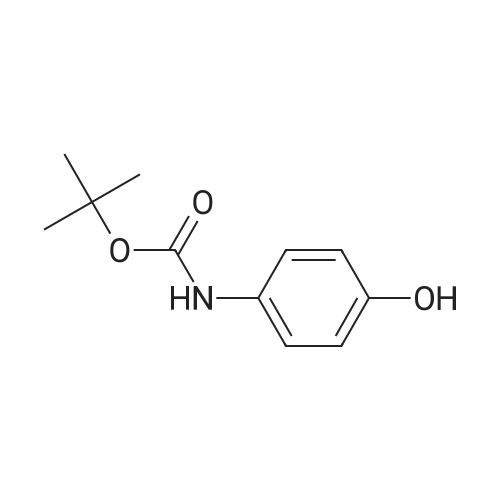
[ 68-12-2 ]
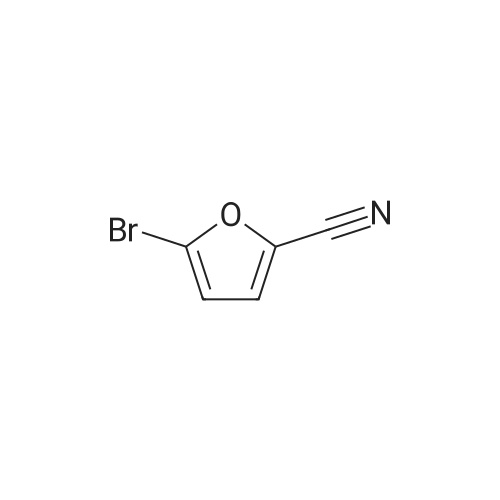
[ 87199-16-4 ]
Yield
Reaction Conditions
Operation in experiment
106.5 g
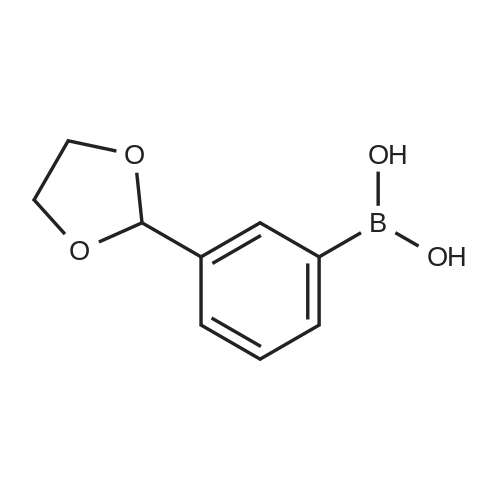
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -60 - 20℃; Inert atmosphere Stage #2: With hydrogenchloride In tetrahydrofuran; hexane; water at 20℃;
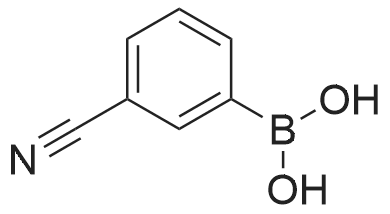
Under nitrogen, the above-obtained m-bromobenzeneboronic acid trimer was added to anhydrous tetrahydrofuran (600 ml)After the addition, transfer to a 2 L three-necked flask and add dimethylformamide (80.4 g, 1.1 mol). Then cool the system to -Below 70 ° C, 440 ml (1.1 moles) of 2.5 M n-butyllithium hexane solution was slowly added dropwise, and the temperatureThe maximum is not more than -60 ° C, and the addition is continued until the stirring is continued for 1 to 3 hours, followed by natural stirring to room temperature3-5 hours. TLC detection reaction is completed, the system cooled to 0 , adding 10percent hydrochloric acid aqueous solution quenching reaction, adjust the PH to1-2 continue at room temperature for 2-3 hours to ensure that the trimer hydrolysis is complete. The reaction solution was distilled, and the organic solvent was distilled and solidPrecipitation, filtration, toluene after recrystallization to be light yellow solid aldehyde phenyl benzene boric acid 106.5 grams, HPLC: 99.5percent, the total yield of two steps71percent
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -80 - 20℃; Inert atmosphere Stage #2: With hydrogenchloride In tetrahydrofuran; hexane; water at 20℃;
Under nitrogen protection, the above-obtained iodobenzeneboronic acid trimer was added to 500 ml of anhydrous tetrahydrofuranAfter the addition, the mixture was transferred to a 2 L three-necked flask and dimethylformamide (65.7 g, 0.90 mol) was added. The system was then cooled to -75 ° C to -80 ° C and a slow dropwise addition of 609 ml (0.98 mol) of 1.6 M n-butyllithium hexane solution was added dropwise. The addition temperature was not higher than -70 ° C during the addition and the temperature The reaction was continued for 1 to 3 hours, followed by spontaneously stirring at room temperature for 3-5 hours. TLC detection reaction is completed, the system cooled to 0 , adding 15percent hydrochloric acid aqueous solution quenching reaction, adjust the PH to 1-2 to room temperature stirring for 3-5 hours to ensure that the trimer hydrolysis complete. The reaction solution was distilled and the organic solvent was distilled, and the residue was precipitated by cooling. After recrystallization from acetonitrile, 86.6 g of aldehyde phenylbenzene borate was obtained as a pale yellow solid, and 99.8percent by HPLC and 77percent in two steps.
Reference:
[1] Arkiv foer Kemi, 1957, vol. 10, p. 507,509
15
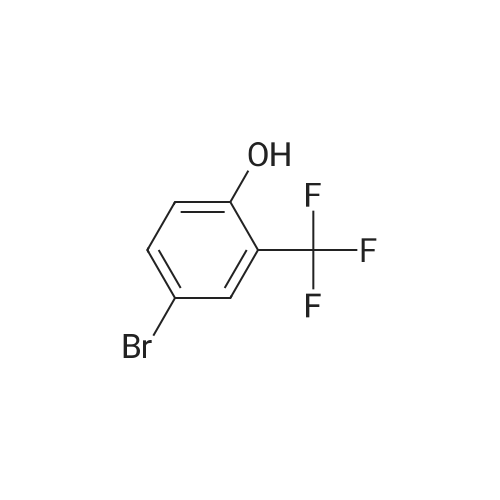
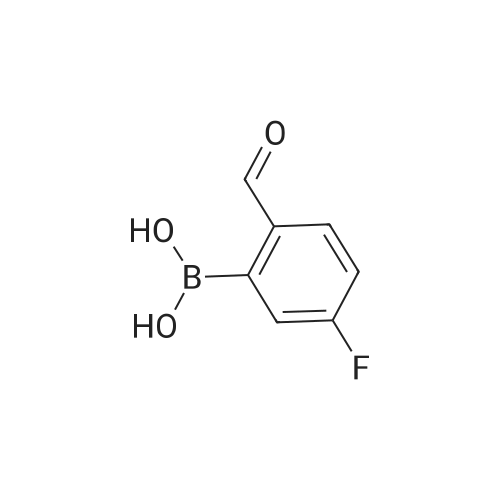
[ 87199-16-4 ]
[ 767-00-0 ]
[ 90178-72-6 ]
Reference:
[1] Journal of Medicinal Chemistry, 2017, vol. 60, # 14, p. 6384 - 6399
16
[ 87199-16-4 ]
[ 150255-96-2 ]
Yield
Reaction Conditions
Operation in experiment
68%
Stage #1: for 8 h; Heating / reflux Stage #2: at 20℃; for 12 h;
In a three-necked flask with a capacity 3 liters, 123.4g of the 3-formylphenylboronic acid prepared in Production Example 2, 68.6g of hydroxylamine hydrochloride, 1050 ml of formic acid and 112.1g of sodium formate are placed and mixed with each other. The resultant mixture liquid was heated for 8 hours while refluxing. The resultant reaction mixture liquid was left to stand in the ambient atmosphere for one night. Thereafter, in the case where a precipitate was generated in the reaction mixture liquid, this mixture liquid was agitated while cooling with ice and, in the case where no precipitate was generated in the reaction mixture liquid, the mixture liquid was mixed with a small amount of precipitation seed particles and agitated. From the resultant reaction mixture liquid, the solid precipitate was collected, by filtration and dried. The target compound, 3-cyanophenylboronic acid was obtained in an amount of 82.0g. The yield thereof was 68percent. The results of the 1H-NMR measurement of the target compound (200 MHz, δ ppm, CDCl3) were shown below. 7.47 (t, J = 7.7 Hz, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.9 - 8.0 (br. d, 1H), 8.0 - 8.1 (br. s, 1H)
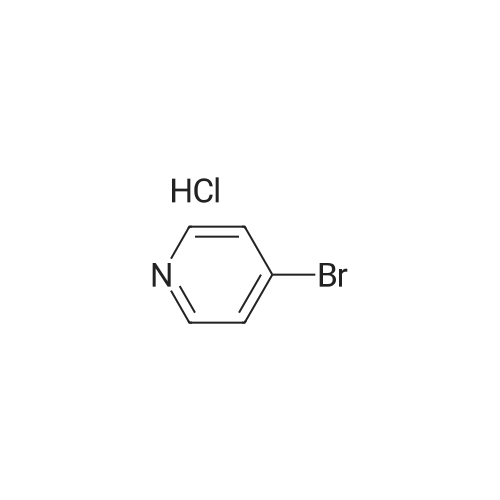
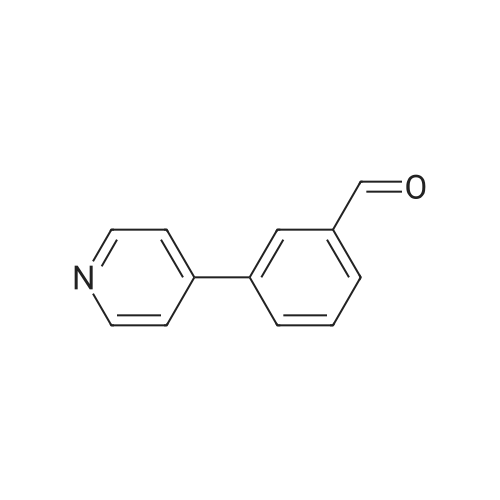
Stage #1: With sodium carbonate In water; toluene at 0 - 20℃; Inert atmosphere Stage #2: With tetrakis(triphenylphosphine) palladium(0) In water; toluene at 85℃; for 18 h; Inert atmosphere
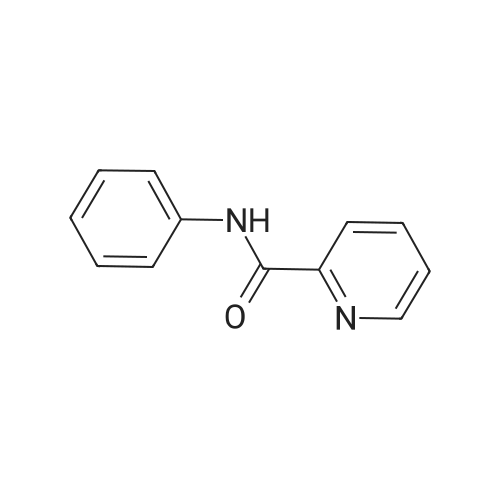
A mixture of 4-bromopyridine hydrochloride 15 (2.00 g, 10.3 mmol, 1.0 equiv) in water (16 ml) and toluene (22 ml) was cooled to 0 °C and a solution of Na2CO3 (2.51 g, 23.8 mmol, 2.3 equiv) in water (26 ml) was added. After warming to rt, 3-formylphenylboronic acid 16 (1.62 g, 10.8 mmol, 1.04 equiv) and Pd(PPh3)4 (0.600 g, 0.515 mmol, 0.05 equiv) were added and the reaction mixture was stirred at 85 °C for 18 h. Then, the cooled solution was diluted with CH2Cl2 (40 ml) and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 16 ml) and the combined organic layers were dried over MgSO4. The solvent was removed under reduced pressure and the crude product was purified by column chromatography (n-hexane/EtOAc 4:1 → 1:4, v/v) to obtain 17 as a white solid (1.77 g, 94percent); mp: 34–36 °C; IR (KBr): ν˜ ν˜ = 3440, 3025, 1689, 1582, 1379, 1188, 789, 690, 653 cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.51–7.53 (m, 2H), 7.63–7.67 (m, 1H), 7.87–7.94 (m, 2H), 8.12–8.13 (m, 1H), 8.68–8.70 (m, 2H), 10.08 (s, 1H); 13C NMR (100 MHz, CDCl3): δ = 121.7, 127.9, 130.0, 130.5, 132.9, 137.2, 139.3, 147.0, 150.6, 191.8; ESI-HRMS m/z calcd for C12H9NO: 206.05764 [M+Na]+, 389.12605 [2M+Na]+, found: 206.05779, 389.12615
Reference:
[1] Bioorganic and Medicinal Chemistry, 2012, vol. 20, # 21, p. 6465 - 6481,17
18
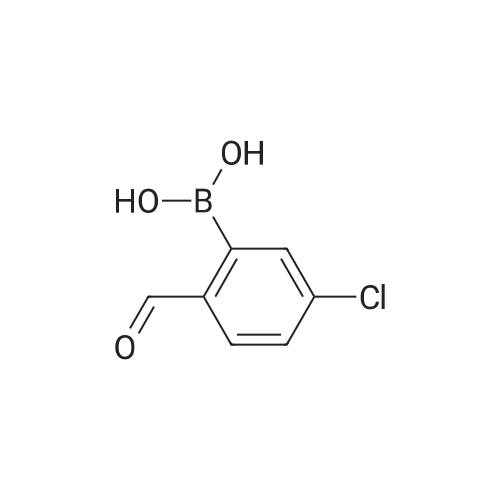
[ 1120-87-2 ]
[ 87199-16-4 ]
[ 208190-04-9 ]
Reference:
[1] Organic Letters, 2004, vol. 6, # 19, p. 3337 - 3340
[2] Beilstein Journal of Organic Chemistry, 2012, vol. 8, p. 841 - 849
19
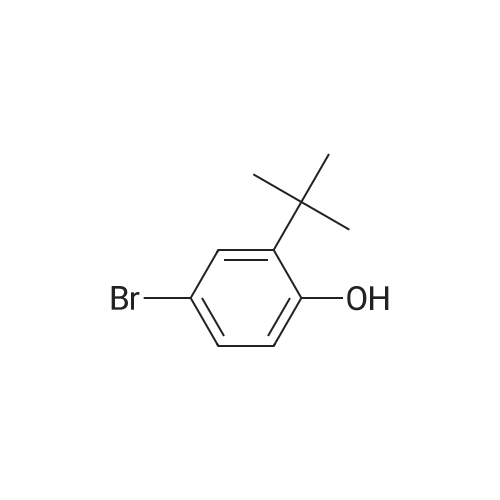
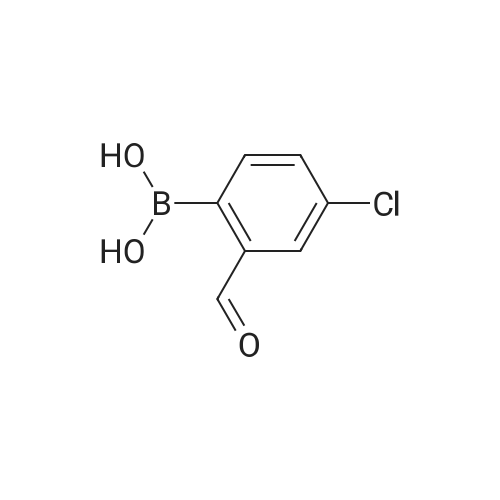
[ 76-09-5 ]
[ 87199-16-4 ]
[ 380151-86-0 ]
Yield
Reaction Conditions
Operation in experiment
100%
at 20℃; for 18 h; Inert atmosphere
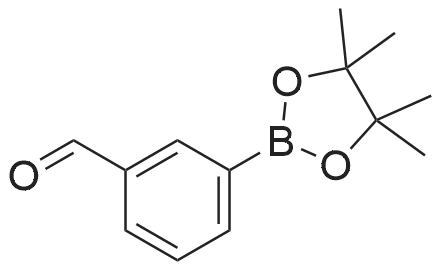
Stage 1. 3-(4,4, 5, 5-Tetramethyl- 1, 3,2-dioxaborolan-2-yl)benzaldehyde To a solution of 3-formyl phenylboronic acid (5 g, 33 mmol) in anhydrous THF (50 mL) was added pinacol (4.34 g, 37 mmol). The reaction mixture was allowed to stir at RT under nitrogen for 18 hrs before concentration in vacuo. The crude residue was dissolved in DCM (150 mL) and washed with water (3 x 100 mL). The organic layer was dried over MgSO4 and evaporated in vacuo to give the title compound as a yellow oil (7.73 g, 100percent).1H NMR (300 MHz, ODd3) ö ppm: 10.06 (1H, 5), 8.31 (1H, 5), 8.07 (1H, d, J=7.4 Hz),7.99 (1H, dt, J=1.5, 7.4 Hz), 7.54 (1H, t, J=7.4 Hz), 1.38 (12H, 5).
Reference:
[1] Patent: WO2014/1802, 2014, A1, . Location in patent: Page/Page column 39; 40
[2] Chemical Biology and Drug Design, 2010, vol. 76, # 1, p. 17 - 24
[3] Canadian Journal of Chemistry, 2001, vol. 79, # 7, p. 1115 - 1123
[4] Canadian Journal of Chemistry, 2001, vol. 79, # 7, p. 1115 - 1123
[5] Molecules, 2013, vol. 18, # 10, p. 12346 - 12367
[6] Organic Letters, 2015, vol. 17, # 12, p. 3086 - 3089
20
[ 4595-59-9 ]
[ 87199-16-4 ]
[ 198084-12-7 ]
Yield
Reaction Conditions
Operation in experiment
60%
Stage #1: With sodium carbonate In water; toluene for 0.333333 h; Stage #2: at 80℃; for 16 h;
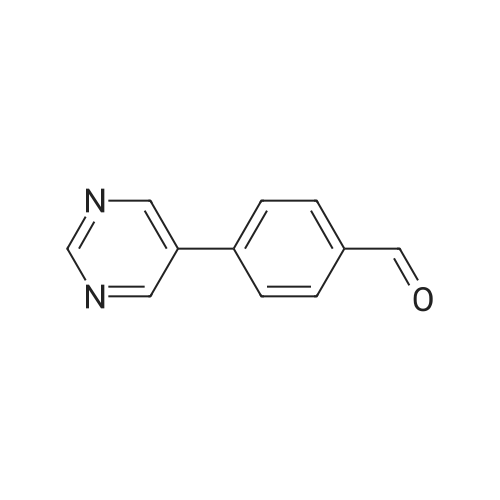
Example 271; 4- (5-pyrimidinyl) benzaldehyde; 5-Bromopyrimidine (159 mg, 1 mmol) was dissolved in toluene (5 mL) and treated with tetrakis (triphenylphosphine)-palladium (0) (116 mg, 0.1 equivalent) and a 2 M solution of sodium carbonate (1 mL, 2 equivalents). The mixture was stirred under an argon atmosphere for 20 min. followed by addition of 3-formylphenyl boronic acid (165 mg, 1.1 equivalensts) in ethanol (1 mL). The reaction was heated to 80 C and stirred for 16 hrs. The mixture was filtered and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried over magnesium sulfate, filtered, and the solvent was removed by evaporation. This material was purified using hexanes: dichloromethane (1: 1) followed by dichloromethane: methanol (97: 3) to give 110 mg (60percent) of the title compound.
60%
Stage #1: With sodium carbonate In water; toluene for 0.333333 h; Stage #2: at 80℃; for 16 h;
EXAMPLE 271 4-(5-pyrimidinyl)benzaldehyde 5-Bromopyrimidine (159 mg, 1 mmol) was dissolved in toluene (5 mL) and treated with tetrakis(triphenylphosphine)-palladium(0) (116 mg, 0.1 equivalent) and a 2 M solution of sodium carbonate (1 mL, 2 equivalents). The mixture was stirred under an argon atmosphere for 20 min. followed by addition of 3-formylphenyl boronic acid (165 mg, 1.1 equivalensts) in ethanol (1 mL). The reaction was heated to 80° C. and stirred for 16 hrs. The mixture was filtered and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried over magnesium sulfate, filtered, and the solvent was removed by evaporation. This material was purified using hexanes:dichloromethane (1:1) followed by dichloromethane:methanol (97:3) to give 110 mg (60percent) of the title compound.
In tetrahydrofuran; at 20℃; for 18h;Inert atmosphere;
Stage 1. 3-(4,4, 5, 5-Tetramethyl- 1, 3,2-dioxaborolan-2-yl)benzaldehyde To a solution of 3-formyl phenylboronic acid (5 g, 33 mmol) in anhydrous THF (50 mL) was added pinacol (4.34 g, 37 mmol). The reaction mixture was allowed to stir at RT under nitrogen for 18 hrs before concentration in vacuo. The crude residue was dissolved in DCM (150 mL) and washed with water (3 x 100 mL). The organic layer was dried over MgSO4 and evaporated in vacuo to give the title compound as a yellow oil (7.73 g, 100%).1H NMR (300 MHz, ODd3) oe ppm: 10.06 (1H, 5), 8.31 (1H, 5), 8.07 (1H, d, J=7.4 Hz),7.99 (1H, dt, J=1.5, 7.4 Hz), 7.54 (1H, t, J=7.4 Hz), 1.38 (12H, 5).
With magnesium sulfate; In methanol; at 20℃; for 6h;
General procedure: To a mixture of a4-formylbenzenboronic acid (1a, 375 mg, 2.50 mmol), pinacol (355 mg, 3.00 mmol) and anhydrous magnesium sulfate (625 mg, 5.00 mmol), methanol was added (12.50 mL). The mixture was stirred at room temperature for 6 h. After the reaction was completed, the crude solution was filtered, and then sodium borohydride (47 mg, 1.25 mmol) was added to the filtrate. Afterwards, the reaction mixture was stirred for an additional 5 h. Once the reaction was completed, the reaction mixture was filtered and the filtrate was concentrated in vacuo to give the desired product 2a as a white solid (m.p. 75-77 C) in88% yield (513 mg). 1H-NMR (CD3OD-d4) delta ppm 7.71 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 7.8 Hz, 2H),4.62 (s, 2H), 1.34 (s, 12H); 13C-NMR (CD3OD-d4) delta ppm 146.23, 135.93, 127.26, 85.19, 65.24, 25.34;11B-NMR (CDCl3) delta ppm 34.82.
590.a Example 590. 3-[3-(2,4-Dichlorobenzoylaminooxy)phenyl]-2-isopropoxypropanoic acidProduction example 590a)3-[1,3,2]Dioxaborinan-2-yl-benzaldehyde
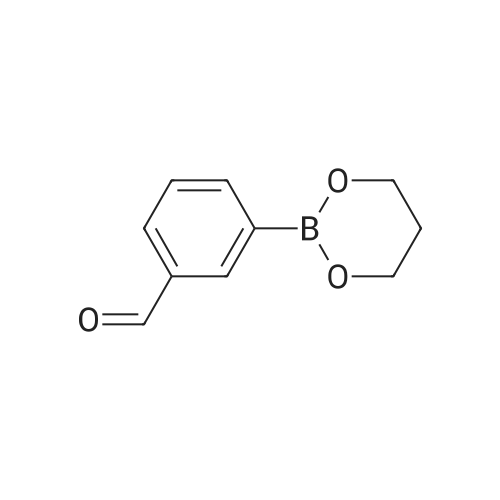
5.0 g of 3-Formylbenzene boronate was suspended in diethyl ether, and 3.7 ml of ethylene glycol was added, and the mixture was stirred at room temperature for 15 minutes. The solvent was concentrated, to give 10.36 g of the title compound as a colorless oil.
With pyridine; copper diacetate; In dichloromethane; at 20℃; for 36h;Molecular sieve;
Intermediate 58 Step A; The compound (a) from Intermediate 55, Step A (1.6 g, 6.722 mmol) and boronic aldehyde (2 g, 13.35 mmol) are added to a suspension of Cu (OAc) 2 (2.45 g, 13.488 mmol) in a mixture of pyridine (5.4 mL) and CH2C12 (40 mL). The mixture is stirred at r. t. under air atmosphere in the presence of sieves for 36 h. It is filtered through Celite (CHzClz washings) and partitioned between CH2C12 and diluted HC1 (3% aqueous solution). The organic layer is dried, filtered and concentrated to give a crude residue that is flash chromatographed on Si02 (5-10% EtOAc/hexanes) affording 0.525 mg of unreacted starting compound (a) and 700 mg of the coupling product (30%, colorless oil).
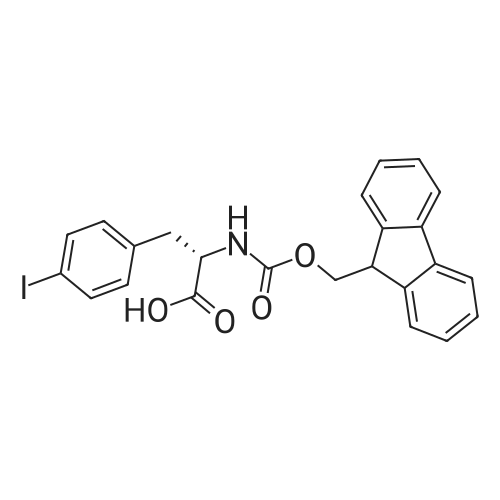
(3R,S)-3-(9-fluorenylmethoxycarbonylamino)-3-phenyl-propanoic acid[ No CAS ]
[ 2905-24-0 ]
[ 87199-16-4 ]
[ 5993-91-9 ]
(3R,S)-3-(3'-[(1H-benzoimidazol-2-ylmethyl)-amino]-methyl}-biphenyl-3-sulfonylamino)-3-phenyl-propanoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
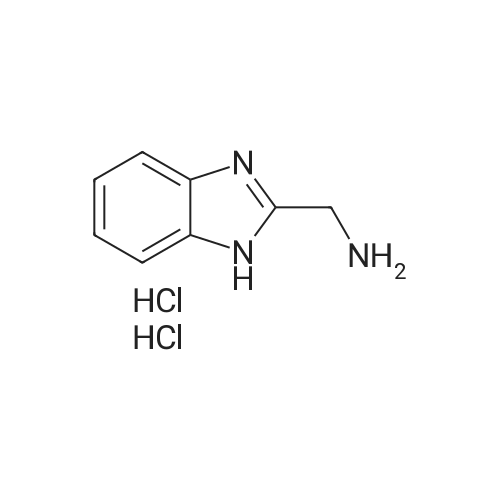
1.2 g of polystyrene Wang resin (Rapp-Polymere, Tuebingen; loading 1.08 mmol/g) are swollen in dimethylformamide (DMF). The solvent is filtered off with suction and a solution of 904 mg of (3R,S)-3-(9-fluorenylmethoxycarbonylamino)-3-phenyl-propanoic acid (amino acid reagent) in 9 ml of dimethylformamide (DMF) is added. After shaking at room temperature for 15 min, the suspension is treated with 345 mul of pyridine and 543 mg of 2,6-dichlorobenzoyl chloride. It is shaken overnight at room temperature. The resin is then washed with dimethylformamide (DMF), methanol and dichloromethane. The resin is treated with 15 ml of a 20% strength piperidine solution in dimethylformamide (DMF) and shaken at room temperature for 10 min. It is then washed 3 times with dimethylformamide (DMF) and a further 15 ml of a 20% strength piperidine solution in dimethylformamide (DMF) are added. After shaking for 20 min, it is washed with dimethylformamide (DMF) and tetrahydrofuran (THF). The resin is treated with a solution of 452 ml of diisopropylethylamine in 5 ml of tetrahydrofuran (THF) and a solution of 431 mg of 3-bromobenzenesulfonyl chloride in 5 ml of tetrahydrofuran (THF). It is shaken overnight at room temperature. The resin is then washed with dimethylformamide (DMF), methanol and tetrahydrofuran (THF). The resin is suspended in 9 ml of xylene, treated with 1.55 g of 3-formylbenzeneboronic acid (boronic acid reagent) and a solution of 2.2 g of sodium carbonate in 9 ml of water and shaken for 5 min at room temperature. 227 mg of bis-(triphenylphosphane)-palladium(II) chloride and 170 mg of triphenylphosphane are then added and the mixture is stirred overnight at 85C. The resin is then washed with tetrahydrofuran (THF)/water 1:1, 0.25 M aqueous hydrochloric acid, is then washed with water, dimethylformamide (DMF), methanol, tetrahydrofuran (THF) and dichloromethane. The resin is treated with a solution of 2.16 g of 2-aminomethylbenzimidazole dihydrochloride (amine reagent), 5.1 ml of diisopropylethylamine (for neutralization) and 2.68 ml of trimethyl orthoformate in 8 ml of dimethylformamide (DMF). After shaking at room temperature for 2 h, a solution of 3.14 g of tetrabutylammonium borohydride and 2.8 ml of acetic acid in 18 ml of dimethylformamide (DMF) is added. The mixture is shaken at room temperature overnight. The resin is then filtered off with suction and washed with dimethylformamide (DMF), methanol, tetrahydrofuran (THF) and dichloromethane. For removal of the product, the resin is shaken with 10 ml of trifluoroacetic acid (TFA)/dichloromethane for 1 h, filtered off, and the filtrate is concentrated in vacuo and purified on silica gel. 190 mg of the title compound are obtained. Mass spectrometry (ESI): 541. Retention time (HPLC): Rt = 7.1.
With potassium phosphate;tetrakis(triphenylphosphine) palladium(0); In water; N,N-dimethyl-formamide; at 80℃; for 2h;
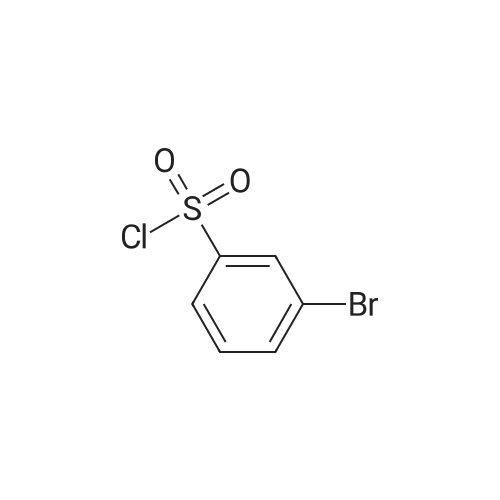
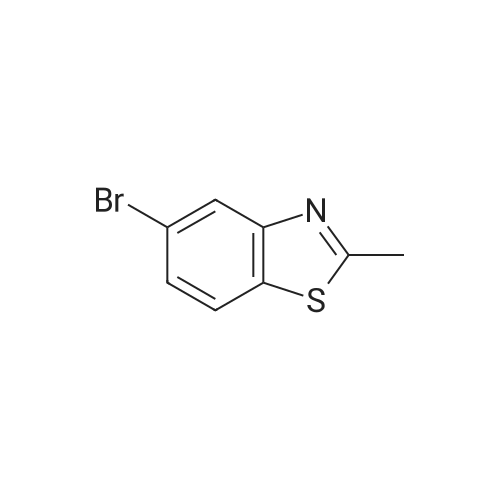
To a stirred suspension of 3-formylphenylboronic acid (0.24 g, 1.61 mniol), 5-bromo-2- methylbenzothiazole (0.35 g, 1.53 mmol) and tetrakis(triphenylphosphine)palladium(0) (5 mg) in DMF (3 mL) was added 2M K3PO4 (aq) (1 mL). The reaction was heated to 80 0C in a ReactiVial for 2 hours. The mixture was cooled to room temperature and the solvent removed in vacuo. The resulting material was partitioned between DCM and H2O, dried over MgSO4 and evaporated to dryness. Material purified by column chromatography eluting with 10% EtOAc in petrol to 20% EtOAc in petrol to give the title compound as a colourless crystalline solid. (0.238 g, 58%). 1H NMR (400 MHz, MeOH-d4): delta 2.78 (s, 3H); 7.53-7.56 (m, 2H); 7.8 (d, IH, J= 7.9); 7.83-7.86 (m, 2H); 7.9 (d, IH, J = 1.53 Hz); 8.05 (m, IH); 9.98 (s, IH). m/z (ES) 254 [M+H].
3-[(3-phenoxyphenyl)[[3-[(3-trifluoromethyl)-2-pyridinyl]phenyl]methyl]amino]-1,1,1-trifluoro-2-propanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
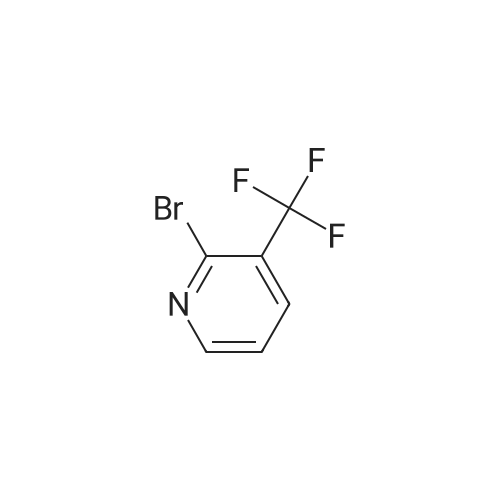
With potassium carbonate;Pd(PPh3)4; In hexane; N,N-dimethyl-formamide; toluene;
3-[(3-phenoxyphenyl)[[3-[(3-trifluoromethyl)-2-pyridinyl]phenyl]methyl]amino]-1,1,1-trifluoro-2-propanol EX-588A) To a toluene (10 mL) solution of <strong>[175205-82-0]2-bromo-3-trifluoromethylpyridine</strong> (1.10 g, 4.87 mmol) was added 3-formylphenylboronic acid (0.90 g, 6.0 mmol) and DMF (4 mL). To the resulting solution was added K2CO3 (1.67 g, 12.1 mmol) and Pd(PPh3)4 (0.35 g, 0.30 mmol). The slurry was heated to reflux under argon for 18 h. The cooled mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried (MgSO4) and evaporated to an oil. Purification by flash chromatography on silica gel eluding with 20% ethyl acetate in hexane gave 0.55 g (45%) of the desired 3-[(3-trifluoromethyl)-2-pyridinyl]benzaldehyde product as a color-less oil which solidified upon standing. HRMS: calcd. for C13H9NOF3: 252.0636 [M+H]+, found: 252.0639. EX-588B) A mixture of solid 3-phenoxyaniline (2.96 g, 16 mmol) and 1,1,1-trifluoro-2,3-epoxypropane (1.30 mL, 15.0 mmol) was placed in a sealed tube and heated to 100 C. giving a dark solution. The stirred solution was heated 18 h and cooled to give a dark oil. Purification by flash chromatography on silica gel eluding with dichloromethane gave 3.15 g (71%) of the desired 3-[(N-3-phenoxy-phenyl)amino]-1,1,1-trifluoro-2-propanol product as a colorless oil. Anal. calcd. for C15H14NO2F3.0.05 CH2Cl2: C, 59.92; H, 4.71; N, 4.64. Found: C, 59.92; H, 4.53; N, 4.73. HRMS calcd. 298.1055 [M+H]+, found: 298.1056. To a 1,2-dichloroethane (8 mL) solution of aldehyde (0.55 g, 2.2 mmol) from EX-588A was added the amine (0.66 g, 2.2 mmol) from EX-588B, NaB(OAc)3H (0.61 g, 2.9 mmol) and acetic acid (0.15 mL, 2.6 mmol). The cloudy solution was stirred at room temperature for 4 h. The reaction mixture was poured into water and extracted with dichloromethane. The organic layer was washed with saturated NaHCO3 and brine, dried (MgSO4) and evaporated to give an oil. Purification by flash chromatography on silica gel eluding with 20% ethyl acetate in hexane gave 0.33 g (29%) of the desired 3-[(3-phenoxyphenyl)[[3-[(3-trifluoromethyl)-2-pyridinyl]phenyl]methyl]amino]-1,1,1-trifluoro-2-propanol product as a white foam, >97% pure by HPLC analysis. Anal. calcd. for C28H22N2O2F6: C, 63.16; H, 4.16; N, 5.26. Found: C, 62.87; H, 4.02; N, 5.33. HRMS: calcd. 533.1664 [M+H]+, found: 533.1658. 1H NMR (C6D6) delta2.97 (d, 1H), 3.26 (dd, 1H), 3.46 (dd, 1H), 3.77 (m, 1H), 4.22 (dd, 2H), 6.31 (dd, 1H), 6.35 (dd, 1H), 6.40 (dd, 1H), 6.54 (t, 1H), 6.80 (t, 1H), 6.9-7.0 (m, 7H), 7.26 (d, 1H), 7.33 (d, 1H), 7.40 (s, 1H), 8.17 (d, 1H). Additional examples of 3-[(3-phenoxyphenyl)[[3-(heteroaryl)phenyl]methyl]-amino]-1,1,1-trifluoro-2-propanols are prepared by one skilled in the art using similar methods, as shown in Example Table 33.
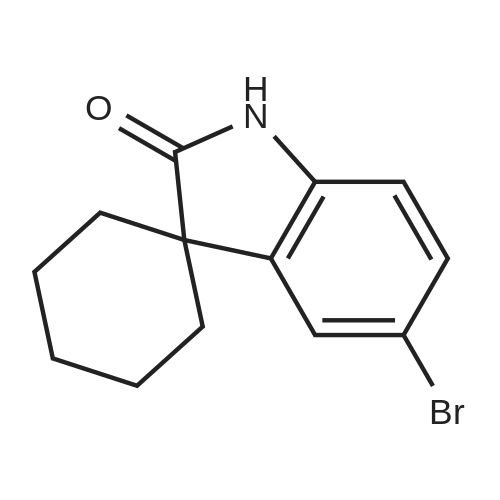
Stage #1: 5'-bromospiro[cyclohexane-1,3'-[3H]indol]-2'(1'H)-one In 1,2-dimethoxyethane for 0.25h;
Stage #2: 3-Formylphenylboronic acid With potassium carbonate In 1,2-dimethoxyethane; water for 20h; Heating/reflux;
To a solution of 5'-bromospiro[cyclohexane-1,3'-[3H]indol]-2'(1'H)-one (1.00 g, 3.57 mmol) in dimethoxyethane (20 cm3) was added tetrakis(triphenylphosphine)palladium (0.20 g, 0.17 mmol). After 15 min. 3-formylphenylboronic acid (1.00 g, 6.93 g) was added followed by potassium carbonate (2.90 g, 21 mmol) in water (10 cm3). After 20 h at reflux, the mixture was cooled poured into water and extracted with EtOAc (×3). The combined organic extract was washed with saturated brine, dried (MgSO4) and evaporated. The residue was purified by column chromatography (SiO2, EtOAc:hexane, gradient elution) to afford the title compound (0.66 g, 2.15 mmol, 60%) as a white solid, 1H NMR (CDCl3) δ 1.65-1.85 (m, 6H), 1.86-2.08 (m, 4H), 7.22 (d, 1H, J=8 Hz), 7.48 (dd, 1H, J=8, 2 Hz), 7.61 (t, 1H, J=8 Hz), 7.66 (d, 1H, J=2 Hz), 7.81-7.88 (m, 2H), 8.06 (t, 1H, J=2 Hz), 8.30 (s, 1H, br), MS ((+)ESI) m/z 306 (M+H)+.
With potassium carbonate In 1,2-dimethoxyethane; water
14 5'-Bromospiro[cyclohexane-13'-[3H]indol]-2'(1'H)-one
To a solution of 5'-bromospiro[cyclohexane-1,3'-[3H]indol]-2'(1'H)-one (1.00 g, 3.57 mmol) in dimethoxyethane (20 cm3) was added tetrakis(triphenylphosphine)palladium (0.20 g, 0.17 mmol) under nitrogen. After 15 min. 3-formylphenylboronic acid (1.00 g, 6.93 g) was added followed by potassium carbonate (2.90 g, 21 mmol) in water (10 cm3). After 20 h at reflux, the mixture was cooled poured into water and extracted with EtOAc (*3). The combined organic extract was washed with sat. brine, dried (MgSO4) and evaporated. The residue was purified by column chromatography (SiO2, EtOAc: hexane, gradient elution) to afford the title compound (0.66 g, 2.15 mmol, 60%) as a white solid, 1H NMR (CDCl3) δ 1.65-1.85 (m, 6H), 1.86-2.08 (m, 4H), 7.22 (d, 1H, J=8 Hz), 7.48 (dd, 1Hz), J=8, 2 Hz), 7.61 (t, 1H, J=8 Hz), 7.66 (d, 1H, J=2 Hz), 7.81-7.88 (m, 2H), 8.06 (t, 1H, J=2 Hz), 8.30 (s, 1H, br); MS ((+)ESI) m/z 306 (M+H)+.
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; water; toluene; at 80℃; for 24h;
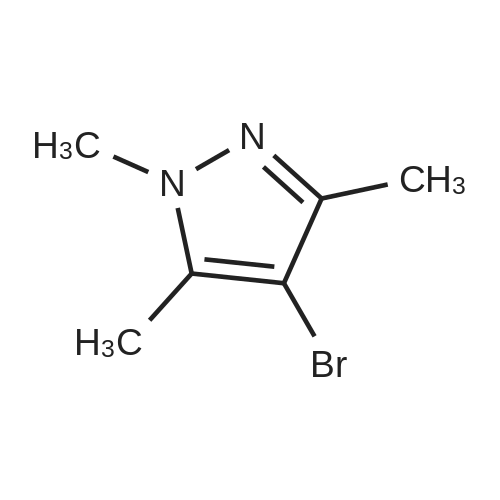
<strong>[15801-69-1]4-Bromo-1,3,5-trimethyl-1H-pyrazole</strong> (2.84 g, 15.0 mmol) and (3-formylphenyl)boronic acid (2.13 g, 15.0 mmol) were dissolved in a mixed solution of 1 M aqueous sodium carbonate solution (30 mL), ethanol (15 mL) and toluene (30 mL), and after argon substitution, tetrakis (triphenylphosphine) palladium (0) (0.867 g, 0.750 mmol) was added. The reaction mixture was stirred under an argon atmosphere at 80C for 24 hr. After cooling the reaction mixture, water and ethyl acetate were added, and the insoluble material was filtered off through celite. The organic layer of the filtrate was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (5%-95% ethyl acetate/hexane) to give the title compound (2.05 g, yield 64%) as a brown oil. MS: m/z 215 (MH+).
48%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; at 80℃; for 12h;Inert atmosphere;
General procedure: The starting materials 1a or 5a (1 equiv) and (3-formylphenyl)boronic acid (1 equiv) were dissolved in a mixture of 1 M sodiumcarbonate aqueous solution (15 mL), EtOH (5 mL) and toluene(15 mL). After nitrogen substitution, Pd(PPh3)4 (0.05 equiv) wasadded. The reaction mixture was stirred at 80 C under nitrogenatmosphere for 12 h. The reaction mixture was cooled, and water(15 mL) was added. The mixture was diluted with AcOEt (15 mL),and the insoluble material was filtered off through Celite. Theorganic layer of the filtrate was washed with brine, dried overanhydrous sodium sulfate, and concentrated in vacuo. The residuewas purified by silica gel column chromatography using a mixtureof petroleum ether/ethyl acetate (10:1, v/v) as eluent to afford thedesired product 2a or 6a as a solid. 4.1.1.2. 3-(1,3,5-trimethyl-1H-pyrazol-4-yl)benzaldehyde (6a). Yield:48%; 1H NMR (300 MHz, DMSO-d6) d: 10.05 (s, 1H), 7.87-7.75 (m,2H), 7.70-7.56 (m, 2H), 3.72 (s, 3H), 2.24 (s, 3H), 2.15 (s, 3H).
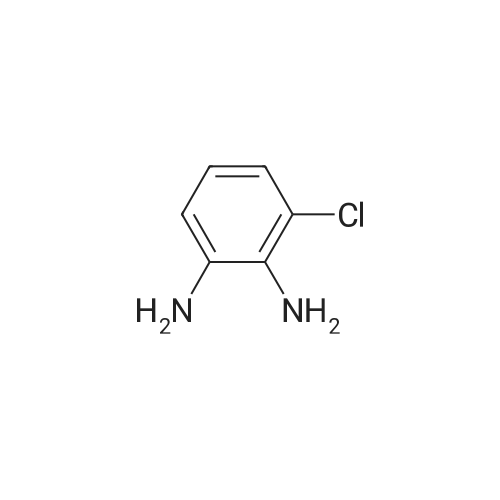
Preparation of [3-(4-Chloro-1H-benzoimidazol-2-yl)-phenyl]boronic acid (13).; To a solution of <strong>[21745-41-5]3-chloro-benzene-1,2-diamine</strong> (11, 12.3 g, 85.9 mmol) and 3-formylphenylboronic acid (12, 14.2 g, 94.7 mmol) (Aldrich) in MeOH (250 mL) was added a solution of sodium metabisulfite (9.0 g, 47.3 mmol) in water (200 mL). The mixture was then heated at 60 C. overnight. The reaction mixture was concentrated to about half of the original volume and the pH adjusted to about 6.5 by glacial acetic acid. The resulting solid was collected by filtration and washed with a small amount of water to afford the desired product as an amber solid.
With sodium hydrogencarbonate;tetrakis(triphenylphosphine) palladium(0); In water; N,N-dimethyl-formamide; at 100℃; for 16h;
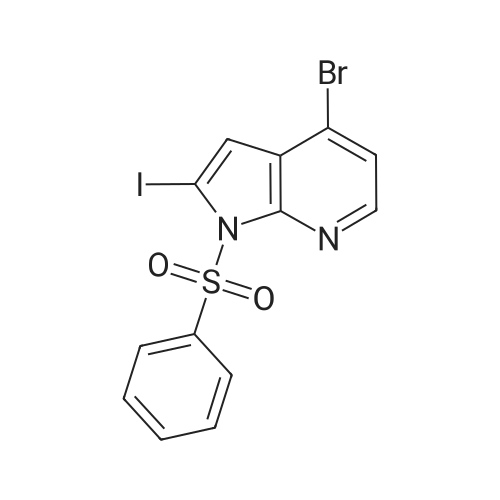
Intermediate 99 ; 4-Bromo-2-(3-formylphenyl)-1-phenylsulfonyl-1H-pyrrolo[2,3-b]pyridine; In a sealed pressure tube was added <strong>[889939-26-8]4-bromo-2-iodo-1-phenylsulfonyl-1H-pyrrolo[2,3-b]pyridine</strong> (2.1 mmol; preparation disclosed in WO03/000690A1), 3-formylbenzeneboronic acid (2.1 mmol), N,N-dimethylformamide (15 mL), aqueous saturated sodium bicarbonate (5 mL) and tetrakis(triphenylphosphine)palladium(0) (0.1 mmol). The reaction was purged with nitrogen, capped and stirred at 100 oC for 16 h. After cooling to room temperature the reaction was concentrated under vacuum, taken up in ethyl acetate, washed with water, brine, dried over sodium sulfate, filtered and evaporated to dryness under vacuum. Purification by flash chromatography by silica gel (0-4% ethyl acetate in methylene chloride) gave the title compound as a white solid (77%). ESMS [M+H]+: 441.2
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; water; toluene; for 1.0h;Heating / reflux;Product distribution / selectivity;
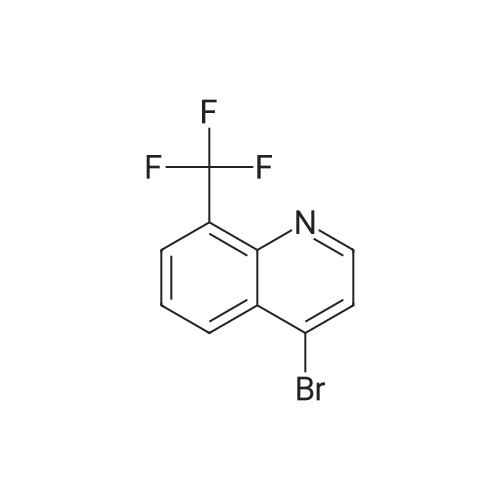
Step 1: 3-[8-(Trifluoromethyl)quinolin-4-yl]benzaldehyde was prepared from <strong>[260973-10-2]4-bromo-8-(trifluoromethyl)quinoline</strong> and 3-formylphenylboronic acid following the procedure of Example 1 Step 3 as a white solid; MS (ES) m/z 301.9;
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; water; toluene; for 1h;Heating / reflux;Product distribution / selectivity;
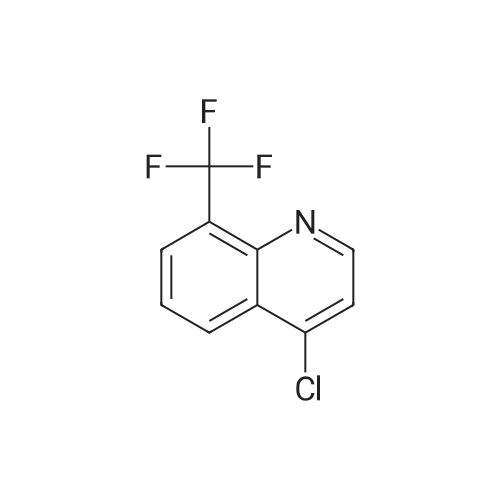
Step 3: A solution of <strong>[23779-97-7]4-chloro-8-(trifluoromethyl)quinoline</strong> (1.65 g, 5.0 mmol), 3-formylphenyl boronic acid (15.0 g, 10 mmol), Pd(PPh3)4 (0.58 g, 0.5 mmol) in 2M aqueous sodium carbonate (10 mL), toluene (20 mL) and ethanol (5 mL) was heated to reflux. After 1 hr, the reaction was cooled, partitioned between water and EtOAc, dried over MgSO4 and concentrated in vacuo, and the residue was purified by chromatography eluding with ethyl acetate/hexanes to give 3-(8-(trifluoromethyl)quinolin-4-yl)benzaldehyde (1.31 g, 86%) as a gummy solid; MS (ESI) m/z 301.9 (M+H)+.
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In tetrahydrofuran; water; toluene; for 24h;Heating / reflux;
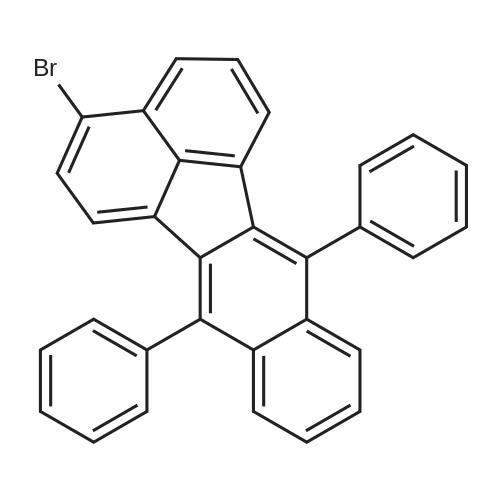
2.5 g of (5.2 mmol) 4-bromo-7,12-diphebenzo[k]fluorantene was dissolved in 20 ml of THF (tetrahydrofuran). Next, 778 mg (5.2 mmol) of 3-formylphenylborate, 300 mg (0.26 mmol) of tetrakis triphenyl phosphine palladium (Pd(PPh3)4) and 3.2 ml of 2M aqueous solution of potassium carbonate (K2CO3) were each dissolved in 20 ml of toluene, added to the THF mixture, and then refluxed for 24 hours. After the reaction was complete, the solvent was removed by evaporation. Next, 500 ml of ethyl acetate and 500 ml of water were each added to wash the resulting product, and an organic layer was collected and dried with anhydride magnesium sulfate. Then the resulting product was separated using silica chromatography to obtain 1.2 g of a compound (yield 34%) represented by Intermediate B above.
With sodium carbonate;bis-triphenylphosphine-palladium(II) chloride; In water; acetonitrile; at 140℃; for 0.5h;Microwave irradiation;
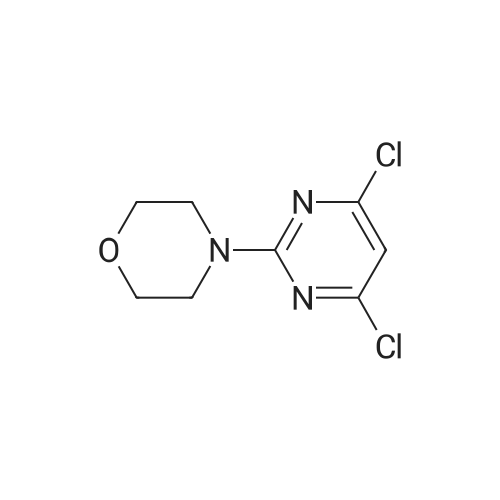
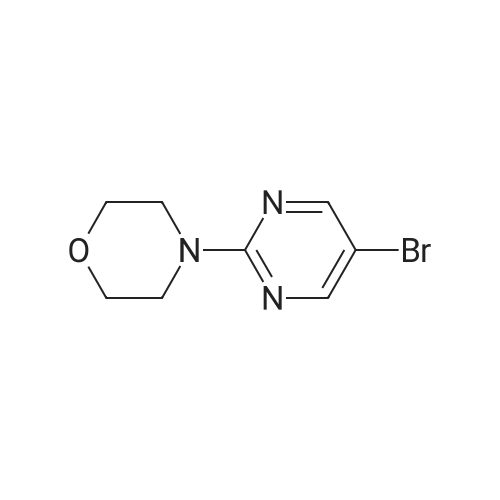
Reference Example 3 3-(6-ChIoro-2-morphoIin-4-yl-pyrimidin-4-yl)- benzaldehyde (Intermediate Cl)Intermediate Al (250 mg, 1.07 mmol) and 3-formylboronic acid (1.0 equiv., 160mg) were taken up in acetonitrile (4ml). To this were added sodium carbonate (3 equiv., 340mg) as a solution in water (ImI) and PdCl2(PPh3)2 (0.05 equiv.). The reaction mixture was heated in microwave at 14O0C for 30 min. Purification using DCM/brine extraction and flash chromatography gave 3-(6-chloro-2-morpholin-4-yl- pyrimidin-4-y l)-benzaldehyde (101 mg) .
With sodium hydrogencarbonate; In ethanol; water; ethyl acetate;
Alternate preparation of Intermediate I-X-9: [3-(3',5'-difluoro-4'-formyl-3-biphenylyl)-1H-1,2,4-triazol-1-yl]methyl 2,2-dimethylpropanoate A flask was charged with PhMe/EtOH (4:1, 50 mL), water (25 mL) and NaHCO3 (3.78 g; 45 mmol) and the mixture was sparged with N2 for 15 min in an ultrasonic bath. [3-(3-bromophenyl)-1H-1,2,4-triazol-1-yl]methyl 2,2-dimethylpropanoate (5.07 g; 15.0 mmol; Ex IV-24), 3,5-difluoro-4-formyl-phenyl boronic acid (3.07 g; 16.5 mmol) and PdCl2(dppf).CH2Cl2 (0.245 g; 0.30 mmol) were added in one portion and the mixture was heated under reflux for 10 h. Upon cooling, solids were collected by filtration, washed (Et2O/hexane), dissolved in hot EtOAc and filtered without delay through a short pad of silica get (1:1 EtOAc/hexanes eluent). Filtrate collected from the reaction mixture was diluted with water/EtOAc, separated and the aqueous layer extracted with EtOAc (*3). Combined organics were washed (water, brine), dried over Na2SO4 and concentrated in vacuo. The residue obtained from the extracts was dissolved in hot EtOAc, filtered through silica (1:1 EtOAc/hexanes eluent), and the filtrate combined with that obtained from the crude reaction mixture solids. Combined filtrates were concentrated in vacuo, affording the title compound as a pale yellow solid. In another preparation of the title compound, Pd(OAc)2/S-Phos (0.005/0.010 equiv) were used as catalyst, NaHCO3 (3 equiv) was used as base, and PhMe/water were used as solvents (without EtOH additive), reaction time 3 h/85 C. Product I-X-9 was isolated in a manner analogous to the above procedure. Choice of base appears to be a key parameter in the cross-coupling of Ex. IV-24 under typical biphasic conditions. In our hands, NaHCO3 was an effective and reliable base for preparation of I-X-9. On some occasions, the use of Na2CO3,as base resulted in stalled reactions; mixtures of IV-24 and I-X-9 were returned, accompanied by varying amounts of deprotected IV-24 (i.e., 3-bromo phenyl-1H-1,2,4-triazole). Intermediate I-X-17: [3-(3'-formyl-4-biphenylyl)-1H-1,2,4-triazol-1-yl]methyl 2,2-dimethylpropanoate To a solution of [3-(4-bromophenyl)-1H-1,2,4-triazol-1-yl]methyl 2,2-dimethyl-propanoate (0.108 g, 0.32 mmol; IV-30), 3-formylphenyl boronic acid (0.099 g, 0.66 mmol), and PdCl2(dppf).CH2Cl2 (0.024 g, 0.029 mmol) was added 2M Na2CO3 (aq) (0.38 mL, 0.76 mmol). The mixture was subjected to microwave heating (135 C./50 min), cooled, and partitioned between EtOAc and water. The aqueous layer was extracted with EtOAc, combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (EtOAc/hexanes), affording the title compound as a colorless solid. 1H NMR (400 MHz, CDCl3) deltappm 1.20 (s, 9H), 6.10 (s, 2H), 7.63 (t, J=7.69 Hz, 1H), 7.73 (d, J=8.06 Hz, 2H), 7.90 (dd, J=14.65, 7.57 Hz, 2H), 8.15 (m, 1H), 8.23 (d, J=8.30 Hz, 2H), 8.39 (s, 1H), 10.10 (s, 1H).
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; at 120℃; for 0.333333h;Sealed tube; Microwave irradiation;
INTERMEDIATE 13 -(Quinoxalin-6-yl)benzaldehydeA mixture of <strong>[50998-17-9]<strong>[50998-17-9]6-bromoquinoxalin</strong>e</strong> (210 mg, 1.01 mmol), 3-formylphenylboronic acid (301 mg, 2.01 mmol), 2M aqueous sodium carbonate solution (1.7 mL, 14.4 mmol) and Pd(PPh3)4 (35 mg, 0.03 mmol) in DME (3.5 mL) was heated to 1200C in a sealed tube, under microwave irradiation, for 20 minutes. After cooling, the organic phase was adsorbed onto silica and purified by column chromatography (SiO2, 10-100% EtOAc in heptane) to give the title compound (340 mg) as a beige solid. 6H (CDCl3) 10.15 (s, IH), 8.91 (d, IH), 8.89 (d, IH), 8.38 (d, IH), 8.26-8.31 (m, IH), 8.24 (d, IH), 8.10 (dd, IH), 8.02-8.07 (m, IH), 7.94-8.00 (dd, IH), 7.72 (t, IH). LCMS (ES+) 235 (M+H)+, RT 2.99 minutes.
With potassium phosphate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; at 140℃; for 0.5h;Sealed tube; Microwave irradiation;
INTERMEDIATE 383-(Pyridor3,4-&1pyrazin-7-ylN)benzaldehyde 7-Chloropyrido[3,4-Z?]pyrazine (140 mg, 0.85 mmol), 3-formylbenzeneboronic acid (127 mg, 0.85 mmol), potassium phosphate (180 mg, 0.85 mmol), water (2 mL), DME (6 mL) and Pd(PPh3)4 (100 mg, 0.085 mmol) were combined in a sealed tube and heated under microwave irradiation to 1400C for 30 minutes. The reaction mixture was then partitioned between ethyl acetate and water. The organic layer was dried (MgSO4) and concentrated to dryness to give the title compound (155 mg, 77%) as an off-white solid. deltaH (DMSOd6) 10.21 (IH, s), 9.69 (IH, s), 9.26 (IH, d, J 1.82 Hz), 9.15 (IH, d, J 1.83 Hz), 8.93 (IH, s), 8.76 (IH, s), 8.70 (IH, d, J 7.80 Hz), 8.07 (IH, d, J 7.61 Hz), 7.84 (lH, t, J7.68 Hz).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In water; toluene; at 80℃; for 12h;Inert atmosphere;
General procedure: Thecompound 30 (1 equiv) and(3-formylphenyl)boronic acid (1 equiv) were dissolved in a mixture of 1 Msodium carbonate aq. (15 mL), EtOH (5mL) and toluene (15 mL). After nitrogensubstitution, Pd(PPh3)4 (0.05equiv) was added. The reaction mixture was stirred at 80 C under nitrogen atmospherefor 12 h. The reaction mixture was cooled, and water (15 mL) was added. Themixture was diluted with ethyl acetate (15 ml), and the insoluble material wasfiltered off through Celite. The organic layer of the filtrate was washed withbrine, dried over anhydrous sodium sulfate, and concentrated in vacuo. Theresidue was purified by silica gel column chromatographyusing a mixture of petroleum ether/ethyl acetate (10:1, v/v) as eluent toafford the desired product as a solid.
92%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; at 80℃; for 12h;Inert atmosphere;
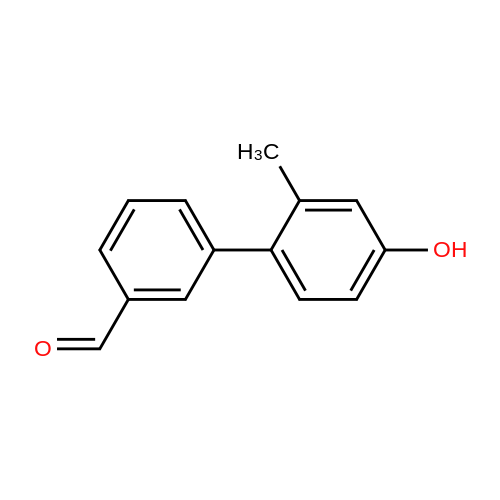
General procedure: The bromobenzene (1 equiv) and (3-formylphenyl)boronic acid (1 equiv) were dissolved in a mixture of 1M sodium carbonate solution (15 mL), EtOH (5 mL) and toluene 15 mL). After nitrogen substitution, Pd(PPh3)4 (0.05 equiv) wasadded. The reaction mixture was stirred at 80 C under nitrogen atmosphere for 12 h. The reaction mixture was cooled, and wate r(15 mL) was added. The mixture was diluted with ethyl acetate(15 mL), and the insoluble material was filtered off through Celite. The organic layer of the filtrate was washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo. The residue was purified by column chromatography using a mixture of petroleum ether/ethyl acetate (10:1, v/v) as eluent to afford the desired product as a solid. 4?-Hydroxy-2?-methyl-3-biphenylcarbaldehyde (5a). Yield:92%; white solid; 1H NMR (300 MHz, CDCl3) d: 10.04 (s, 1H), 7.87 (d,J 7.6 Hz, 1H), 7.66 (t, J 1.7 Hz, 1H), 7.59e7.53 (m, 2H), 7.42 (d,J 7.4 Hz, 1H), 6.89 (d, J 1.5 Hz, 1H), 6.76e6.72 (m, 1H), 4.67 (s,1H), 1.98 (s, 3H).
92%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; at 80℃; for 12h;Inert atmosphere;
General procedure: The bromobenzene (1 equiv) and (3-formylphenyl)boronic acid (1 equiv) were dissolved in a mixture of 1M sodium carbonate solution (15mL), EtOH (5mL) and toluene (15mL). After nitrogen substitution, Pd(PPh3)4 (0.05 equiv) was added. The reaction mixture was stirred at 80C under nitrogen atmosphere for 12h. The reaction mixture was cooled, and water (15mL) was added. The mixture was diluted with ethyl acetate (15mL), and the insoluble material was filtered off through Celite. The organic layer of the filtrate was washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo. The residue was purified by column chromatography using a mixture of petroleum ether/ethyl acetate (10:1, v/v) as eluent to afford the desired product as a solid.4.1.1.4. 4?-Hydroxy-2?-methyl-3-biphenylcarbaldehyde (11a).Yield: 92%; white solid; 1H NMR (300MHz, CDCl3) delta: 10.04 (s, 1H), 7.87 (d, J=7.6Hz, 1H), 7.66 (t, J=1.7Hz, 1H), 7.59-7.53 (m, 2H), 7.42 (d, J=7.4Hz, 1H), 6.89 (d, J=1.5Hz, 1H), 6.76-6.72 (m, 1H), 4.67 (s, 1H), 1.98 (s, 3H).
20%
With bis-triphenylphosphine-palladium(II) chloride; sodium carbonate; In water; N,N-dimethyl-formamide; at 120℃; for 0.166667h;Microwave irradiation;
Step 1 a Synthesis of 4'-Hydroxy-2'-methyl-[1 ,1 '-biphenyl]-3-carbaldehyde To a solution of <strong>[14472-14-1]4-bromo-3-methylphenol</strong> (300 mg. 1 .604 mM) and (3- formylphenyl)boronic acid (361 mg, 2.406 mMI) in DMF:H20 (2 ml:0.2 ml), sodium carbonate (340 mg, 3.21 mM) was added. To the resulting solution PdCI2(PPh3)2 (56 mg, 0.08 mM) was added and the mixture was heated at 120 QC for 10 min in microwave. The reaction mixture was diluted with ethyl acetate (100 ml) and water (10 ml), organic layer was separated and washed with brine, dried and concentrated. The crude compound was purified by column chromatography to obtain the compound 4'-hydroxy-2'-methyl-[1 ,1 '-biphenyl]-3-carbaldehyde (68 mg, 0.320 mM). Yield: 20 %; 1 H NMR (300 MHz, DMSO-d6): delta 10.07 (s, 1 H), 7.86-7.83 (m, 2H), 7.59 (d, J= 4.2 Hz, 2H), 7.14 (d, J= 8.1 Hz, 2H), 6.80-6.75 (m, 2H), 2.25 (s, 3H); MS: (m/z) 213 (M+1 ).
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; water; toluene; at 80℃; for 13h;Inert atmosphere;
Production Example 11 Under a nitrogen atmosphere, to a mixture of sodium carbonate (5.67 g), water (28 ml), <strong>[14472-14-1]4-bromo-3-methylphenol</strong> (5.00 g), (3-formylphenyl)boronic acid (4.40 g), ethanol (20 ml) and toluene (40 ml), tetrakistriphenylphosphinepalladium (1.54 g) was added, stirred at 80C for 13 hours, and cooled to room temperature. To the reaction mixture, activated carbon (0.5 g) was added, stirred for 5 minutes, filtrated over Celite, and washed with ethyl acetate and water. The obtained filtrate was separated, and the aqueous layer was extracted with ethyl acetate. The organic layer was combined and washed with water and a saturated aqueous solution of sodium chloride, and then anhydrous magnesium sulfate and activated carbon (0.5 g) was added. The desiccant and activated carbon were removed by filtration, the filtrate was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane-ethyl acetate) to obtain 4.42 g of 4'-hydroxy-2'-methylbiphenyl-3-carbaldehyde.
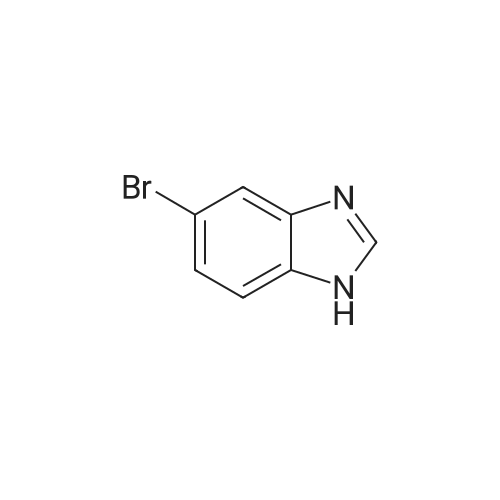
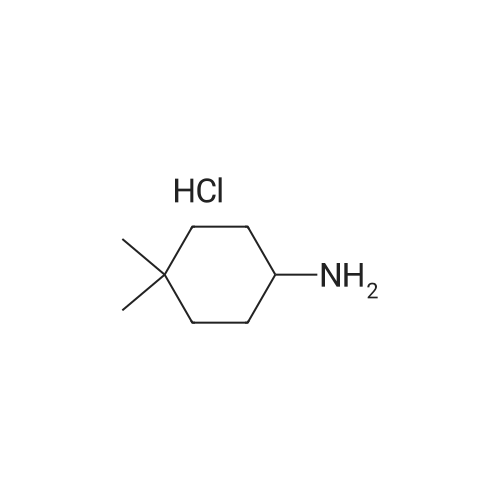
To a solution of 3-[1-(triphenylmethyl)-1H-benzimidazol-5-yl]benzaldehyde and 3-[1-(triphenylmethyl)-1H-benzimidazol-6-yl]benzaldehyde (0.22 g, 0.47 mmol) in dichloromethane (5 mL) was added 1 drop of acetic acid, followed by 4,4-dimethylcyclohexylamine hydrochloride (78 mg, 0.47 mmol) and triethylamine (66 muL, 0.47 mmol). NOTE: If a free amine was used, triethylamine was not added. After stirring 60 min at room temperature, sodium triacetoxyborohydride (0.40 g, 1.9 mmol) was added and the reaction was stirred an additional 3 h. The reaction mixture was diluted with H2O and CH2Cl2. The layers were separated and the aqueous layer was extracted 2× CH2Cl2. The combined organics were washed with brine, dried over Na2SO4, and concentrated in vacuo. Chromatography provided the reductive amination product as a mixture of trityl regioisomers, (4,4-dimethylcyclohexyl)({3-[1-(triphenylmethyl)-1H-benzimidazol-6-yl]phenyl}methyl)amine and (4,4-dimethylcyclohexyl)({3-[1-(triphenylmethyl)-1H-benzimidazol-5-yl]phenyl}methyl)amine. (M+1) 576.4 ES, 2.40 min and 2.51 min (LC/MS Method A).
To a solution of (4,4-dimethylcyclohexyl)({3-[1-(triphenylmethyl)-1H-benzimidazol-6-yl]phenyl}methyl)amine and (4,4-dimethylcyclohexyl)({3-[1-(triphenylmethyl)-1H-benzimidazol-5-yl]phenyl}methyl)amine (0.18 g, 0.31 mmol) in tetrahydrofuran (3 mL) were added water (3 mL) and acetic acid (3 mL). The reaction mixture was heated at reflux for 1 h, then was allowed to cool to room temperature and was treated with 10% HCl (aq) until pH 2. The mixture was washed 2× EtOAc, then the aqueous phase was treated with solid K2CO3 until it reached pH 10, at which point it was extracted 3× CHCl3. The combined CHCl3 extracts were dried (Na2SO4) and concentrated in vacuo. The residue was taken up in Et2O and enough acetone to dissolve completely, then 4M HCl in dioxane was added until no more solid crashed out. The white solid was collected by filtration and dried under vacuum, providing the product [3-(1H-benzimidazol-5-yl)phenyl]methyl}(4,4-dimethylcyclohexyl)amine hydrochloride (M+1) 334.2 ES, 1.30 min (LC/MS Method A).
To a solution of 5-bromo-1-(triphenylmethyl)-1H-benzimidazole and 6-bromo-1-(triphenylmethyl)-1H-benzimidazole (1:1 mixture) (6.0 g, 13.7 mmol) in toluene (30 mL), was added tetrakis(triphenylphosphine) palladium(0) (0.8 g, 0.7 mmol), followed by a solution of sodium bicarbonate (2.9 g, 34.2 mmol) in water (15 mL). A solution of 3-formylphenylboronic acid (2.3 g, 15.0 mmol) in ethanol (8 mL) was then added and the reaction was heated at reflux overnight. After cooling, the bulk of the solvent was removed in vacuo, and the residue was partitioned between ethyl acetate and water. The organics were washed with brine, dried (Na2SO4), and concentrated in vacuo. Chromatography of the residue on silica gel (35-50% EtOAc/hexanes) provided the product as an inseparable mixture of trityl regioisomers, 3-[1-(triphenylmethyl)-1H-benzimidazol-5-yl]benzaldehyde and 3-[1-(triphenylmethyl)-1H-benzimidazol-6-yl]benzaldehyde. (M+1) 465.2 ES, 3.01 min and 3.05 min (LC-MS Method A).
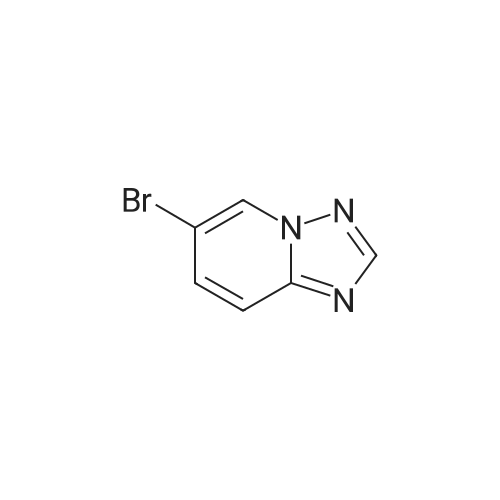
With caesium carbonate;[1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II); In water; dimethyl sulfoxide; at 45℃;
Step C: To a mixture of 3-formylphenylboronic acid (21.41 g, 143 mmol),6-bromo-[l,2,4]triazolo[l,5-a]pyridine (28.27 g, 143 mmol) in DMSO (600 mL) and water (50 mL) was added Pd(dppf)Cl2 (5.83 g, 7.14 mmol) and Cs2CO3 (116 g, 357 mmol). The reaction temperature reached 45 0C after the addition. HPLC showed that starting materials were consumed after 15 min. The reaction was diluted with water (400 mL). The black precipitate was collected by filtration and dissolved in DCM (300 mL), and washed with brine (200 mL). The aqueous layer was back extracted with DCM (100 mL). The combined organic layers were filtered through a Celite pad and the filtrate was concentrated to give a black solid mixture. The product was recrystallized in methanol to give 3-([l,2,4]triazolo[l,5-a]pyridin-6-yl)benzaldehyde (27.4 g, 123 mmol, 86 % yield) as a pale grey solid: m/z = 224.0 [M+l]; 1H NMR (400 MHz, DMSO-D6) delta ppm 7.74 (t, J=7.68 Hz, 1 H), 7.91 - 8.02 (m, 2 H), 8.11 (dd, J=9.19, 1.89 Hz, 1 H), 8.17 (d, J=7.81 Hz, 1 H), 8.36 (s, 1 H), 8.57 (s, 1 H), 9.45 (s, 1 H), 10.11 (s, 1 H).
86%
With caesium carbonate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In water; dimethyl sulfoxide; at 45℃; for 0.25h;
To a mixture of 3-formylphenylboronic acid (21.41 g, 143 mmol),6-bromo-[l,2,4]triazolo[l,5-a]pyridine (28.27 g, 143 mmol) in DMSO (600 mL) and water (50 mL) was added Pd(dppf)Cl2 (5.83 g, 7.14 mmol) and Cs2CO3 (116 g, 357 mmol). The reaction temperature reached 45 0C after the addition. HPLC showed that starting materials were consumed after 15 min. The reaction was diluted with water (400 mL). The black precipitate was collected by filtration and dissolved in DCM (300 mL), and washed with brine (200 mL). The aqueous layer was back extracted with DCM (100 mL). The combined organic layers were filtered through a Celite pad and the filtrate was concentrated to give a black solid mixture. The product was recrystallized in methanol to give 3-([l,2,4]triazolo[l,5-a]pyridin-6-yl)benzaldehyde (27.4 g, 123 mmol, 86 % yield) as a pale grey solid: m/z = 224.0 [M+l]; 1H NMR (400 MHz, DMSO-D6) delta ppm 7.74 (t, J=7.68 Hz, 1 H), 7.91 - 8.02 (m, 2 H), 8.11 (dd, J=9.19, 1.89 Hz, 1 H), 8.17 (d, J=7.81 Hz, 1 H), 8.36 (s, 1 H), 8.57 (s, 1 H), 9.45 (s, 1 H), 10.11 (s, 1 H).
86%
With caesium carbonate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In water; dimethyl sulfoxide; at 45℃; for 0.25h;
To a mixture of 3-formylphenylboronic acid (21.41 g, 143 mmol),6-bromo-[l,2,4]triazolo[l,5-a]pyridine (28.27 g, 143 mmol) in DMSO (600 mL) and water (50 mL) was added Pd(dppf)Cl2 (5.83 g, 7.14 mmol) and Cs2CO3 (116 g, 357 mmol). The reaction temperature reached 45 0C after the addition. HPLC showed that starting materials were consumed after 15 min. The reaction was diluted with water (400 mL). The black precipitate was collected by filtration and dissolved in DCM (300 mL), and washed with brine (200 mL). The aqueous layer was back extracted with DCM (100 mL). The combined organic layers were filtered through a Celite pad and the filtrate was concentrated to give a black solid mixture. The product was recrystallized in methanol to give 3-([l,2,4]triazolo[l,5-a]pyridin-6-yl)benzaldehyde (27.4 g, 123 mmol, 86 % yield) as a pale grey solid: m/z = 224.0 [M+l]; 1H NMR (400 MHz, DMSO-D6) delta ppm 7.74 (t, 7=7.68 Hz, 1 H), 7.91 - 8.02 (m, 2 H), 8.11 (dd, 7=9.19, 1.89 Hz, 1 H), 8.17 (d, 7=7.81 Hz, 1 H), 8.36 (s, 1 H), 8.57 (s, 1 H), 9.45 (s, 1 H), 10.11 (s, 1 H).
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; water; at 100℃; for 4h;Inert atmosphere;
Step 1: Synthesis of N-((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-3'-formyl-4-methyl-[1,1'-biphenyl]-3-carboxamide To a stirred solution of <strong>[1403257-80-6]5-bromo-N-((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methylbenzamide</strong> (400 mg, 0.84 mmol) and (3-formylphenyl)boronic acid (189 mg, 1.26 mmol) in dioxane (2 mL), aqueous 2M Na2CO3 solution (1.5 mL, 3.03 mmol) was added and solution was purged with argon for 15 min. Then Pd(PPh3)4 (97 mg, 0.08 mmol) was added and argon was purged again for 15 min. Reaction mass was heated at 100 C. for 4 h. On completion, reaction mixture was diluted with water and extracted with 10% MeOH/DCM (3 times). Combined organic layer was dried over sodium sulphate. Removal of the solvent under reduced pressure followed by column chromatographic purification afforded the title compound (270 mg, 64%).
A mixture of 4-bromopyridine hydrochloride 15 (2.00 g, 10.3 mmol, 1.0 equiv) in water (16 ml) and toluene (22 ml) was cooled to 0 C and a solution of Na2CO3 (2.51 g, 23.8 mmol, 2.3 equiv) in water (26 ml) was added. After warming to rt, 3-formylphenylboronic acid 16 (1.62 g, 10.8 mmol, 1.04 equiv) and Pd(PPh3)4 (0.600 g, 0.515 mmol, 0.05 equiv) were added and the reaction mixture was stirred at 85 C for 18 h. Then, the cooled solution was diluted with CH2Cl2 (40 ml) and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 16 ml) and the combined organic layers were dried over MgSO4. The solvent was removed under reduced pressure and the crude product was purified by column chromatography (n-hexane/EtOAc 4:1 ? 1:4, v/v) to obtain 17 as a white solid (1.77 g, 94%); mp: 34-36 C; IR (KBr): nu? nu? = 3440, 3025, 1689, 1582, 1379, 1188, 789, 690, 653 cm-1; 1H NMR (400 MHz, CDCl3): delta = 7.51-7.53 (m, 2H), 7.63-7.67 (m, 1H), 7.87-7.94 (m, 2H), 8.12-8.13 (m, 1H), 8.68-8.70 (m, 2H), 10.08 (s, 1H); 13C NMR (100 MHz, CDCl3): delta = 121.7, 127.9, 130.0, 130.5, 132.9, 137.2, 139.3, 147.0, 150.6, 191.8; ESI-HRMS m/z calcd for C12H9NO: 206.05764 [M+Na]+, 389.12605 [2M+Na]+, found: 206.05779, 389.12615
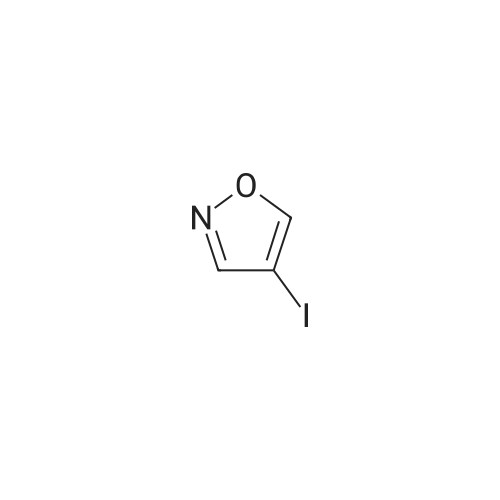
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; ethanol; water; toluene; at 80℃; for 3h;Inert atmosphere;
[00460] Step 2: Synthesis of 3-(isoxazol-4-yl)benzaldehyde: To a mixture of 4- iodoisoxazole (1 g, 5.13 mmol), 3-formylphenylboric acid (923 mg, 6.15 mmol) and sodium carbonate in DME/H20/Toluene/EtOH (15 mL, 3/1/10/6, V/V) was added Pd(PPh3)4 (200 mg). The mixture was purged with N2 for 30 min and heated to 80 C for 3 h. The reaction mixture was cooled followed by a standard aqueous/EtOAc workup and purified by prep- TLC (EA : PE = 1 : 5) to give Intermediate 46 (12 mg, 1.5%).
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium carbonate; In 1,4-dioxane; water; at 80℃; for 5h;Inert atmosphere;
a) Preparation of intermediate 63 A mixture of 5-bromo-2-fluorobenzotrifluoride (4 g, 16.46 mmol), 3-formyl- phenylboronic acid (2.96 g, 19.75 mmol), PdCl2(dppf)) (0.602 g, 0.823 mmol) and K2C03 (7.571 g, 32.92 mmol) in dioxane/water 5:1 (50 mL) was degassed for a few min with N2. The r.m. was then heated at 80 C for 5 h. The dioxane was removed under vacuum and the residual extracted with DCM. The organic layer was dried over Na2S04, filtered and the solvent was evaporated. The residue was purified by flash chromatography (silica; EtO Ac/petroleum ether 0/100 to 1/15). The desired fractions were collected and the solvent was evaporated to give intermediate 63 (3.4 g, 77%).
With [bis(acetoxy)iodo]benzene; triethylamine; 1,1'-carbonyldiimidazole; In dichloromethane; at 20℃; for 3h;
General procedure: To a mixture of benzoic acid (1 mmol), carbonyldiimidazole (1mmol), triethylamine, (5 mmol) and boronic acid (1 mmol) in dichlorormethane (5mL) were charged to PhI(OAc)2 (0.38g, 1.2 mmol). The reaction mixture was stirred at room temperature for 3h. After complete conversion, as indicated by TLC (9:1 Hexane:EtOAc), the reaction mixture was evaporated under reduced pressure and theresidue was purified by flash column chromatography on silica gel (2% ethylacetate inpetroleum ether) to give the product.
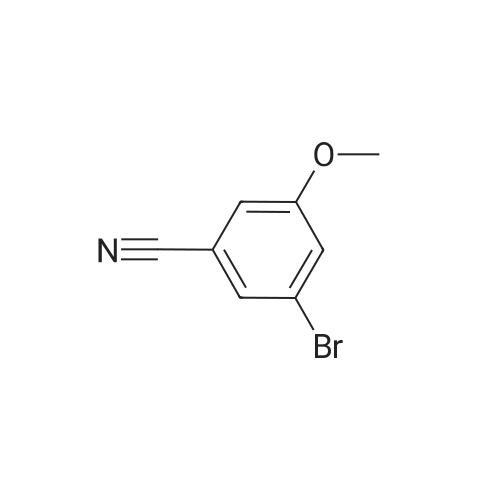
3'-formyl-5-methoxy-[1,1'-biphenyl]-2-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: A suspension of 3-bromobenzaldehyde (200 mg; commercial) and (3-cyanophenyl)boronic acid neopentyl glycol ester (325 mg; commercial) in toluene/EtOH (2.3 mL; 1 : 1) was treated with sat. aq. Na2C03 (2.3 mL) and degassed by bubbling with nitrogen for 5 min. The suspension was treated with Pd(PPh3)4 (28 mg) and refluxed overnight in a sealed tube. The reaction mixture was allowed to reach rt and diluted with water and EA. The aq. layer was extracted with EA and the combined org. layers were washed with brine, dried over MgS04, filtered and evaporated under reduced pressure. After purification by CC (Hept/EA 2: 1 to 1 : 1), the title compound was obtained as an off-white solid (325 mg; quantitative yield). Starting from <strong>[127666-99-3]2-chloro-4-methoxybenzonitrile</strong> (223 mg; commercial) and 3-formylphenylboronic acid (200 mg; commercial) and proceeding in analogy to Preparation A, the title compound was obtained as a beige solid (325 mg; 100% yield as crude material). 1H MR (CDCI3) delta: 10.10 (s, 1H); 8.03 (s, 1H); 7.98 (m, 1H); 7.85 (m, 1H); 7.69 (m, 3H); 6.98 (s, 1H); 3.91 (m, 3H).
3'-formyl-5-methoxy-[1,1'-biphenyl]-3-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
55%
General procedure: A suspension of 3-bromobenzaldehyde (200 mg; commercial) and (3-cyanophenyl)boronic acid neopentyl glycol ester (325 mg; commercial) in toluene/EtOH (2.3 mL; 1 : 1) was treated with sat. aq. Na2C03 (2.3 mL) and degassed by bubbling with nitrogen for 5 min. The suspension was treated with Pd(PPh3)4 (28 mg) and refluxed overnight in a sealed tube. The reaction mixture was allowed to reach rt and diluted with water and EA. The aq. layer was extracted with EA and the combined org. layers were washed with brine, dried over MgS04, filtered and evaporated under reduced pressure. After purification by CC (Hept/EA 2: 1 to 1 : 1), the title compound was obtained as an off-white solid (325 mg; quantitative yield). Starting from <strong>[867366-91-4]3-bromo-5-methoxybenzonitrile</strong> (1 13 mg; commercial) and 3-formylphenylboronic acid (80 mg; commercial) and proceeding in analogy to Preparation A, the title compound was obtained, after CC purification (Hept/EA 2: 1 to 1 : 1), as a colourless solid (69 mg; 55% yield). 1H NMR (CDCI3) delta: 10.10 (s, 1H); 8.07 (m, 1H); 7.93 (m, 1H); 7.82 (m, 1H); 7.66 (m, 1H); 7.49 (m, 1H); 7.36 (m, 1H); 7.17 (m, 1H); 3.90 (m, 3H).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In acetonitrile; for 1.83333h;Inert atmosphere;
General procedure: A mixture of 4-bromobenzaldehyde (2.78 g; 15.0 mmol), [3-(aminocarbonyl)-phenyl]boronic acid (2.72 g; 16.5 mmol), PdCl2(dppf).CH2Cl2(0.306 g; 0.38 mmol), DME (25 mL) and Na2CO3(25 mL of a 2M solution) was sparged 20 min with N2and heated under reflux for 90 min (consumption of aryl bromide observed by LC/MS). Upon cooling, the mixture was partitioned between EtOAc/H2O, layers were separated, and the aqueous layer was extracted with EtOAc (×2). Combined organics were washed (H2O, brine), dried over Na2SO4and concentrated in vacuo. The residue was purified by flash chromatography (EtOAc/hexanes), affording the title compound as a tan solid. Used IV-4 and 3- formylphenyl boronic acid. Note 2, 4. Note 2 Used Pd(PPh3)4as catalyst (instead of PdCl2(dppf).CH2Cl2). Note 4 Used acetonitrile/0.4M Na2CO3instead of DME/2M Na2CO3.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; at 80℃;Inert atmosphere;
2 -Bromo- 1,3 -dimethyl benzene (20. Og, 108.04 mmol) and 3-formylphenylboronic acid (19.43g, 129.64 mmol) were dissolved in mixture of 1M aqueous solution of sodium carbonate (292 mL), ethanol (100 mL) and toluene (300 mL). The reaction mixture was degassed with argon gas for 10 min and Pd(PPh3)4 (6.24 g, 5.402mmol) was added. Then the reaction was heated to 80C for 18 h under inert atmosphere. The solution was cooled to RT and filtered through celite. The filtrate was concentrated and the aq. layer extracted with ethyl acetate. The organic layer was dried over Na2S04, and concentrated. The crude compound was purified by column chromatography on silica gel (230-400 mesh) using 2-4% of ethyl acetate in pet-ether as an eluent to afford Compound l-2a (7.5g, 32.8%). LC-MS: 2.36mins, [M+H]+ 211
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; at 80℃; for 12h;Inert atmosphere;
General procedure: Step 1: The substituted bromobenzene (1 equiv) and (3-formylphenyl)boronic acid or (4-formylphenyl) boronic acid (1 equiv) were dissolved in a mixture of 1M sodium carbonate aqueous solution (15mL), EtOH (5mL) and toluene (15mL). After nitrogen substitution, Pd(PPh3)4 (0.05 equiv) was added. The reaction mixture was stirred at 80C under nitrogen atmosphere for 12h. The reaction mixture was cooled, and water (15mL) was added. The mixture was diluted with AcOEt (15mL), and the insoluble material was filtered off through Celite. The organic layer of the filtrate was washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo. The residue was purified by silica gel column chromatography using a mixture of petroleum ether/ethyl acetate (10:1, v/v) as eluent to afford the desired product 2a-r, 5a and 5b as a solid. Step 2: To a solution of 2a-r, 5a or 5b (1 equiv) in MeOH (10mL) and THF (20mL) was added portionwise sodium borohydride (3 equiv) at 0C and the mixture was stirred at 0C for 1h. The reaction mixture was pouring into ice water (10mL), and extracted with ethyl acetate (3×15mL), the organic fractions were combined, washed with saturated brine (2×15mL) prior to drying over anhydrous sodium sulfate. After filtration and concentrate using a rotary evaporator, the residue was used in next step without further purification. To a solution of the obtained solid (1 equiv) in dichloromethane (20mL) was slowly added thionyl chloride (6 equiv) and a catalytic amount of DMF at room temperature. After stirring at 40C for 4h, the reaction was concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a mixture of petroleum ether/ethyl acetate (20:1, v/v) as eluent to afford the desired product. 4.1.2 3'-(chloromethyl)-2,6-dimethyl-1,1'-biphenyl (3a) (0020) Yield: 65%; 1H NMR (300MHz, CDCl3) δ: 7.44-7.39 (m, 1H), 7.34 (d, J=7.9Hz, 1H), 7.07-7.19 (m, 5H), 4.52 (s, 2H), 1.97 (s, 6H).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; at 80℃; for 12h;Inert atmosphere;
General procedure: Step 1: The substituted bromobenzene (1 equiv) and (3-formylphenyl)boronic acid or (4-formylphenyl) boronic acid (1 equiv) were dissolved in a mixture of 1M sodium carbonate aqueous solution (15mL), EtOH (5mL) and toluene (15mL). After nitrogen substitution, Pd(PPh3)4 (0.05 equiv) was added. The reaction mixture was stirred at 80C under nitrogen atmosphere for 12h. The reaction mixture was cooled, and water (15mL) was added. The mixture was diluted with AcOEt (15mL), and the insoluble material was filtered off through Celite. The organic layer of the filtrate was washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo. The residue was purified by silica gel column chromatography using a mixture of petroleum ether/ethyl acetate (10:1, v/v) as eluent to afford the desired product 2a-r, 5a and 5b as a solid. Step 2: To a solution of 2a-r, 5a or 5b (1 equiv) in MeOH (10mL) and THF (20mL) was added portionwise sodium borohydride (3 equiv) at 0C and the mixture was stirred at 0C for 1h. The reaction mixture was pouring into ice water (10mL), and extracted with ethyl acetate (3×15mL), the organic fractions were combined, washed with saturated brine (2×15mL) prior to drying over anhydrous sodium sulfate. After filtration and concentrate using a rotary evaporator, the residue was used in next step without further purification. To a solution of the obtained solid (1 equiv) in dichloromethane (20mL) was slowly added thionyl chloride (6 equiv) and a catalytic amount of DMF at room temperature. After stirring at 40C for 4h, the reaction was concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a mixture of petroleum ether/ethyl acetate (20:1, v/v) as eluent to afford the desired product.
3-[2-(morpholin-4-yl)pyrimidin-5-yl]benzaldehyde[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,2-dimethoxyethane; water; at 80℃; for 2.5h;Inert atmosphere;
General procedure: To a solution of <strong>[84539-22-0]4-(5-bromopyrimidin-2-yl)morpholine</strong> (17;4.10 g, 16.8 mmol) and ethyl [3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]acetate (6.34 g, 21.8 mmol) in DME(82 mL)/H2O (41 mL) were added Na2CO3 (5.34 g, 50.4 mmol),and Pd(PPh3)4 (1.94 g, 1.68 mmol) under an argon atmosphere.The mixture was stirred at 80 C for 2.5 h. After being cooled toroom temperature, the mixture was diluted with H2O and EtOAc.The mixture was filtered, and the filtrate was separated. Theorganic layer was washed with H2O and brine, dried over Na2SO4,and concentrated in vacuo. The residue was purified by columnchromatography on silica gel (hexane/EtOAc = 17:3 to 3:2) and columnchromatography on amino functionalized silica gel (hexane/EtOAc = 9:1 to 7:3) to give the product (5.29 g, 96%) as a colorlesssolid. 1H NMR (DMSO-d6): d 1.19 (3H, t, J = 7.1 Hz), 3.64-3.79 (8H,m), 3.72 (2H, s), 4.10 (2H, q, J = 7.1 Hz), 7.25 (1H, d, J = 7.6 Hz),7.35-7.45 (1H, m), 7.50-7.58 (2H, m), 8.70 (2H, s); MS (ESI) m/z[M+H]+ 328.
3-((ferrocenylimino)methyl)phenylboronic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In ethanol; toluene;
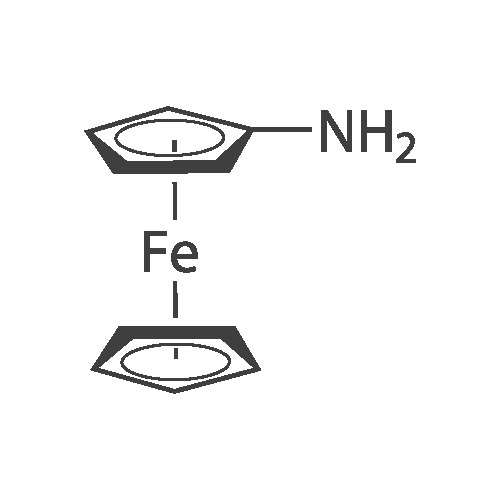
General procedure: Aminoferrocene (1, 11.0 mg, 0.0547 mmol) was dissolved in 9 cm3 toluene. Formylphenylboronic acid (2, 8.2 mg,0.0547 mmol) was dissolved in 1 cm3 dry ethanol. Both reagent solutions were mixed in an evaporating flask. The solvents were removed under reduced pressure on a rotary vacuum evaporator (the water bath temperature strictly below 40 C) to give [(ferrocenylimino)methyl]phenylboronic acid 3 as a violet/red powder; 18.2 mg (quant.). The products were used as prepared without need of a further purification (Fig. 5).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; at 80℃; for 12h;Inert atmosphere;
General procedure: The starting materials 1a or 5a (1 equiv) and (3-formylphenyl)boronic acid (1 equiv) were dissolved in a mixture of 1 M sodiumcarbonate aqueous solution (15 mL), EtOH (5 mL) and toluene(15 mL). After nitrogen substitution, Pd(PPh3)4 (0.05 equiv) wasadded. The reaction mixture was stirred at 80 C under nitrogenatmosphere for 12 h. The reaction mixture was cooled, and water(15 mL) was added. The mixture was diluted with AcOEt (15 mL),and the insoluble material was filtered off through Celite. Theorganic layer of the filtrate was washed with brine, dried overanhydrous sodium sulfate, and concentrated in vacuo. The residuewas purified by silica gel column chromatography using a mixtureof petroleum ether/ethyl acetate (10:1, v/v) as eluent to afford thedesired product 2a or 6a as a solid. 4.1.1.1. 3-(3-methylthiophen-2-yl)benzaldehyde (2a). Yield: 56%; 1HNMR (300 MHz, DMSO-d6) d: 10.08 (s, 1H), 7.97 (d, J = 1.3 Hz, 1H), 7.88 (d, J = 7.6 Hz, 1H), 7.79 (d, J = 1.3 Hz, 1H), 7.70 (d, J = 7.6 Hz,1H), 7.54 (d, J = 5.1 Hz, 1H), 7.05 (d, J = 5.1 Hz, 1H), 2.32 (s, 3H).
methyl 5-bromo-1-(3-formylphenyl)-6-oxo-1,6-dihydropyridine-3-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
297 mg
With pyridine; copper diacetate; at 80℃; for 1h;
Methyl 5-bromo-6-oxo-1,6-dihydropyridine-3-carboxylate (450 mg)(3-formylphenyl) boronic acid (400 mg),A mixture of copper (II) acetate (400 mg) and pyridine (10 ml) was stirred at 80 C. for 1 hour.The mixture was cooled to room temperature and extracted with ethyl acetate and water. The obtained organic layer was washed with 1N hydrochloric acid and saturated brine, dried over magnesium sulfate and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (297 mg).
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium carbonate; In 1,2-dimethoxyethane; dichloromethane; at 85℃; for 6h;
A mixture of (3-formylphenyl)boronic acid (2.1 g, 14 mmol) and 4-bromomethylpyridine hydrochloride (2.4g, 12 mmol) in 1,2-dimethoxy ethane (48mL) was degassed with nitrogen for 10 minutes. Aqueous potassium carbonate (2.5 M, 14 mL) was added, followed by [1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with DCM (0.38g, 0.46 mmol). The reaction mixture was heated to 85C for 6 hours under nitrogen. The reaction mixture was diluted with EtOAc (100 mL), and washed with water (60 mL) and brine (60 mL). The organic layer was dried over sodium sulfate, filtered and concentrated in vacuo. Purification by silica gel chromatography (20 - 90% EtOAc/hexanes) gave Compound 35A (1.35 g, 50%). 1 NMR (CDC13) delta 10.10 (s, 1H), 8.57 (s, 1H), 8.53 (d, 1H), 7.95 (m, 1H), 7.87 (m, 1H), 7.63 (m, 2H), 7.18 (d, 1H), 2.30 (s, 3H).
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; potassium carbonate; In 1,4-dioxane; water; at 80℃; for 12h;Inert atmosphere;
Compound BB-4D-2 (1.00 g, 4.42 mmol) and 3-formylbenzeneboronic acid (663.13 mg, 4.42 mmol) were dissolved in 1,4-dioxane (10 mL) under nitrogen atmosphere, followed by addition of (1,1'-bis(diphenylphosphino)ferrocene)palladium dichloride dichloromethane complex (360.95 mg, 4.42 mmol), potassium carbonate (916.33 mg, 6.63 mmol) and water (3 mL). The reaction mixture was heated to 80 C and stirred for 12 hours under nitrogen atmosphere. After completion of the reaction, the mixture is cooled to room temperature and concentrated under reduced pressure. The obtained residue was purified by column chromatography (eluent: ethyl acetate/petroleum ether = 1/100 - 1/20) to obtain the title compound BB-4D (yellow liquid, 489.00 mg, yield: 44.02%). 1H NMR (400MHz,CDCl3) delta: 10.08 (s, 1H), 8.07 (s, 1H), 7.83 (d, J = 7.8 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.58 - 7.52 (m, 3H), 6.66 (d, J = 8.8 Hz, 2H), 3.39 - 3.32 (m, 4H), 2.05 (td, J = 3.4, 6.5 Hz, 4H).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; at 75℃;Inert atmosphere;
In 25 mL round bottom flask equipped with reflux condenser, 0.0004 mol of 4-chloro-3,5-dimethylphenol mixed with appropriate quantity of arylboronic acid (1-6) and 0.05 g of Pd(PPh3)4 were added into the flask containing 20 mL of absolute ethanol under inert atmospheric condition (N2) gas. The mixture was stirred and heated and 5 mL of solution (5% Na2CO3) were added into the flask.The reaction mixture was stirred and heated at 75 C for 5-6 h then followed by TLC and monitor reactions using (n-hexane and ethyl acetate) in 2:3 ratio v/v. The mixture was filtrated off to remove the inorganic salt and the residue of pallidum catalyst, washed by cooled ethanol and the solvent was removed and decanted by cooled ether (Scheme-I). 4'-Hydroxy-2',6'-dimethyl-[1,1'-biphenyl]-3-carbaldehyde (7): Prepared from 4-chloro-3,5-dimethyl phenol (0.20 g, 0.001 mol) and 3-(formylphenyl)boronic acid (0.28 g, 0.001mol) solid white; yield 85 %; Rf = 0.88; m.p. 217-220 C. FT-IR (KBr, cm-1): 3393, 2750, 1692, 1596-1551. 1H NMR (DMSO-d6): 2.26(t, 6H), 6.48-6.90 (m, 4H), 7.02-7.85 (m, 4H), 9.07 (s, 1H), 10.43 (bs, 1H). Anal. for C15H14O2 calcd. (found) %: C, 79.62 (79.51); H, 6.24 (6.10).
2',6'-bis(trifluoromethyl)-[1,1'-biphenyl]-3-carbaldehyde[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1: 67.3%
2: 22%
With dicyclohexyl-(2',6'-dimethoxybiphenyl-2-yl)-phosphane; palladium diacetate; sodium carbonate In 1,4-dioxane; water at 90℃; for 2h; Sealed tube; Inert atmosphere;
General CC procedure employing SPhos/Pd(OAc)2 as catalyst (Scheme 5):
General procedure: To a 20 mL crimp-cap vial with stir-bar was charged 95.7 mg (0.327 mmol) of 2-Br-1,3-bis(trifluoromethyl)benzene, 97.9 mg (0.653 mmol) of (4-formylphenyl)boronic acid, 69.2 mg (0.653 mmol) ofsodium carbonate, 2.2 mg (0.010 mmol) of Pd(OAc)2, and 8.3 mg (0.020 mmol) of SPhos. The vial was thensealed with a crimp-cap septum, and made inert under nitrogen. To the vial was charged degassed 1,4-dioxane(0.7 mL) and degassed water (0.25 mL). The vial was then immersed in an oil bath at 90°C for 2 h. The vial wasallowed to cool to room temperature, and water was then charged, followed by extraction with CH2Cl2 (2x4mL).To the combined organics was charged 60.0 μL (0.488 mmol) of ,,-trifluorotoluene as analytical standard,and an aliquot was diluted in CDCl3 for quantitative 19F NMR spectroscopy. The organics were then dried withNa2SO4 and concentrated in vacuo. Products were isolated by purification by flash chromatography on silica gelusing hexanes/EtOAc mixtures.
1: 59.3%
2: 17%
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; sodium carbonate In 1,4-dioxane; water at 90℃; for 2h; Sealed tube; Inert atmosphere;
Biphenyl-4,4′-dicarbaldehyde (14a); Typical HC Procedure
General procedure: A crimp-cap vial equipped with magnetic stirrer bar wascharged with (dppf)PdCl2·CH2Cl2 (8.2 mg, 0.010 mmol), (4-formylphenyl)boronic acid (2a; 97.9 mg, 0.653 mmol), Na2CO3(69.2 mg, 0.653 mmol), and 2-bromo-1,3-bis(trifluoromethyl)benzene (6; 95.7 mg, 0.327 mmol). The vial was sealedwith a crimp-cap septum and filled with Ar by using threevacuum-purge cycles. Degassed (Ar sparge) 1,4-dioxane (0.70mL) and H2O (0.25 mL) were added, and the vial was immersedin an oil bath at 90 °C for 2 h, then cooled to r.t. H2O was addedand the mixture was extracted with CH2Cl2 (2 × 4 mL). The combinedorganics were mixed with PhCF3 (60.0 L, 0.488 mmol) asan added standard, and an aliquot was diluted in CDCl3 forquantitative 19F NMR spectroscopy. The organics were thendried (Na2SO4) and concentrated in vacuo. Purification by flashchromatography [silica gel, hexanes-EtOAc (0-25%)] gave awhite solid; yield: 53.2 mg (77%); mp 141-143 °C; Rf = 0.26 (25%EtOAc-hexanes).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; at 80℃; for 24h;Inert atmosphere;
General procedure: The solution of 3a (600mg, 2.79mmol), (3-formylphenyl)boronic acid (460mg, 3.07mmol), Pd(PPh3)4 (390mg, 0.33mmol) and sodium carbonate (870mg, 8.37mmol) in toluene/ethanol/H2O (35mL, 3/1/3) was refluxed under nitrogen atmosphere for 24h. Then the mixture was diluted with saturated ammonium chloride solution and ethyl acetate, and the insoluble material was filtered through Celite. The organic layer of the filtrate was washed with water (25mL) and brine (25mL), dried over anhydrous sodium sulfate and evaporated in vacuo. The residue was purified by column chromatography using a mixture of petroleum ether /ethyl acetate (10:1, v/v) as eluent to afford the desired product 4a 450mg (yield: 67%) as a white solid.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In ethanol; water; toluene at 80℃; for 24h; Inert atmosphere;
General procedure: The solution of 3a (600mg, 2.79mmol), (3-formylphenyl)boronic acid (460mg, 3.07mmol), Pd(PPh3)4 (390mg, 0.33mmol) and sodium carbonate (870mg, 8.37mmol) in toluene/ethanol/H2O (35mL, 3/1/3) was refluxed under nitrogen atmosphere for 24h. Then the mixture was diluted with saturated ammonium chloride solution and ethyl acetate, and the insoluble material was filtered through Celite. The organic layer of the filtrate was washed with water (25mL) and brine (25mL), dried over anhydrous sodium sulfate and evaporated in vacuo. The residue was purified by column chromatography using a mixture of petroleum ether /ethyl acetate (10:1, v/v) as eluent to afford the desired product 4a 450mg (yield: 67%) as a white solid.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In ethanol; water; toluene at 80℃; for 24h; Inert atmosphere;
General procedure: The solution of 3a (600mg, 2.79mmol), (3-formylphenyl)boronic acid (460mg, 3.07mmol), Pd(PPh3)4 (390mg, 0.33mmol) and sodium carbonate (870mg, 8.37mmol) in toluene/ethanol/H2O (35mL, 3/1/3) was refluxed under nitrogen atmosphere for 24h. Then the mixture was diluted with saturated ammonium chloride solution and ethyl acetate, and the insoluble material was filtered through Celite. The organic layer of the filtrate was washed with water (25mL) and brine (25mL), dried over anhydrous sodium sulfate and evaporated in vacuo. The residue was purified by column chromatography using a mixture of petroleum ether /ethyl acetate (10:1, v/v) as eluent to afford the desired product 4a 450mg (yield: 67%) as a white solid.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping