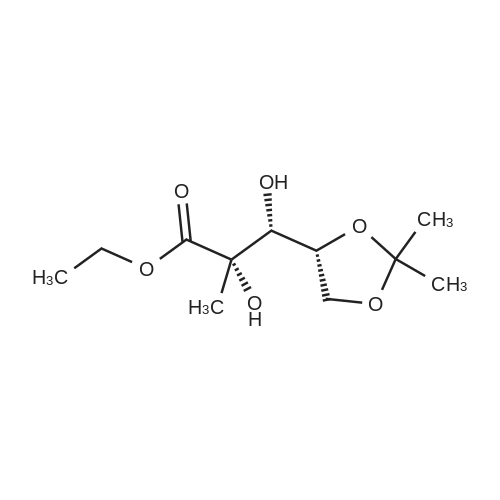
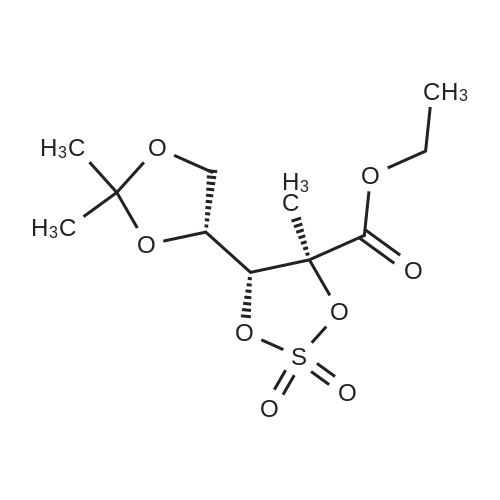
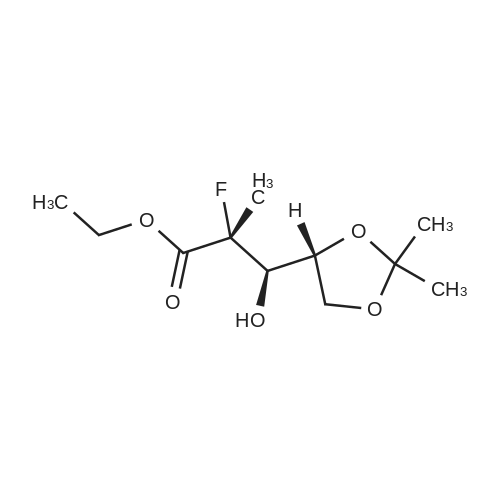
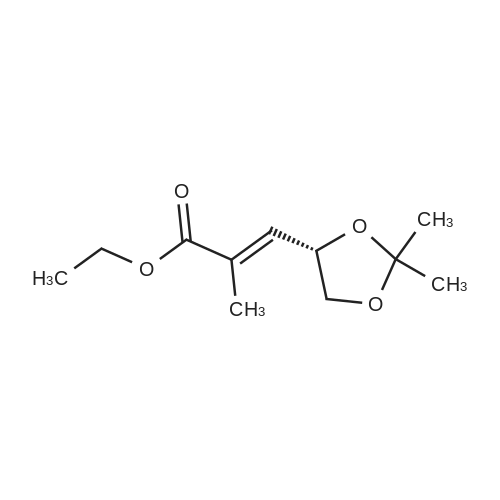
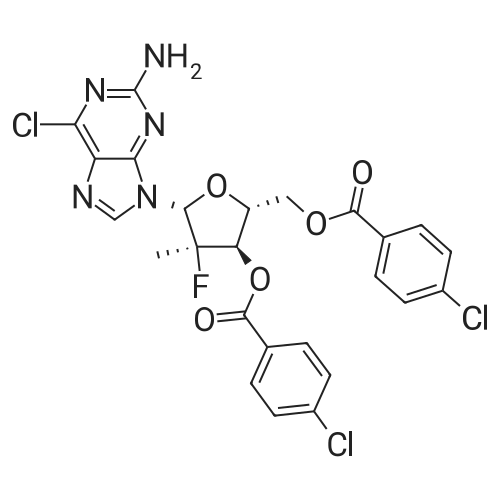
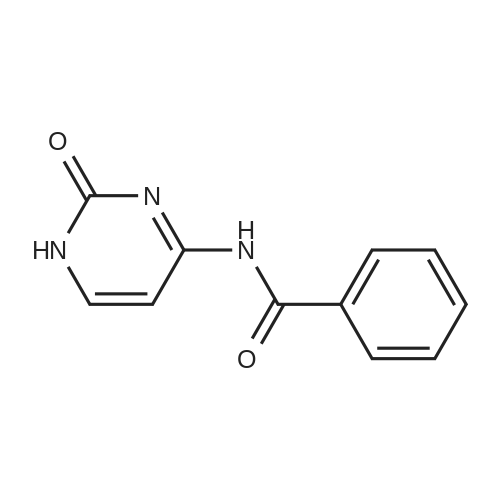
| 84% |
With dmap; triethylamine In acetonitrile at 20 - 25℃; for 4h; Inert atmosphere; |
12F Synthesis of intermediate (I)
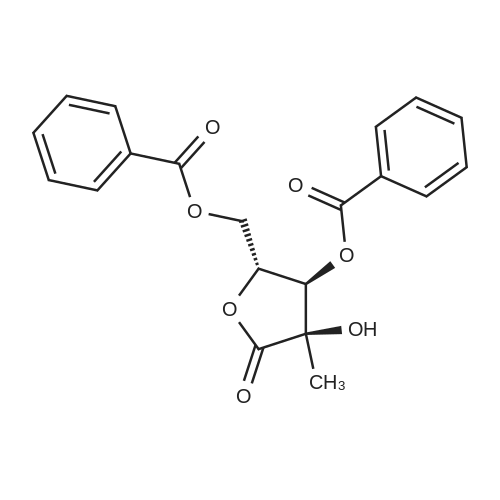
Under nitrogen, acetonitrile (160 mL), DMAP (0.74 g, 0.006 mol) and benzoyl chloride (51.6 g, 0.365 mol) were added to a solution of compound of formula IX (20 g, 0.122 mol) and cooled to about 20 ° C. Triethylamine (37.0 g, 0.365 mol) was added at a controlled temperature not higher than 40 ° C. Add finished, cooled to 20-25 ° C, the reaction was stirred for 4 hours. The reaction was completed, cooled to 0-5 ° C, add water (lOOmL) quenched. Ethyl acetate (300 mL) was added and the organic phase was separated. The aqueous phase was extracted once more with ethyl acetate (150 mL). The combined organic phases were washed with saturated sodium bicarbonate and brine respectively, dried and concentrated. The resulting crude product is recrystallized from isopropanol to give the intermediate of formula I. |
| 83.9% |
With dmap; triethylamine In tetrahydrofuran at 0 - 40℃; for 2h; |
4 Preparation of ((3R,4R)-3-(benzoyloxy)-4-fluoro-4-methyl-5-oxotetrahydrofuran-2- yl)methyl benzoate
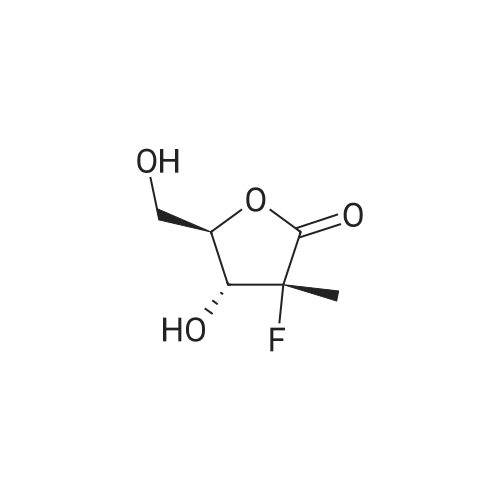
(3R,4R)-3-fluoro-4-hydroxy-5-(hydroxymethyl)-3-methyldihydrofuran-2(3H)-one (25.5 g, 0.155 mol) obtained from example 3 was dissolved in 200 ml of THF. 4-(Dimethylamino)- pyridine (8.3 g, 0.067 mol) and triethylamine (35 g, 0.35 mol) were added and the reaction mixture was cooled to 0°C. Benzoyl chloride (46.7 g, 0.33 mol) was added, and the mixture was warmed to 35-40 °C in the course of 2 hrs. Upon completion of the reaction (TLC check) water (100 ml) was charged and the mixture was stirred for 30 min. Phases were separated and to the aqueous phase methyl-tert-butyl ether (100 ml) was added and the mixture was stirred for 30 min. Phases were separated and the organic phase was washed with saturated NaCl solution (100 ml). The combined organic phases were dried over Na2SC>4 (20 g) filtered and the filtrate was evaporated to dryness. The residue was taken up in i-propanol (250 ml) and the mixture was warmed to 50 °C and stirred for 60 min, then cooled down to 0°C and further stirred for 60 min. The solid was filtered and the wet cake was washed with i-propanol (50 ml) and then dried under vacuum. The title compound ((3R,4R)-3-(benzoyloxy)-4-fluoro-4-methyl-5-oxotetrahydrofuran- 2-yl)methyl benzoate (48.3 g, 83.9 % yield) was obtained. 'H-NMR (CDCI3, 400 MHz): 58.10 (d, 7=7.6 Hz, 2H), 8.00 (d, 7=7.6 Hz, 2H), 7.66 (t, 7=7.6 Hz, IH), 7.59 (t, 7=7.6 Hz, IH), 7.50 (m, 2H), 7.43 (m, 2H), 5.53 (dd, 7=17.6, 5.6 Hz, IH), 5.02 (m, IH), 4.77 (dd, 7=12.8, 3.6 Hz, IH), 4.62 (dd, 7=12.8, 5.2 Hz, IH), 1.77(d, 7=23.2 Hz, 3H). |
| 82.6% |
With dmap; triethylamine In tetrahydrofuran at 0 - 40℃; for 2h; |
17 Example 17: preparation of ((3R,4R)-3-(benzoyloxy)-4-fluoro-4-methyl-5-oxotetra hydro furan-2- yl)methyl benzoate
(3R,4R)-3-fluoro-4-hydroxy-5-(hydroxymethyl)-3-methyldihydrofuran-2(3H)-one(19)(25.4 g, 0.154 mol) obtained from example 3 was dissolved in 200 ml ofTHF. 4-(dimethylamino)pyridine(8.2 g, 0.066 mol) and triethylamine (35 g, 0.35 mol) were added and the reaction mixturewas cooled to 0 'C. Benzoyl chloride (46.0 g, 0.33 mol) was added, and the mixture was warmed to35-40 'C in the course of2 hs. Upon completion of the reaction, water (100 mL) was charged andthe mixture was stirred for 30 min. Phases were separated and to the aqueous phase methyl-tertbutylether (100 mL) was added and the mixture was stirred for 30 min. Phases were separated andthe organic phase was washed with saturated NaCl solution (100 mL). The combined organicphases were dried over Na2S04 (20 g) filtered and the filtrate was evaporated to dryness. Theresidue was taken up in iso-propanol (250 mL) and the mixture was warmed to 50 'C and stirred for60 min, then cooled down to 0 'C and further stirred for 60 min. The solid was filtered and the wetcake was washed with i-propanol (50 mL) and then dried under vacuum. The title compound( (3R,4R)-3-(benzoyloxy)-4-fluoro-4-methyl-5-oxotetrahydrofuran- 2-yl)methyl benzoate ( 4 7.5 g,82.6% yield) was obtained. 'H-NMR (CDCl3, 400 MHz): 8.10 (d, 7=7.6 Hz, 2H), 8.00 (d, 7=7.6Hz, 2H), 7.66 (t, 7=7.6 Hz, 1H), 7.59 (t, 7=7.6 Hz, 1H), 7.50 (m, 2H), 7.43 (m, 2H), 5.53 (dd, 7=17.6,5.6 Hz, 1H), 5.02 (m, 1H), 4.77 (dd, 7=12.8, 3.6 Hz, 1H), 4.62 (dd, 7=12.8, 5.2 Hz, 1H), 1.77(d,7=23.2 Hz, 3H). |
| 70.7% |
With dmap; triethylamine In acetonitrile at 10 - 40℃; |
4.4
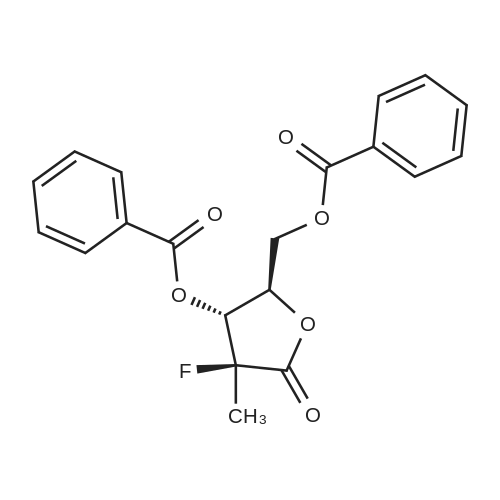
Step 4; - To the residue from step 3 containing 28 was added MeCN (35 kg) and ca. 15L was distilled out under atmospheric pressure. The reaction mixture was cooled to ca. 100C and then benzoyl chloride (8.27 kg) and DMAP (0.14 kg) are added. TEA (5.84 kg) was added slowly to the reaction mixture while cooling to maintain temperature below 4O0C. The batch was aged at ca. 200C and the progress of the benzoylation is monitored by HPLC. After completion of the reaction, EtOAc (30 kg) was added to the mixture and the resulting suspension is stirred for about 30 min. The reaction mixture was filtered through a CELITE pad (using a nutsche filter) to remove inorganic salts. The solid cake was washed with EtOAc (38 kg). The combined filtrate and washes were washed successively with water (38 kg), saturated NaHCθ3 solution (40 kg) and saturated brine (44 kg). The organic phase was polish-filtered (through a cartridge filter) and concentrated under modest vacuum to minimum volume. IPA (77 kg) was added to the concentrate and ca. 25 L of distillate was collected under modest vacuum allowing the infernal batch temperature to reach ca 75°C at the end of the distillation. The remaining solution was then cooled to ca. 5°C over 5 h and optionally aged overnight. The precipitate was filtered and washed with of cold (ca. 5°C) IPA (24 kg). The product was dried under vacuum at 60-700C to afford 6.63 kg (70.7% theory of 10 which was 98.2% pure by HPLC. |
| 61.2% |
With pyridine at 0 - 20℃; for 0.75h; |
48 Example 48
Compound 66-7 (110 g) was dissolved in anhydrous pyridine (1 L). Benzoyl chloride (200 mL, 1.67 mol) was added slowly at 0-5° C. The mixture was stirred at ambient temperature for 45 mins. The reaction was quenched with ice and MeOH to form a precipitate. After filtration, the filtrate was washed with MeOH to give 66-8 (200 g, 61.2%) as a white solid. |
| 61.2% |
With pyridine at 0 - 20℃; for 0.75h; |
1
Compound 66-7 (1 10 g) was dissolved in anhydrous pyridine (1 L). Benzoyl chloride (200 ml_, 1.67 mol) was added slowly at 0-5°C. The mixture was stirred at ambient temperature for 45 mins. The reaction was quenched with ice and MeOH to form a precipitate. After filtration, the filtrate was washed with MeOH to give 66-8 (200 g, 61.2%) as a white solid. |
| 60% |
With pyridine at 0 - 20℃; for 24h; |
5; IV.iv; III.A.iv; III.B.iv
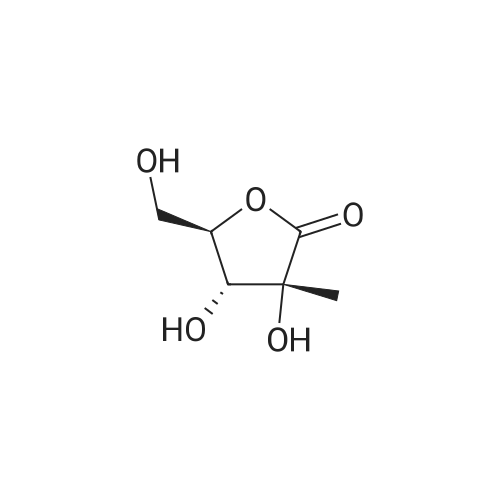
2-Deoxy-2-fluoro-2-C-methyl-D-ribono-l,4-lactone (200mg, 1.219mmol) was dissolved in anhydrous pyridine (2.4ml) and cooled to O0C under an atmosphere of argon. Benzoyl chloride (353μl, 3.05mmol) was added dropwise over 5min. The solution was allowed to warm to room temperature and stirred for 4h. Benzoyl chloride (142μl, 1.219mmol) was added dropwise and the mixture was stirred for 16h after which time a further portion of benzoyl chloride (142μl, 1.219mmol) was added. Having stirred the solution for a further 3h, a final portion of benzoyl chloride (142μl, 1.219mmol) was added and left for a further Ih. T.l.c. (ethyl acetate/heptane, 2:3) indicated complete conversion of the starting material (Rf 0.16) to faintly stained, but UV active products (Rf 0.67, 0.63). The reaction was quenched with water (ImI) and the solids dissolved. After stirring at room temperature for 5min crystals precipitated from the solution which were filtered and washed with water (2ml x 2) and found by t.l.c. to contain two UV active components and pyridine. The dried, sticky solid (400mg) was therefore dissolved in dichloromethane (10ml) and subjected to a IM HClaq (4ml x 2) wash. The organic layer was dried (magnesium sulfate), filtered and concentrated in vacuo. The crude off-white residue (340mg) was purified by flash column chromatography (pre-adsorbed onto silica from dichloromethane, eluted with ethyl acetate/heptane, 1:4) to give 3,5-di-benzoyl-2-deoxy-2-fluoro-2-C-methyl-D- jribono-l,4-lactone (272mg, 60%) as fine, white needles.Data for SjS-di-benzoyl^-deoxy^-fluoro^-C-methyl-D-ribono-l^-lactone C2OHi7FO6 372.34SmOl"1; Rf = 0.62, ethyl acetate/heptane, 2:3; m.p. : 123-125°C; [α]D20 : +102.943 (c, 0.8728 in CH3CN); vmax (thin film) : 1793cm"1 (O=O, α-fluoro- γ-lactone), 1733cm"1, 1717cm"1 (C=O, Bz); 1H NMR δH (400 MHz, CD3CN) : 1.74 (3H, d, JH,F 24.1, CH3), 4.62 (IH, dd, J5 A- 12.7, J5,4 5.6, H-5), 4.75 (IH, dd, Jy,5 12.7, Js- A 3.5, H-5'), 5.07 (IH, ddd, J4j3 7.2, J4)5 5.6, J4,y 3.5, H-4), 5.62 (IH, dd, 3JH,F 17.7, J3,4 7.2, H-3); 7.48, 7.55 (4H, 2 x t, 4 x Hmeta), 7.64, 7.70 (2H, 2 x t, 2 x Hpara), 8.01, 8.09 (4H, 2 x t, 4 x Hortho); 13C NMR δc (100 MHz, CD3CN) : 18.88 (d, 2JC,F 24.5, CH3), 63.51 (C-5), 73.07 (d, 2JC,P 14.6, C-3), 78.84 (C-4), 92.61 (d, 1Jc1F 185.6, C-2), 129.51, 130.53 (2 x Cipso), 129.64, 129.78 (2 x Cmeta), 130.43, 130.76 (2 x Colλo), 134.46, 135.00 (2 x Cpara), 166.22, 166.63 (2 x CO2Bz), 170.64 (d, 2Jc1F 21.0, C=O); 19F NMR δF (376 MHz, CD3CN) : -164.77 (IF, m, 3JF>H 24.4, F). NMR assignments confirmed using COSY, HMQC, HMBC and nOe experiments; Mass Spec m/z (ESI- ) : 373.1 ([M+Hf, 50%), 390.2 ([M+NH4]+, 100%); HPLC (272ntn) R4 = 6.17 (98%); Microanalysis : C2OHnFO6 calculated C 64.51%, H 4.60%, found C 64.56%, H 4.66%. |
| 51.2% |
With dmap; triethylamine In ethyl acetate at -5℃; for 2h; |
10
Compound V-1 (2.56 g) was dissolved in 50 mL of ethyl acetate, and then adding triethylamine (8.75 mL, 4eq) and 4-dimethylaminopyridine (DMAP) (0.75g, 0.4 eq.). Benzoyl chloride (5.45 mL, 3eq) was slowly added dropwise at the temperature of -5 ° C.5° C. The reaction completed two hours later indicating by TLC, the solid was filtered, and the filter cake was washed with 20 mL of ethyl acetate to give 3 g white flocculent solid Compound VI-1. HPLC purity was 97.5%. Yield: 51.2%. 1HNMR(300 MHz, DMSO-d6) δ1.68 (d,3H, J=24.2 Hz), 4.62-4.74 (m, 2H), 5.11-5.15 (m, 1H), 5.76 (dd, 1H, J=7.0, 18.4 Hz), 7.46 (m, 2H), 7.55 (m, 2H), 7.62 (m, 1H), 7.70 (m, 1H), 7.93 (m, 2H), 8.06 (m, 2H), 8.08 (m, 2H). |
| 48.4% |
With dmap; triethylamine In acetonitrile at 10 - 30℃; for 5h; Large scale; |
8
8) Into the reactor. add the above-mentioned oily matter, 360L acetonitrile, stirring 20 minutes, the reaction liquid cooling to 10 °C, and into the reaction system, add 4-dimethylaminopyridine, control temperature to below 10 °C. Slowly add dropwise 82.7 kg benzoyl chloride. After dropwise addition is complete, heat up to 20 °C, control temperature at 25-30 °C slowly add dropwise 74.2 kg triethylamine, dropping complete, maintain temperature and react for 5 hours. Reaction is completed. the reaction liquid centrifugal, the filtrate is concentrated under reduced pressure to remove the 2/3 of the acetonitrile, cake 200L washing twice ethyl acetate, the combined organic phase, the organic phase for respectively 200L purified water, 200L saturated sodium bicarbonate, 200L saturated salt is washed with water, dried with anhydrous sodium sulfate, 45 - 50 °C lower concentrated under reduced pressure to dry, to obtain a yellow solid of about 70 kg, residue by adding 300L methanol heated to 30 - 35 °C stirring 2 hours, the temperature slowly drops to 0 - 5 °C stirring 2 hours, cooling crystallization, filtration, 50 - 60 °C decompression drying shall be 3,5-di-O-benzoyl-2-deoxy-2-fluoro-2-methyl-D-ribono-γ-lactone 101 kg, yield: 48.4%, purity in ethyl (E)-3-(2,2-dimethyl-1,3-dioxolan-4-yl)-2-methyl-2-propenoate idea, product purity 98.7%. |
| 24 g |
With pyridine at 0 - 20℃; for 0.5h; |
|
|
With pyridine at 20℃; for 0.5h; Cooling with ice; |
1 Example 1
[0073]The whole of the above-obtained crude product (regarded as 660 μmol) was mixed with 2.45 g (31.0 mmol, 47.0 eq) of pyridine, followed by adding thereto 750 mg (5.34 mmol, 8.09 eq) of benzoyl chloride under ice cooling. The resulting liquid was stirred for 30 minutes at room temperature. To the thus-obtained reaction-terminated liquid, 2 mL of water was added under ice cooling. The reaction-terminated liquid was further stirred for 10 minutes at room temperature and concentrated under reduced pressure. The concentration residue was admixed with 10 mL of saturated aqueous sodium hydrogencarbonate solution and extracted with 20 mL of ethyl acetate. The recovered aqueous layer was again extracted with 20 mL of ethyl acetate. The recovered organic layers were combined. The combined organic layer was dried with anhydrous sodium sulfate, concentrated under reduced pressure and dried under vacuum. There was thus obtained a crude product of (2R)-2-fluoro-2-C-methyl-D-ribono-γ-lactone of the following formula.It was confirmed by 19F-NMR quantification analysis (internal standard method) of the crude product that the target compound of the above formula was contained in an amount of 380 μmol. The total yield from the 1,2-diol substrate was 38%.[0074]The 19F-NMR analysis results of the crude product are indicated below. 19F-NMR [standard material: C6F6, deuterated solvent: CDCl3] δ ppm: -5.44 (m, 1F). |
| 5.49 g |
With pyridine In acetonitrile at 20℃; for 2h; Cooling with ice; |
11
Represented by the formula [6a] obtained above (2R)-2-fluoro-2-C-methyl-D-Ribono-γ-lactone 7.55g (46.0mmol), acetonitrile 46ml (1.0L / mol) and pyridine 8.37g (105.8mmol, 2.30eq) was added, and benzoyl chloride 14.17g (100. 8mmol, 2.19eq) is added to under ice-cooling, and the mixture was stirred at room temperature for 2 hours. It was added to the reaction-terminated liquid water 180ml under ice-cooling, followed by stirring at room temperature for 10 minutes, and extracted with ethyl acetate 350 ml, and collected organic layers were washed with 5% aqueous sodium hydrogencarbonate solution 100ml, and washed with 5% brine 100ml , the following formula by which concentrated under reduced pressure, and dried under vacuum [1a]: Shown in (2R)-2-fluoro-2-C-methyl-D-Ribono-γ-lactone 1a It was obtained 5.49g |
| 1.6 g |
With dmap; triethylamine In dichloromethane at -5℃; for 2h; Inert atmosphere; |
2.2.3. Preparation of 3,5-Di-O-benzoyl-2-deoxy-2-chloro-2-C-methyl-D-ribono-γ-lactone 5a
General procedure: To a 50 mL three-necked flask equipped with a nitrogen inlet, a magnet stirrer and a thermometer was charged with 4a (740 mg, 4.10 mmol, 1.0 eq.) and DMAP (200 mg, 1.64 mmol, 0.4 eq.), after three times vacuum/nitrogen cycle, DCM (10 mL) was charged followed by triethylamine (2.3 mL, 16.4 mol, 4.0 eq.). The reactor was cooled to an internal temperature of -5°C followed by adding benzoyl chloride (1.4 mL, 12.40 mmol, 3.0 eq.) slowly with the internal temperature not higher than 5°C and reaction mixture was stirred at -5°C for 2h. The insoluble substance was filtered and washed by DCM (10 mL). The combined organic phase was washed by water (10 mL) and brine (10 mL), then dried over Na2SO4 and evaporated. The residue was purified by flash column chromatography (petroleum ether/dichloromethane, from 5/1 to 1/2) to give product 5a as white solid (1.29 g, 3.32 mol, 81% yield). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping