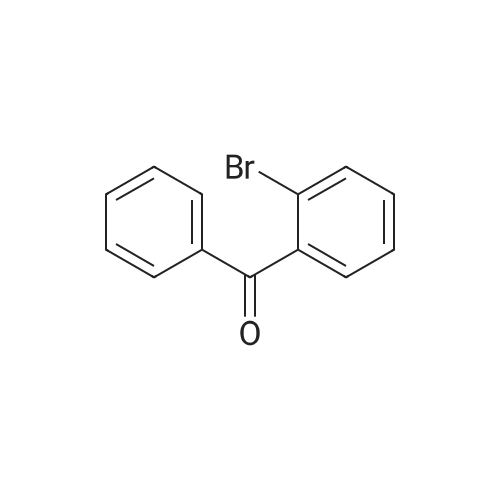
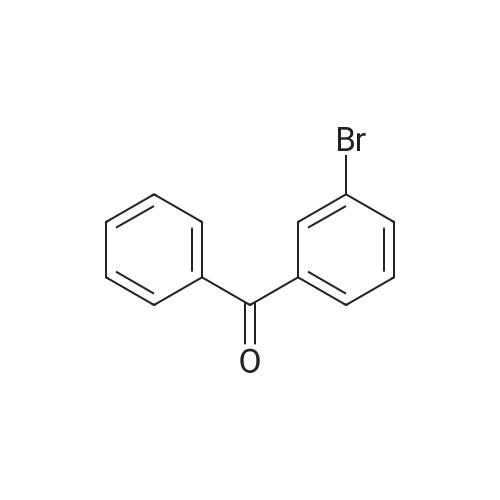
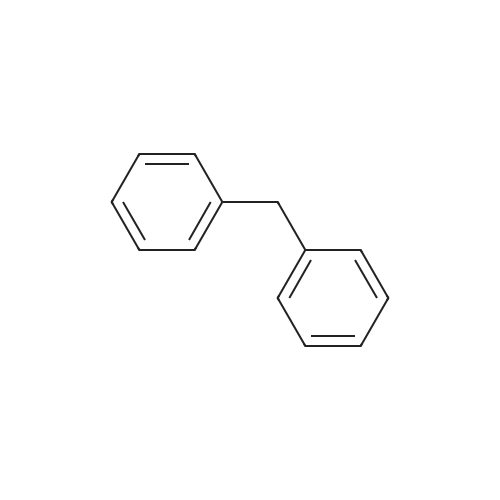
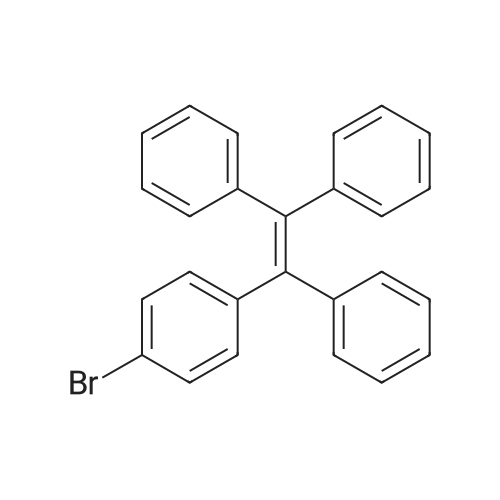
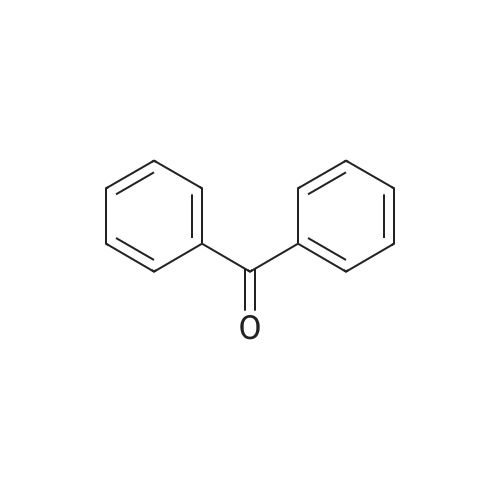
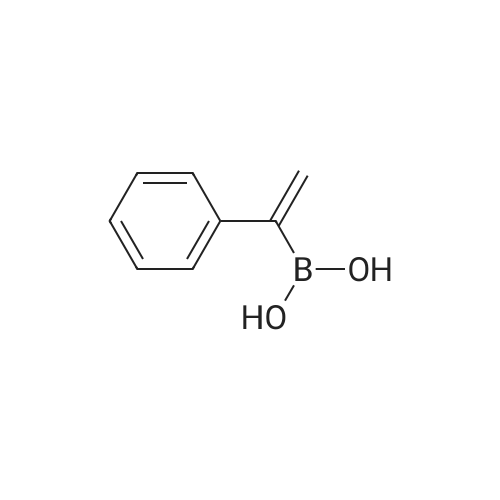
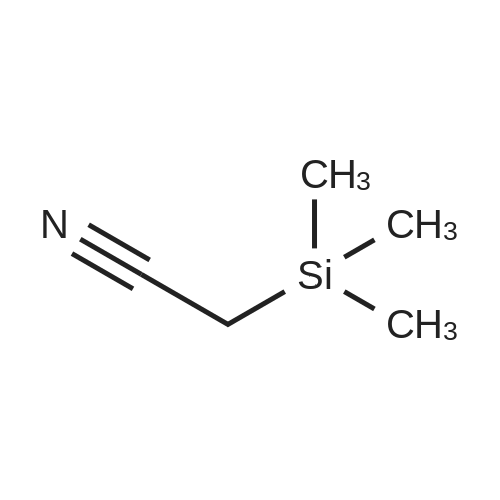
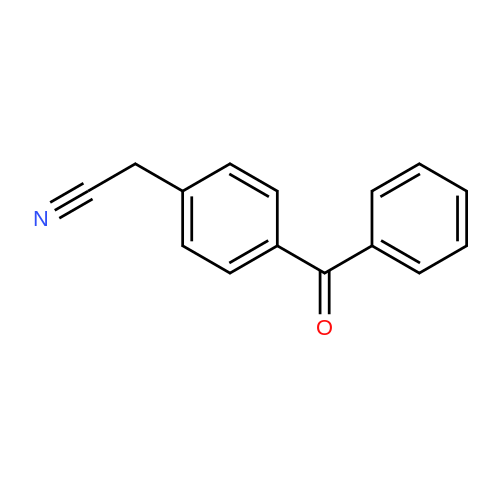
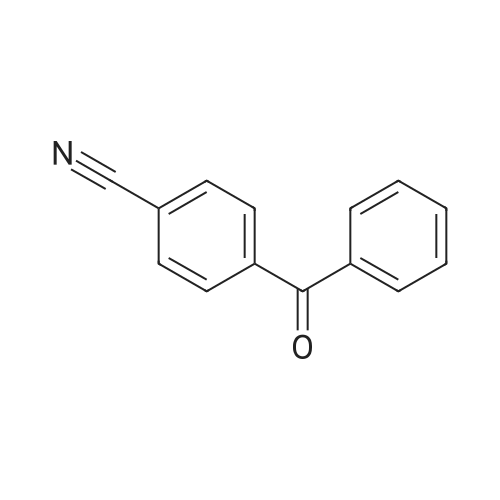
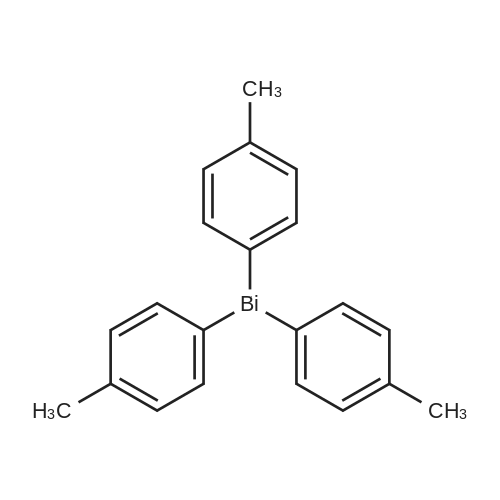
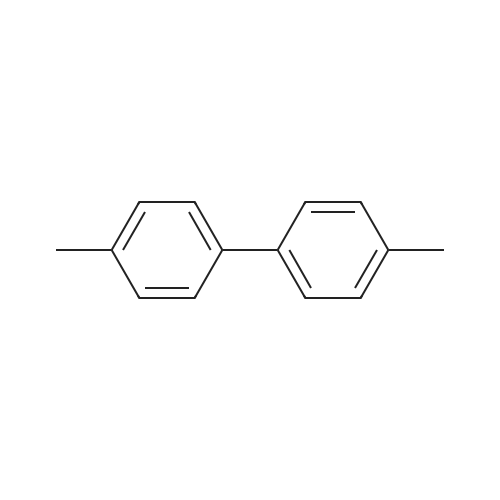
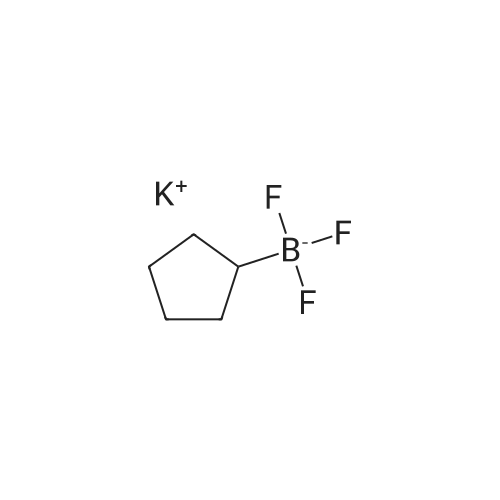
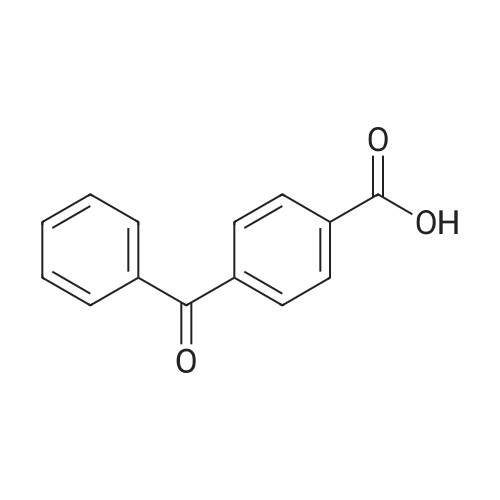
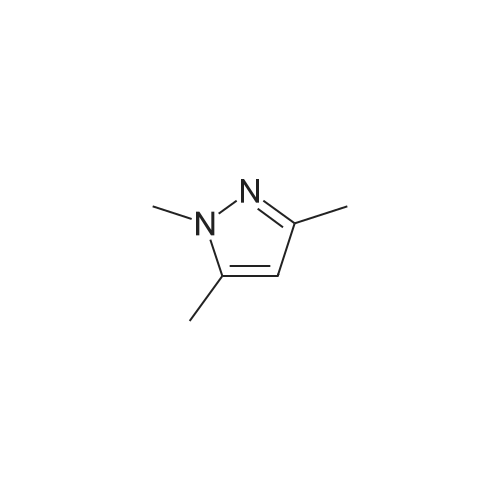
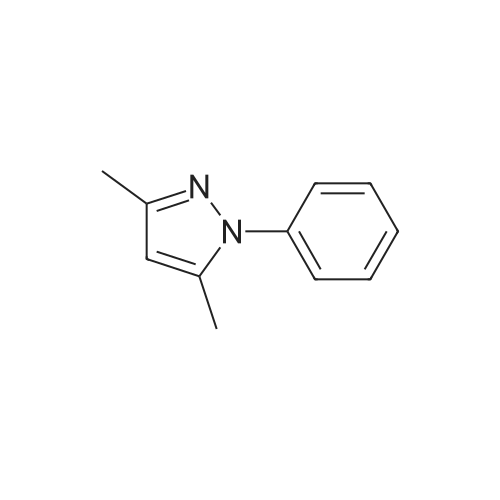
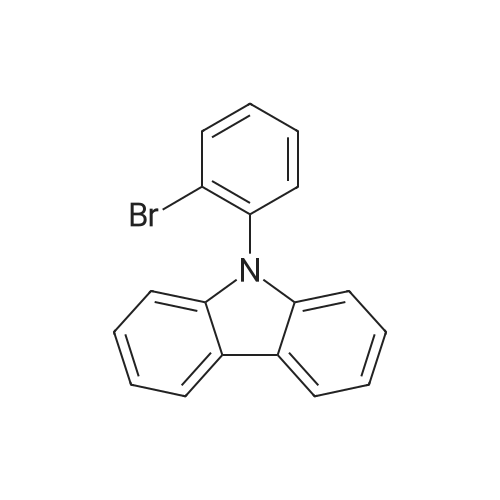
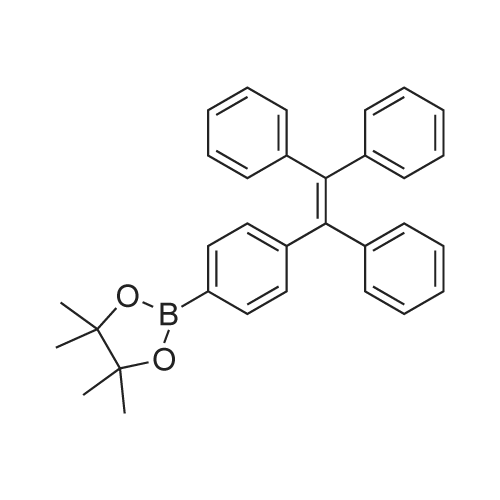
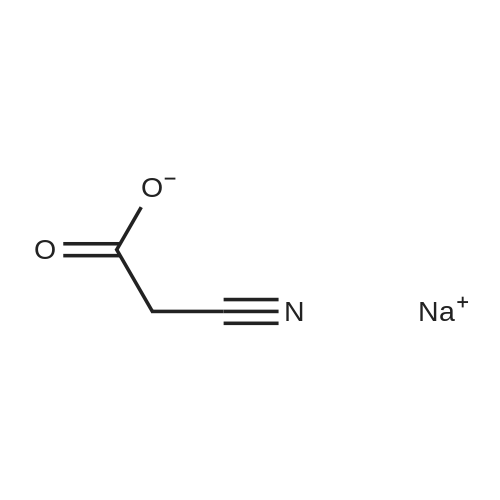
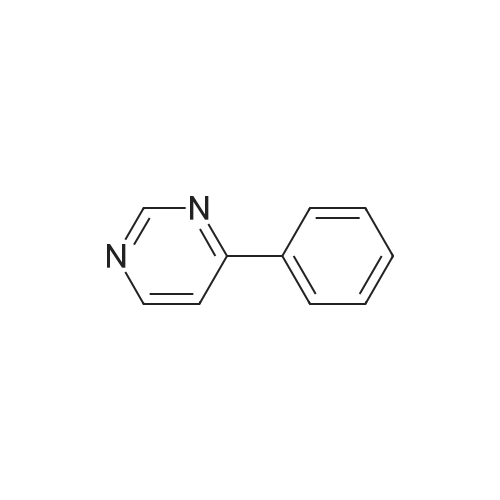
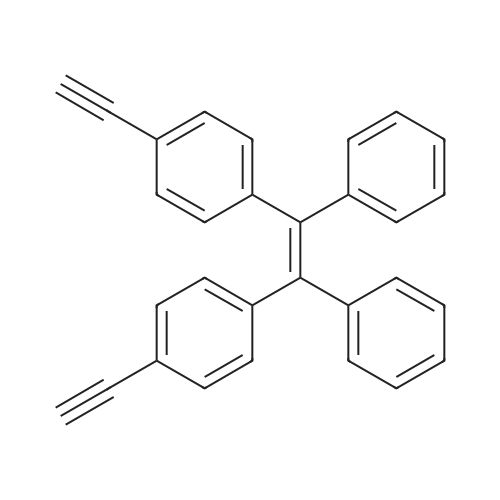
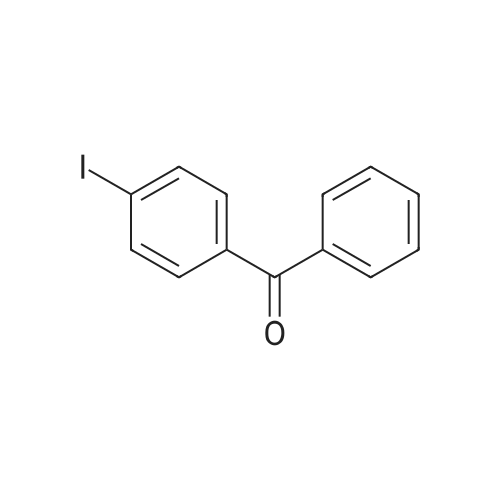
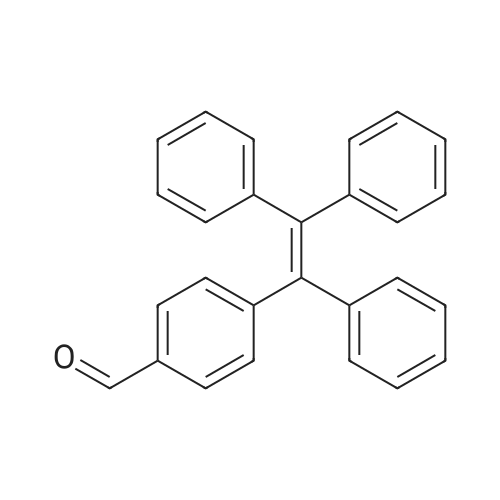
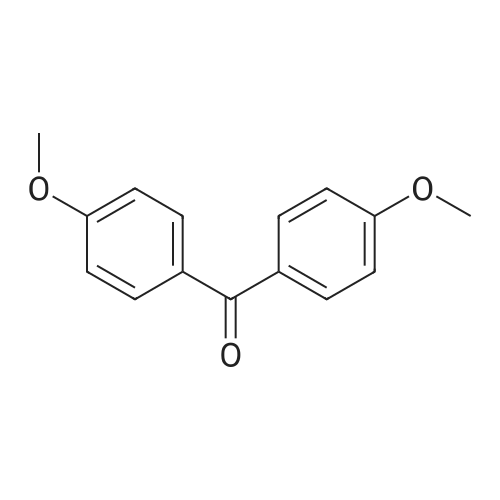
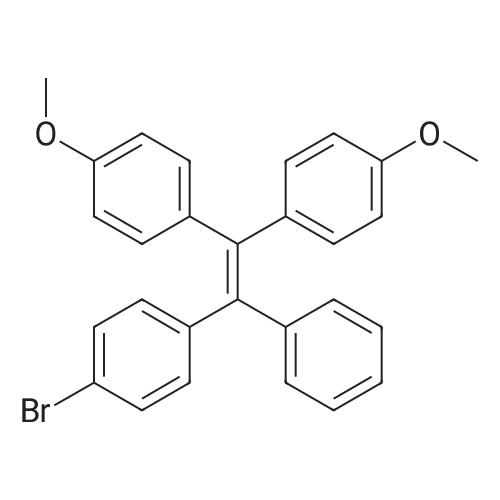
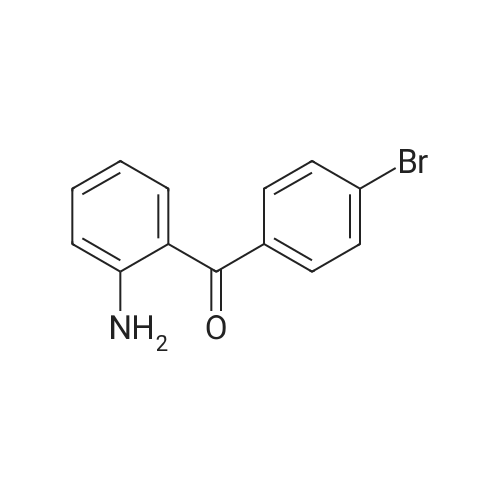
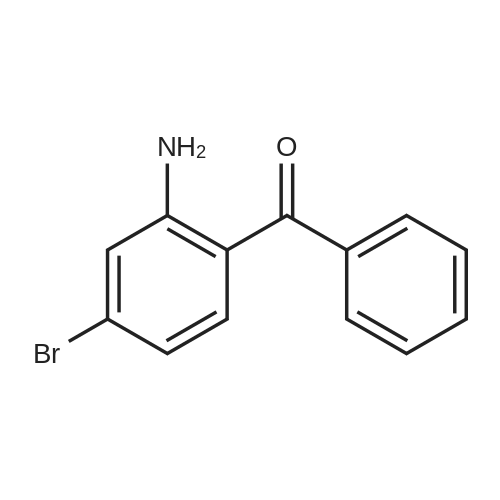
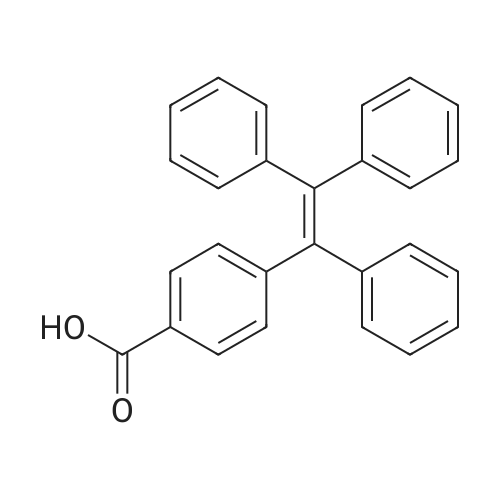
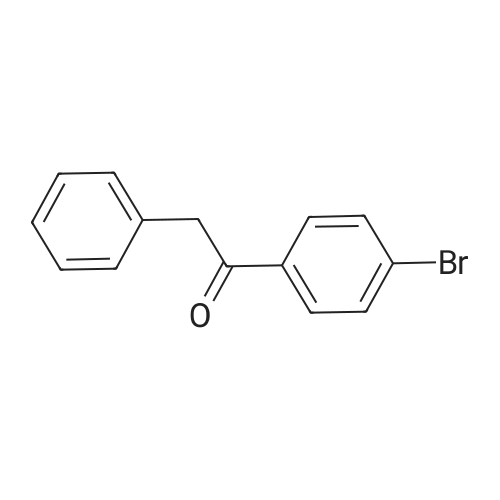
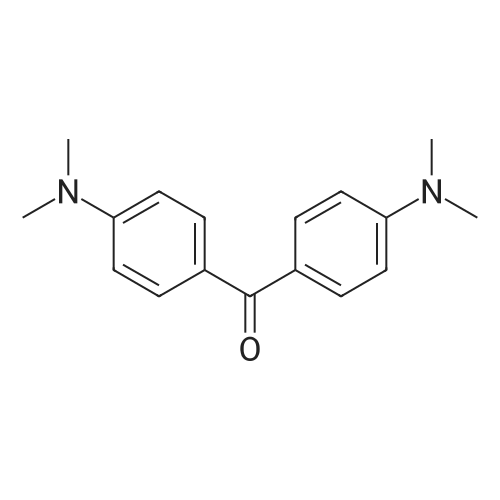
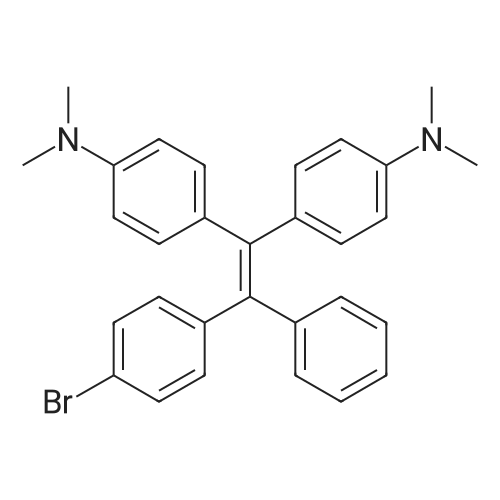
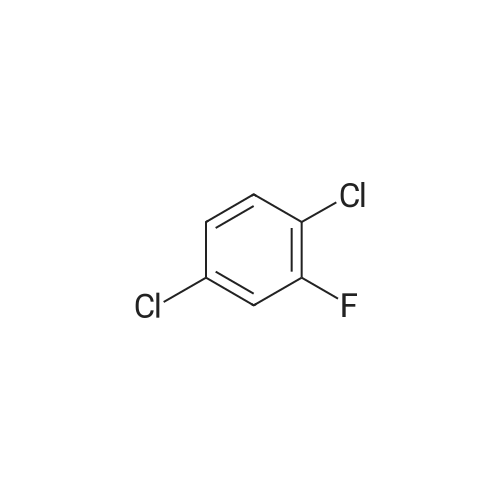
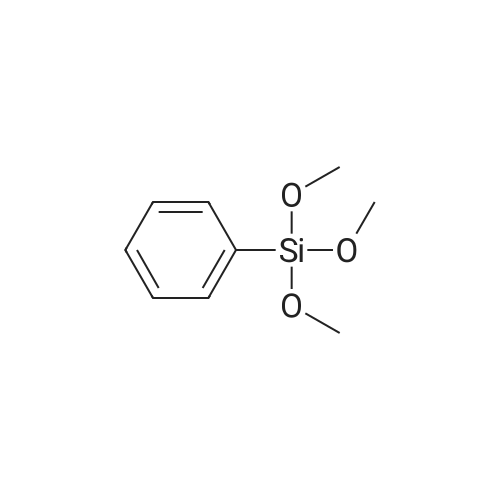
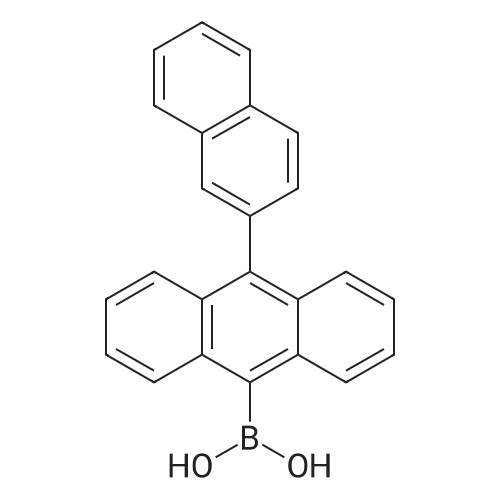
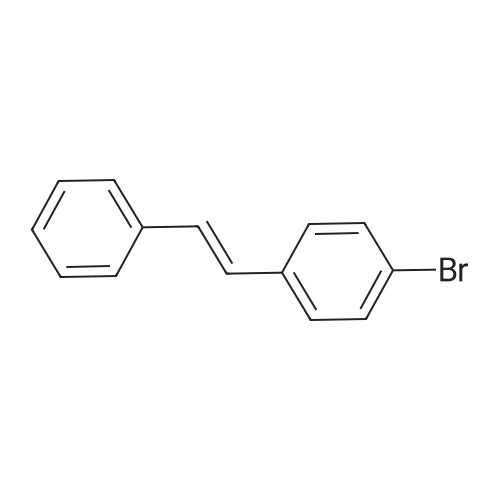
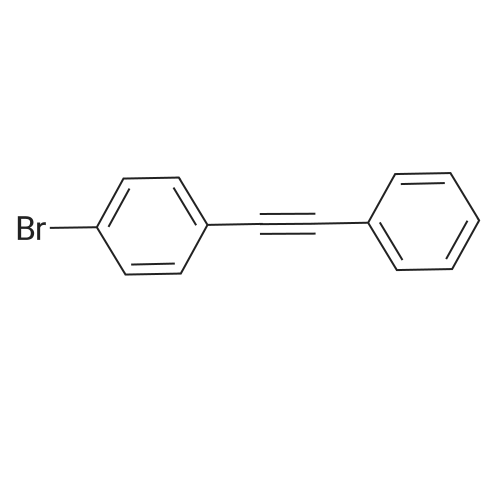
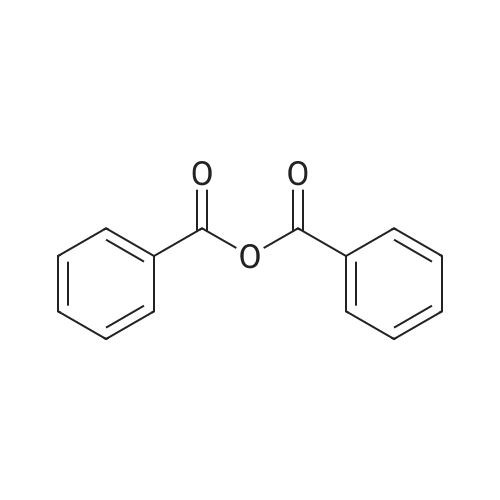
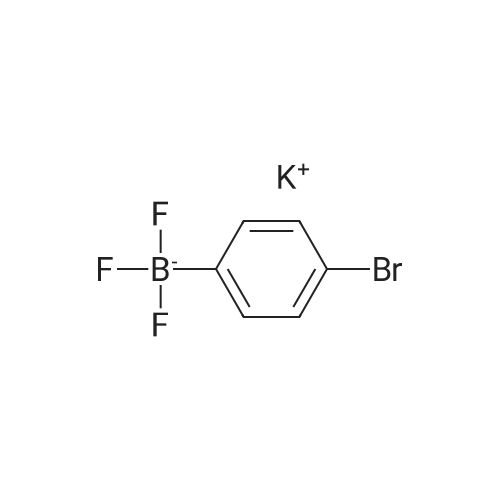
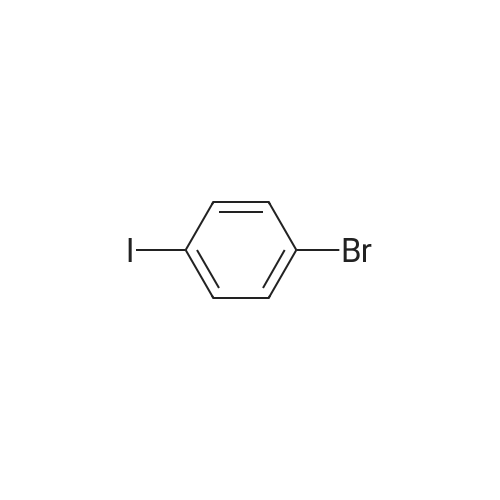
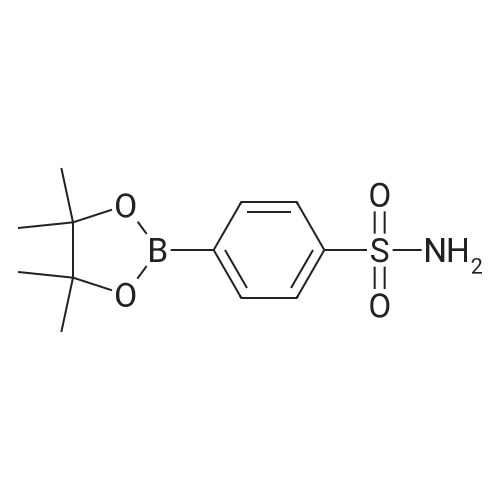
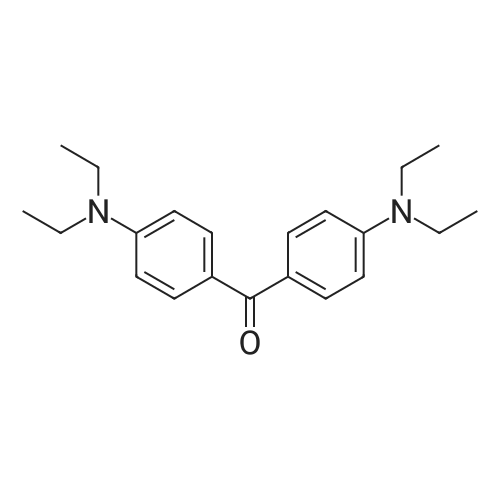
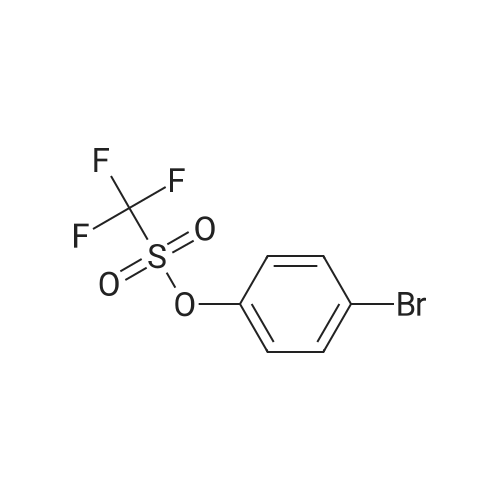
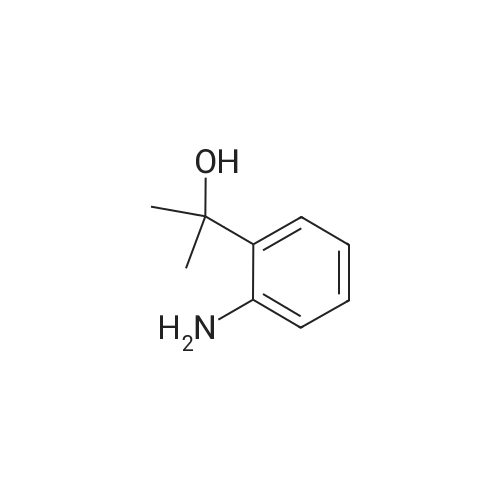
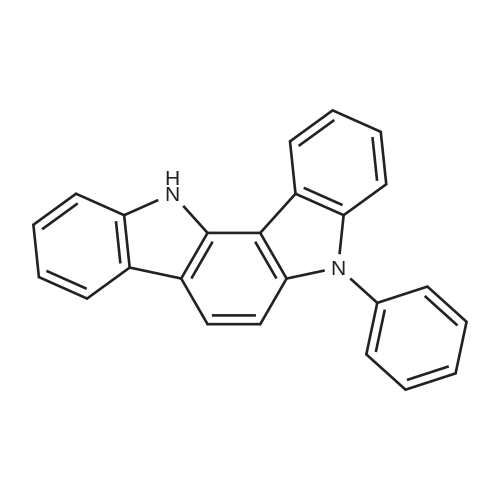
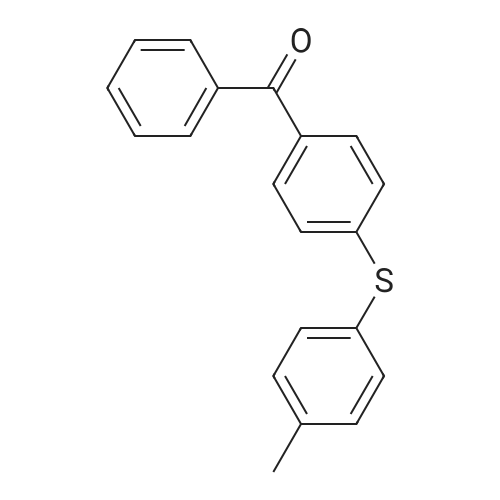
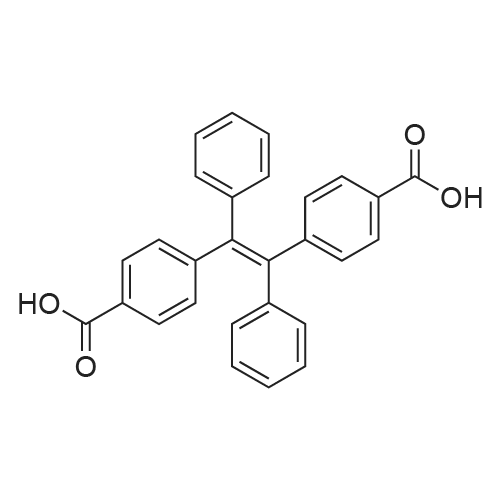
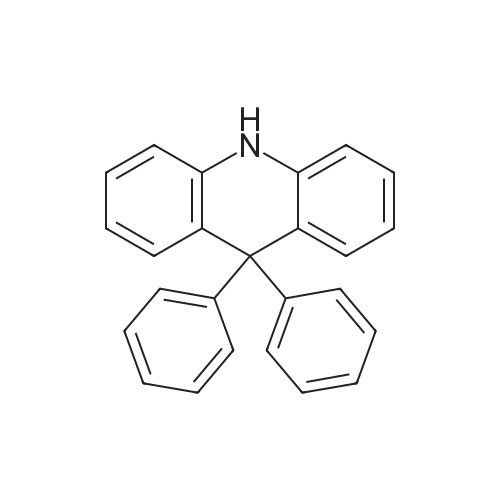
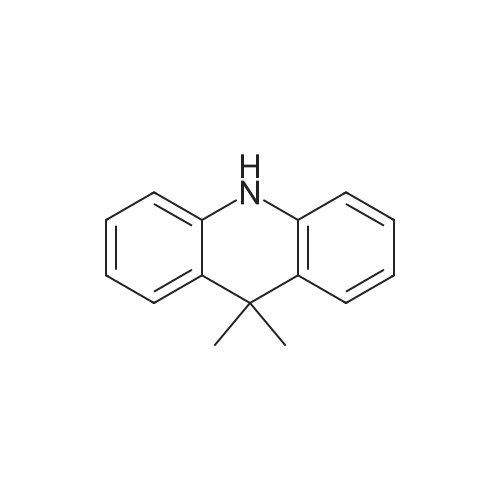
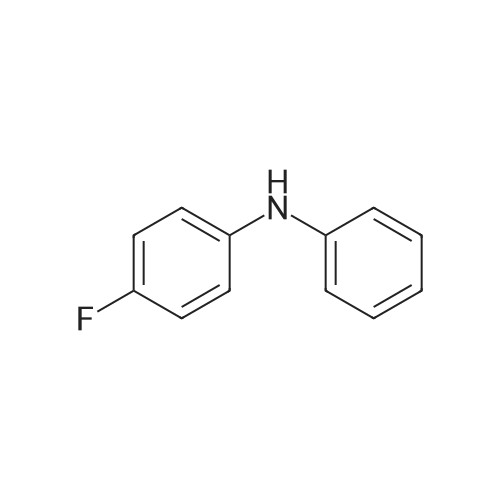
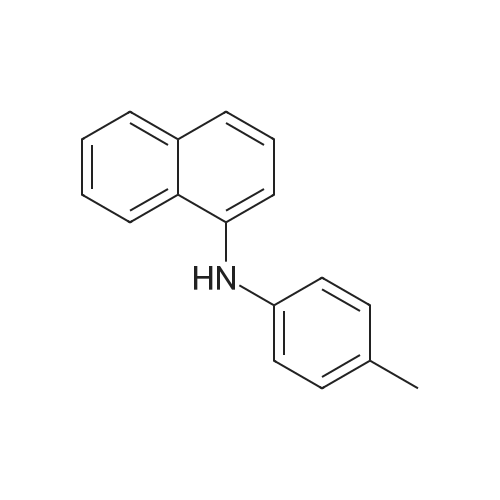
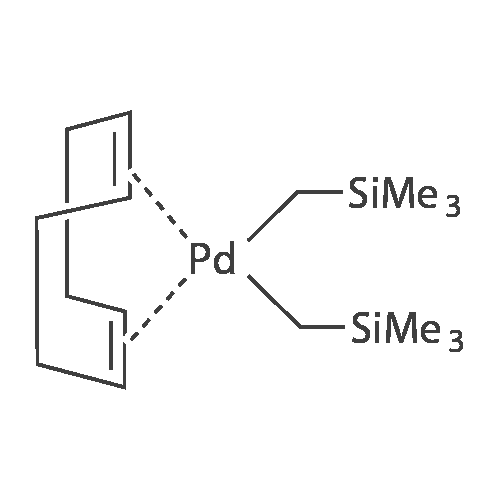
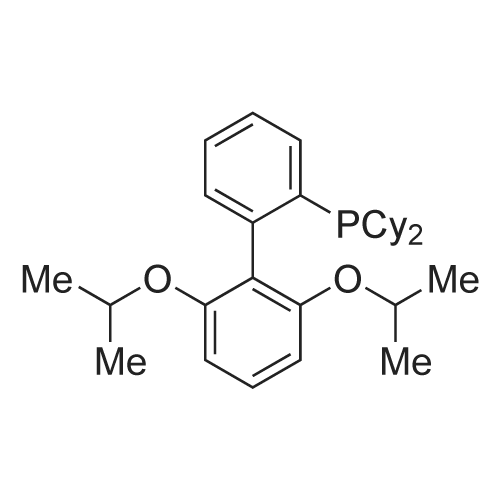
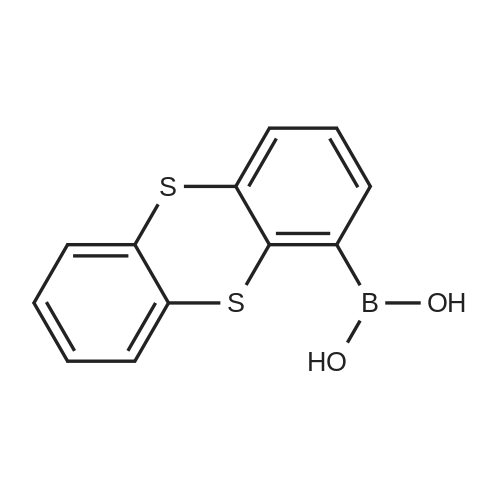
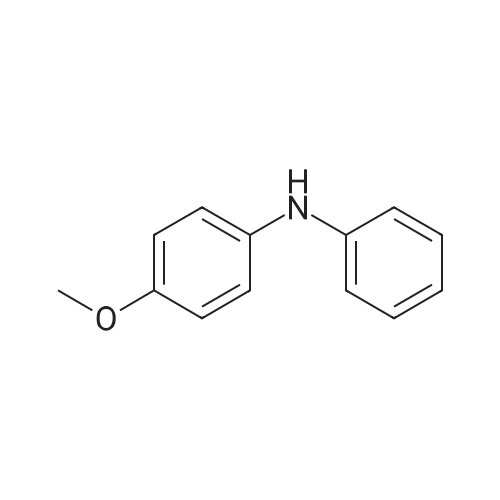
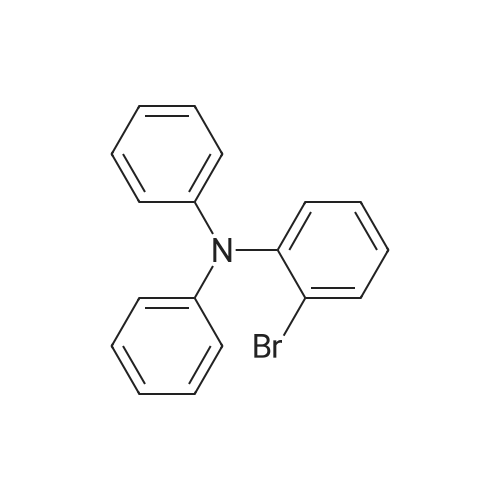
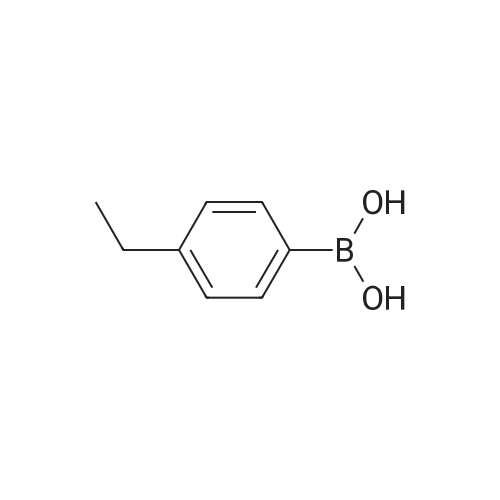
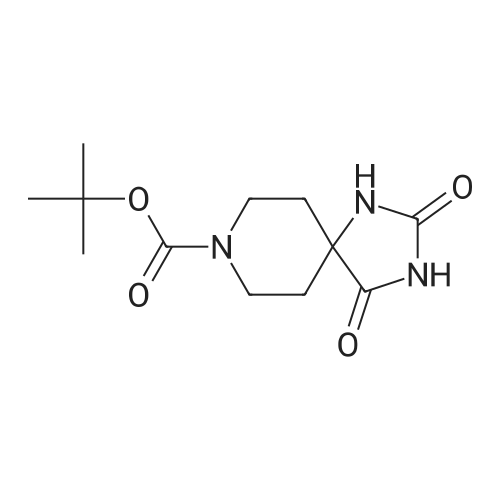
| 98% |
With potassium carbonate; cis,cis,cis-1,2,3,4-tetrakis(diphenylphosphinomethyl)cyclopentane In xylene at 130℃; for 72h; |
|
| 98% |
With sodium hydroxide In water monomer at 80℃; for 0.333333h; Inert atmosphere; Green chemistry; |
6. General procedure for Suzuki-Miyaura reaction
General procedure: 5 mL distilled water was taken in a 25 mL round bottom flask, equipped with a magnetic bar and a water condenser, and deoxygenated with nitrogen gas for 5 min. Aryl halide (1 mmol), arylboronic acid (1.2 mmol), NaOH (1.2 mmol) and Pd-γ-Fe2O3 (20 mg) were added to it and allowed to stir at 80 °C under nitrogen atmosphere. The progress of the reaction was monitored by TLC. After completion of the reaction, the catalyst was recovered using an external magnet. The reaction mixture was extracted with EtOAc (3 x 10 mL) and the combined organic layer was washed with water, brine solution and dried over anhydrous Na2SO4. The crude product was concentrated in a rotary evaporator and purified by column chromatography. The purified compounds were characterized by 1H and 13C NMR. |
| 98% |
With palladium nanoparticles-supported custard apple peels-ash catalyst In ethanol; water monomer at 20℃; for 0.166667h; Green chemistry; |
2.4 General Experimental Procedure forSuzuki-Miyaura Coupling Reaction
General procedure: All the Suzuki-Miyaura coupling reactions were carriedout under air atmosphere in dried glassware. In a 25 mLround bottom flask equipped with a magnetic stirrer, arylbromides (1.0 mmol), arylboronic acids (1.2 mmol), Pd/CAP-ash (5 wt%) and water:ethanol (3 mL) were placed.The resulting reaction mixtures were stirred at room temperaturefor appropriate time. The progress of reactionswas monitored by TLC. After completion of reactions,the reaction mixtures were extracted with ethyl acetate(2 × 10 mL). The organic layers were washed with brine(2 × 10 mL). The combined organic layer was collected,dried over Na2SO4and concentrated in vacuo. The residuewas purified by silica gel column chromatography usingn-hexane:EtOAc (9:1 v/v) to give the corresponding biarylcompound. The desired products were characterized bycomparing 1H, 13C NMR spectral data with authenticsamples. |
| 98% |
With palladium diacetate; potassium carbonate; dicyclohexyl(2’,4’,6’-triisopropyl-[ 1,1’-bi-phenyl]-2-yl)phosphane In ethanol at 60℃; for 5h; Inert atmosphere; Green chemistry; |
Suzuki coupling
Under an argon atmosphere, two parallel oven-dried Schlenk tubes were each charged with 4-bromobenzophenone (0.5 mmol), phenylboronic acid (0.55 mmol), Pd(OAc)2 (0.005 mmol), XPhos (0.01 mmol), K2CO3 (1 mmol), EtOH/H2O (3.6 mL/0.4 mL) was added by syringe. The reaction was stirred at 60 C for 5 h and monitored by TLC. After the reaction was completed, the reaction was worked-up using the following methods: LLE-based workup: The reaction mixture was quenched with water (15 mL) and extracted with EtOAc (3 x 20 mL); the combined organic layers were dried over Na2SO4 and filtered, the excess solvent in the filtrate was removed under reduced pressure. The residue was subjected to silica gel chromatography to afford the desired product 122.9 mg (95%). Fast2Flash-based workup: The reaction mixture was worked-up and purified according to the direct method to afford the desired product 126.3 mg (98%). Eluent: PE/EA=20/1 1H NMR (600 MHz, CDCl3) δ 7.90 (d, J = 8.2 Hz, 2H), 7.84 (d, J = 7.7 Hz, 2H), 7.71 (d, J = 8.2 Hz, 2H), 7.65 (d, J = 7.3 Hz, 2H), 7.60 (t, J = 7.4 Hz, 1H), 7.50 (dt, J = 12.5, 7.7 Hz, 4H), 7.41 (t, J = 7.4 Hz, 1H). Its spectroscopic data is consistent with the literature report. |
| 97% |
With potassium carbonate In water monomer; N,N-dimethyl-formamide at 80℃; for 0.333333h; Schlenk technique; |
19. General procedure for the Suzuki-Miyaura reaction:
General procedure: An oven-dried Schlenk flask, equipped with a magnetic stir bar, septum, and a condenser was charged with aryl halide (1.0 mmol), arylboronic acid (1.2 mmol), K2CO3 (2 mmol), 4 (0.143 g, 1 mol %), and 5 mL of solvent. The flask was immersed in an oil bath and stirred at 80 °C. Upon complete consumption of starting materials as determined by TLC analysis, the reaction mass was filtered and the solid washed with water (2Χ5 mL), and extracted with diethyl ether (3Χ5 mL). The combined organic layers were collected, dried over anhydrous Na2SO4, and concentrated in vacuum to afford product which was purified by silica gel column chromatography (n-hexane/EtOAc = 9:1) |
| 97% |
With Cs2CO3 In water monomer; N,N-dimethyl-formamide at 80℃; for 2h; |
Suzuki-Miyaura Reaction; General Procedure:
General procedure: Arylhalide 19 or 22 (1.0 mmol), phenylboronic acid (20; 1.2mmol, 0.146 g), 18 (0.015 mmol, 0.002 g), Cs2CO3 (1.5mmol, 0.489 g), DMF (2.5 mL), and H2O (2.5 mL) wereplaced in a 25 mL round-bottomed flask equipped with amagnetic stirrer. The flask was immersed in an oil bathregulated at 80 or 100 °C for the reaction time indicated inTable 1 or Table 2. After the reaction mixture was cooled tor.t., Et2O (8 mL) and H2O (8 mL) were added to the flask.The resulting mixture was vigorously stirred for 5 min, andthen filtered. The solid 18 collected on the filter was washedwith H2O (10 mL) and Et2O (10 mL) and then dried. Thefiltrate was transferred to a separation funnel and the organicphase was separated and washed with H2O (5 × 25 mL) andbrine (10 mL), and then dried over MgSO4. The solvent wasremoved under reduced pressure and the resulting residuewas analyzed by 1H and 13C NMR spectroscopy. When arylchlorides were used as substrates, the crude products werepurified by silica gel column chromatography. |
| 97% |
With C56H52N12O12Pd4S4; potassium carbonate In ethanol; water monomer at 62℃; for 2h; |
|
| 96% |
With Pd/SiO2; potassium hydroxide In water monomer for 0.116667h; Microwave irradiation; Green chemistry; |
|
| 95% |
With potassium fluoride; [(ferrocenyl)(phenyl)methyl]diphenylphosphane; palladium diacetate In tetrahydrofuran at 20℃; for 48h; |
|
| 95% |
With C34H32Cl2FeP2Pd In ethanol at 80℃; for 2h; Schlenk technique; |
General procedure for the Suzuki-Miyaura reaction
General procedure: An oven-dried Schlenk flask, equipped with a magneticstir bar, a septum and a condenser was charged with arylhalide (1.0 mmol), arylboronic acid (1.2 mmol), the gelentrappedbase (1 g, 2 mmol), Pd(dppf)Cl2 (0.0085 g,1 mol%) and 5 mL of 95% ethanol. The flask was immersedand stirred in an oil bath at 80 8C. Upon completeconsumption of starting materials as determined by TLCanalysis, the gel was separated by filtration and water(10 mL) was added. The filtrate was extracted with diethylether (3 5 mL). The combined organic layer was collected,dried over anhydrous Na2SO4 and concentratedunder vacuum to afford the product, which was purified bysilica gel column chromatography (n-hexane:ethyl acetate9:1) |
| 95% |
With potassium carbonate In ethanol at 80℃; for 2.5h; Schlenk technique; |
|
| 95% |
With potassium carbonate In water monomer at 60℃; for 0.583333h; Schlenk technique; Green chemistry; |
|
| 94% |
With tripotassium phosphate tribasic; 1,2,3,4,5-pentaphenyl-1′-(di-tert-butylphosphino)ferrocene; Palladium(0) bis(dibenzylideneacetone) In toluene at 100℃; for 1h; |
|
| 94% |
With {2-[1-(benzyloxyimino)ethyl]benzothiazole-k2N,N'}dichloropalladium(II); tetrabutylammonium bromide; potassium hydroxide In water monomer at 160℃; for 0.1h; Microwave irradiation; |
|
| 94% |
With potassium carbonate In ethanol at 80℃; for 2h; Schlenk technique; |
|
| 94% |
With potassium carbonate In ethanol at 80℃; for 2h; Schlenk technique; |
4.4 Typical experimental procedure for the Suzuki-Miyaura reaction
General procedure: In a typical procedure 50mL Schlenk tube containing magnetic stirring bar and equipped with reflux condenser was charged aryl bromide (1.0mmol), arylboronic acid (1.2mmol), K2CO3 (2.0mmol), and 0.009g catalyst (0.02mol%) in ethanol (5mL). The reaction mixture was vigorously stirred at 80°C. After completion of the reaction as monitored by TLC, the catalyst was separated out by filtration, followed by washing with water and diethyl ether. The filtrate was extracted with diethyl ether (3×10mL). The combined organic layers were collected, dried over anhydrous Na2SO4 and concentrated in vacuum to afford crude product, which was purified by silica gel column chromatography (n-hexane:EtOAc=9:1). |
| 93% |
With C56H44N2OP2Pd(2+)*2ClO4(1-); potassium carbonate In water monomer at 20℃; for 20h; |
|
| 93% |
Stage #1: (4-bromophenyl)(phenyl)methanone; phenylboronic acid With sodium 2,5-dimethylbenzene sulfonate In water monomer at 28℃; for 0.0833333h;
Stage #2: With palladium 10% on activated carbon; potassium carbonate In water monomer at 28℃; for 2h; |
|
| 93% |
With 2Na(1+)*C26H18N2O8PdS2(2-); potassium carbonate In water monomer at 20℃; for 10h; |
General procedure: The Suzuki reaction was performed in a 50 mL round-bottomed flask, aryl halide (0.5 mmol), arylboronic acid (0.65 mmol), K2CO3 (1 mmol), Complex 1 (0.2-1 mol%) and water (4 mL) were charged and stirred for the required time at room temperature for aryl bromides or at 100 °C for aryl chlorides. After completion, the mixture was cooled down to room temperature, diluted with water (10 mL) and extracted with diethyl ether (3 × 15 mL). The organic layer was washed with brine (3 × 15 mL), dried over anhydrous Na2SO4. The crude products were chromatographed on silica gel (ethyl acetate/hexane). |
| 92% |
With dichloro[N-hydroxy-1-(1-methyl-1H-benzimidazol-2-yl-κN3)ethanamine-κN]palladium; tetrabutylammonium bromide; potassium hydroxide In water monomer at 160℃; for 0.0666667h; Microwave irradiation; |
|
| 92% |
With potassium carbonate In ethanol at 20℃; for 3h; Schlenk technique; |
General procedure for the Suzuki coupling reaction
General procedure: Aryl halide (1.0 mmol), arylboronic acid (1.2 mmol), K2CO3 (2.0 mmol), Pd(at)Al2O3-agarose (100 mg) and EtOH (5 mL) were added to a Schlenk flask. The mixture was stirred at room temperature under air. Upon complete consumption of starting materials as determined by TLC analysis, the solid was filtered and washed with acetone (3 × 5 mL). The combined organic solvents were concentrated in vacuum to afford product which was purified by silica gel column chromatography (petroleum ether/EtOAc = 10:1). |
| 92% |
With tetrabutylammonium bromide; potassium carbonate In water monomer at 100℃; for 0.666667h; Green chemistry; |
2.6. General procedure for the Pd(0)-EDA/SC-2 catalyzed Suzukireaction
General procedure: To a mixture of aryl halide (1 mmol), aryl/heteroaryl boronicacid (1.2 mmol), TBAB (0.25 mmol), K2CO3(0.25 mmol) and Pd(0)-EDA/SC-2 (0.2 g, 2.5 mol% Pd), double distilled water (5 mL) wasadded and the reaction mixture was stirred in a microwave syn-thesizer at 100C for an appropriate time (monitored by TLC)(Scheme 2). After completion, the reaction mixture was cooledto room temperature and filtered. The catalyst was washed withEtOAc (3 × 5 mL) followed by double distilled water (3 × 10 mL). Itwas dried at 100C for 1 h and could be used in subsequent reac-tions. The organic layer was washed with water and dried overanhydrous Na2SO4. Finally, the product was obtained after removalof the solvent under reduced pressure followed by crystallizationfrom a suitable solvent or passing through column of silica gel(EtOAc-pet. ether). |
| 92% |
With [PdCl2(2-(pyridine-2-ylmethyl)sulfanylbenzoic acid)]; tetrabutylammonium bromide; potassium carbonate In water monomer at 110℃; for 8h; |
|
| 91% |
With potassium carbonate In water monomer; isopropanol at 60℃; for 4h; |
|
| 91% |
Stage #1: (4-bromophenyl)(phenyl)methanone With N2,N6-dibenzylpyridine-2,6-dicarboxamidopalladium(II)triphenylphosphine; potassium carbonate In ethanol; water monomer for 0.0833333h;
Stage #2: phenylboronic acid In ethanol; water monomer at 82℃; for 16h; |
2.3 Suzuki-Miyaura cross-coupling reaction
General procedure: A mixture of aryl halide (1mmol), catalyst (0.005mmol) and K2CO3 (2mmol) was stirred in EtOH-H2O (4:1) (5mL) for 5min. Phenylboronic acid (1.5mmol) was added to the above mixture and stirring was continued for required time at 82°C. Then, reaction mixture was diluted with ethyl acetate and water, and the catalyst was separated by centrifugation. The centrifugate was dried over anhydrous sodium sulphate, filtered and evaporated. Then the product was analyzed by GC/GCMS. |
| 90% |
With potassium carbonate In ethanol at 80℃; for 1h; Green chemistry; |
|
| 89% |
With chloro[4-tert-butyl-benzaldehyde 4-(β-d-glucopyranosyl)thiosemicarbazone]palladium(II) dimer; potassium carbonate In N,N-dimethyl-formamide at 100℃; for 24h; |
2.4. General experimental procedure for the Suzuki-Miyaura coupling
General procedure: Aryl bromide (1.0 mmol), phenylboronic acid (183 mg,1.5 mmol), K2CO3 (276 mg, 2.0 mmol), complex 2 in DMF(0.25 mM, 2 mL, 0.5 lmol) and dodecane (70 lL, 0.3 mmol) asinternal standard were stirred at 100 C in air for 24 h, and thenallowed to cool to room temperature. After addition of water(5 mL) and extraction with dichloromethane (2 10 mL), theorganic phase was washed with brine (10 mL), dried overNa2SO4, filtered, passed through Celite and analyzed by GC andGC-MS. After evaporation of the volatiles, isolation of the purebiaryl was achieved by column chromatography on silica gel usinghexane/AcOEt as eluent. All biaryls are known compounds andwere characterized by 1H NMR spectra. |
| 89% |
With dicyclohexyl({2’,6’-dimethoxy-[1,1‘-biphenyl]-2-yl})phosphane; palladium diacetate; potassium carbonate In 1,4-dioxane; water monomer at 100℃; Schlenk technique; |
|
| 88% |
With (1-(2,6-diisopropylphenyl)-2,3-dihydro-1H-imidazol-2-yl)(methyl(λ1-oxidaneyl)diphenyl-λ5-phosphaneyl)palladium(II) chloride; Cs2CO3 In methanol; dichloromethane; toluene at 100℃; Glovebox; |
|
| 87% |
With C40H56Cl2FeN2O4P2; anhydrous sodium carbonate In water monomer; propan-2-one at 20℃; for 4h; |
|
| 87% |
With PdCl(2-HO-C6H4-CH(Ph)-NH-(CH2)3-SeC6H5); potassium carbonate In water monomer; N,N-dimethyl-formamide for 5h; Heating; Aerobic conditions; |
|
| 87% |
With palladium diacetate; potassium carbonate |
|
| 85% |
With potassium carbonate In ethanol; water monomer at 20℃; for 2h; |
|
| 85% |
With palladium; potassium carbonate In ethanol; water monomer at 50℃; for 1h; |
|
| 83% |
With potassium carbonate In water monomer at 20℃; for 6h; |
Suzuki-Miyaura cross-coupling reaction
General procedure: For Suzuki-Miyaura reaction, appropriate amount of the catalyst, PdNP-NMe2SiO2, was added to a mixture of aryl halide (0.5 mmol), arylboronic acid (0.65 mmol), K2CO3 (1.5 mmol) in 6 mL solvent. The reaction was then stirred under desired temperature for the required time. The initial progress of the reaction was monitored by TLC using aluminum coated TLC plates (Merck) under UV light and the product formation was determined using GC-MS. After completion, the catalyst was collected by filtration and washed with isopropanol-water. The filtrate was diluted with water and extracted with ether and then dried over Na2SO4. After evaporation of the solvent under reduced pressure, the residue was chromatographed (silica gel, ethyl acetate-hexane, 1:9) to obtain the desired product. |
| 81% |
With sodium phosphate tribasic dodecahydrate In ethanol; water monomer at 20℃; for 22.5h; Green chemistry; |
|
| 80% |
With palladium diacetate; triphenylphosphine-3,3',3''-trisulfonic acid trisodium salt; isopropylamine In water monomer; acetonitrile at 80℃; for 1h; |
|
| 78% |
With tripotassium phosphate tribasic; C35H43ClN7Ni(1+)*BF4(1-) In acetonitrile at 80℃; for 8h; |
|
| 75% |
With potassium carbonate In ethanol; water monomer at 20℃; for 8h; Green chemistry; |
2.8 General Procedure for Suzuki-Miyaura Cross-Coupling Reaction
General procedure: A mixture of aryl halide (0.27 mmol), phenylboronicacid (1 mmol), NHC-PdGO heterogeneous catalyst(0.01g) and potassium carbonate (1.1mmol) was stirred inEtOH:H2O (1:1) solvent system at room temperature for designated hours. The progress of the reaction was monitoredby TLC. After reaction completion, the mixture was cooledto room temperature and NHC-PdGO heterogeneous catalystwas separated through centrifugation. To the filtrate,dichloromethane and water were added. Dichloromethanelayer was separated from the water layer using a separatoryfunnel and dried with magnesium sulphate. The drieddichloromethane was concentrated in vacuum and obtainedcrude product was purified from column chromatographyover silica gel using hexane and ethyl acetate as eluting solventto get the corresponding products in good to excellentyields. All the coupled products were known molecules andwere confirmed by comparing the 1H NMR spectral datawith those of authentic samples. |
| 74% |
With potassium carbonate In water monomer at 100℃; for 4.25h; |
2.3. General procedure for the CoGO/Fe3O4/L-dopa catalyzed Suzukicross-coupling
General procedure: A mixture of aryl halide (1 mmol), phenyl boronic acid (1.2 mmol),K2CO3 (1.2 eq.) and CoGO/Fe3O4/L-dopa (0.05 g, 1.84 mol% Co) in double distilled water (5 mL) was stirred in a round bottom flask (50 mL) at 100 °C till the completion of reaction (monitored by TLC) (Table 3). After that, the reaction mixture was cooled to room temperature.The catalyst was removed via external magnet and washed with EtOAc (3×5 mL) followed by deionized water (3×10 mL). It was dried under vacuum for 2 h. The organic fraction was washed with brine solution and dried over anhydrous Na2SO4. Finally, the product was obtained either by the exclusion of the solvent under reduced pressure or by passing through column of silica gel using EtOAc-pet.ether as eluting solvent. |
| 72% |
With potassium carbonate In ethanol; water monomer at 20℃; for 2h; Green chemistry; |
2.4 General procedure for Suzuki-Miyaura cross-coupling reaction
General procedure: In a 10 mL glass vial equipped with a cap containing 5 mL of ethanol:water (1:1) mixture, aryl halide (1 equiv), phenylboronic acid (1.1 equiv), K2CO3 (2.5 equiv) were added followed by dipping of the dip catalyst into the reaction mixture which was then stirred magnetically at room temperature for required time. The progress of the reaction was monitored by thin layer chromatography (TLC). After reaction completion, the dip catalyst was simply removed from the reaction mass and washed with ethanol (1 x 5 mL) and water (1 x 5 mL) and was reused without purifying further. The product was extracted using dichloromethane (2 x 10 mL) and the combined organic layer was subjected to water wash (2 x 10 mL) followed by drying of the organic layer over Na2SO4. The dried organic layer was concentrated in vacuo, and the product was purified by column chromatography using n-hexane and ethyl acetate as eluents to afford the corresponding products in good to excellent yields. All the coupled products were known molecules and were confirmed by comparing with our previous standards (Kandathil et al., 2017; Vishal et al., 2017). |
| 70% |
With Ni0.90Pd0.10; potassium carbonate In ethanol; water monomer at 50℃; for 24h; |
|
| 67% |
With potassium carbonate In ethanol; water monomer at 100℃; for 15h; Green chemistry; |
|
| 61% |
With potassium carbonate In ethanol; water monomer at 20℃; for 12h; Green chemistry; |
2.3.5. General procedure for Suzuki-Miyaura cross-coupling reactionscatalyzed by MNPs(at)SB-Pd nanomagnetic catalyst
General procedure: An oven-dried flask was charged with aryl halide (0.27 mmol),phenylboronic acid (0.036 g, 0.30 mmol), MNPsSB-Pd nanomagneticcatalyst (0.05 mol% Pd) and K2CO3 (0.082 g, 0.60 mmol).EtOH:H2O (1:1, 10 mL) was added and the reaction mixture wasstirred at room temperature for designated time. The progress ofthe reaction was monitored using TLC. Then the reaction mixturewas allowed to cool to room temperature and quenched by addingdichloromethane (20 mL) and the MNPsSB-Pd nanomagnetic catalystwas separated using a permanent magnet. Dichloromethanelayer was separated from water layer through separatory funneland dried with anhydrous MgSO4. The dried dichloromethane layerwas concentrated in vacuum and purified through column chromatographyusing hexane and ethyl acetate as eluting solvent toget the corresponding products in excellent yields. |
|
With potassium carbonate In toluene at 110℃; for 15h; Yield given; |
|
| 99 % Chromat. |
With potassium fluoride; [(μ-PPh2CH2PPh2)Co2((CO)4][μ,η-PhCCP(cy)2] In toluene at 40℃; for 16h; |
|
|
With Merrifield resin-supported salen-type palladium(II); N-ethyl-N,N-diisopropylamine In water monomer; N,N-dimethyl-formamide at 100℃; |
|
|
With potassium carbonate In methanol; toluene for 16h; Reflux; |
|
| 95.6 %Chromat. |
With potassium carbonate In water monomer; N,N-dimethyl-formamide at 50℃; for 3h; |
|
|
With potassium carbonate In ethanol; water monomer at 70℃; for 1h; Green chemistry; |
The general procedure for the PdIRA-900 catalyzed Suzuki coupling reaction was as follows. A 25 mL three necked flask was charged with PdIRA-900 (50 mg). The solvent (5 mL) and phenylboronic acid (1.1 mmol) were added followed by bromobenzene (1.0 mmol) and K2CO3 (2.2 mmol). The flask was stirred and maintained at 70 °C in a water bath for an appropriate time. The reaction products were analyzed by GC-MS. |
|
With C55H66N2O2; palladium diacetate; potassium carbonate In methanol; dichloromethane at 30℃; for 0.5h; |
|
| 88 %Chromat. |
With C39H39ClN2NiP; Cs2CO3 In toluene at 80℃; Inert atmosphere; Schlenk technique; Glovebox; |
|
| 80 %Chromat. |
With palladium dibromo{N,N-bis(diphenylphosphanyl)tert-butylamine}; Cs2CO3 In N,N-dimethyl-formamide at 80℃; for 1h; Inert atmosphere; Schlenk technique; |
4.5 General experimental procedure for the Suzuki-Miyaura coupling
General procedure: Aryl bromide (1.0mmol), phenylboronic acid (183mg, 1.5mmol), Cs2CO3 (652mg, 2.0mmol), palladium(II) complex [Pd(P,P)Br2] in DMF (0.5mM, 2mL, 1.0μmol) and dodecane (70μL, 0.3mmol) as internal standard were stirred over a preheating oil bath at 80°C under argon for 1h. After cooling to room temperature, addition of water (5mL) and extraction with dichloromethane (2×10mL), the organic layer was washed with brine (10mL), dried over Na2SO4, filtered, passed through celite and analyzed by GC. GC retention times were compared with those of authentic samples. After evaporation of the any volatile residue, isolation of the pure biaryl was achieved by column chromatography on silica gel using a mixture of hexane/AcOEt as eluent. All biaryls are known compounds [25c], and were characterized by 1H and 13C NMR spectra. |
| 86 %Chromat. |
With potassium carbonate In ethanol; water monomer at 70℃; for 1h; |
|
|
With potassium carbonate In ethanol at 50℃; Sonication; |
|
|
With C42H26Cl2N2PdS2; potassium carbonate at 90℃; for 8h; |
|
|
With tripotassium phosphate tribasic In toluene |
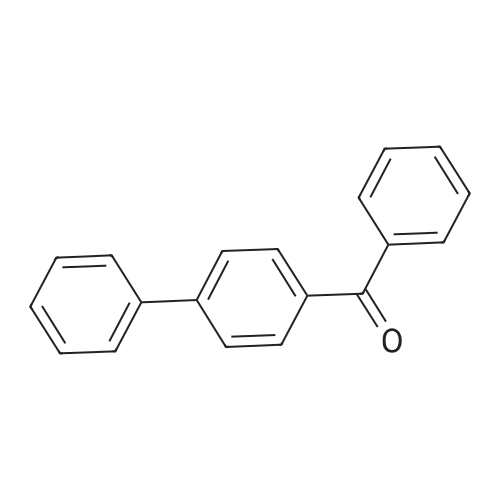
74 4-phenylbenzophenone (Table 10, Entry 2)
Example 74 4-phenylbenzophenone (Table 10, Entry 2) According to the general procedure described above, 4-Bromobenzophenone (131 mg, 0.50 mmol) reacted with phenylboronic acid (92 mg, 0.75 mmol) using 0.0005 mol % of Pd(dba)2, 0.002 mol % of Ph5FcP(t-Bu)2, and K3PO4 (318 mg, 1.50 mmol) in toluene solvent at 100° C. for 1 hr to title compound (121 mg, 94%) as a solid: 1H-NMR (400 MHz, CDCl3): δ 7.92 (d, 2H, J=8.4 Hz), 7.87 (d, 2H, J=7.2 Hz), 7.73 (d, 2H, J=8.4 Hz), 7.18 (d, 2H, J=6.8 Hz), 7.63 (m, 1H), 7.51 (m, 3H), 7.43 (m, 2H). 13C{1H}-NMR (100 MHz, CDCl3): δ 196.30, 145.16, 139.89, 137.68, 136.15, 132.34, 130.69, 129.96, 128.92, 128.26, 128.14, 127.25, 126.91. GC/MS(EI): m/z 181 (M-77+). Anal. Calcd for C19H14O: C, 88.34; H, 5.46. Found: C, 88.26; H, 5.62. |
|
With C25H22Cl2NPPd; potassium carbonate In water monomer; N,N-dimethyl-formamide at 105℃; for 10h; |
2.5.1. Suzuki coupling (i.e., C-C coupling) reaction
General procedure: A 100 mL round bottom flask (equipped with a refluxing con- denser) was charged with aryl halide (1.0 mmol), phenylboronic acid (1.1 mmol, 0.133 g), K 2 CO 3 (2.0 mmol, 0.276 g), Pd(II) complex (0.01 or 0.001 mol%) and aqueous DMF (5.0 mL). The reaction mix- ture was refluxed at 105 °C for 10 h and the progress of catalytic reaction was monitored using thin layer chromatography (TLC). Af- ter completion of ten hours, the resultant mixture was cooled to room temperature and water was added to it. Thereafter, the ex- traction of cross-coupled product was carried out using diethyl ether. The organic layer was dried over anhydrous Na 2 SO 4 and the solvent was evaporated using rotary evaporator to obtain the prod- uct. The % conversion was estimated using 1 H NMR and GC stud- ies. Thereafter, the product was subjected to silica gel column chro- matography for purification using ethyl acetate and n -hexane mix- ture as an eluent. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping