* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
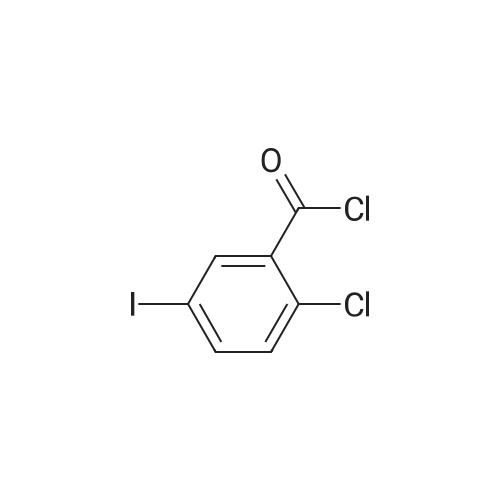
With aluminum (III) chloride In dichloromethane at -5℃; for 6 h;
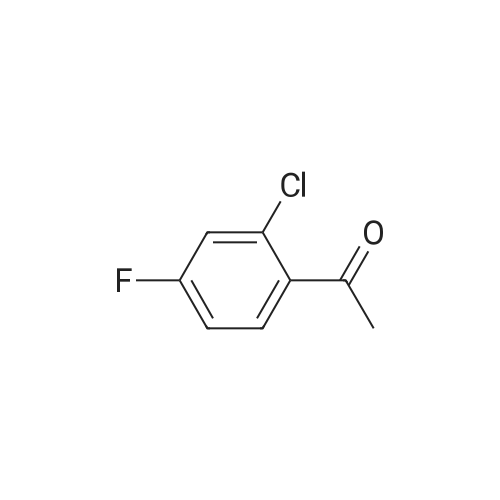
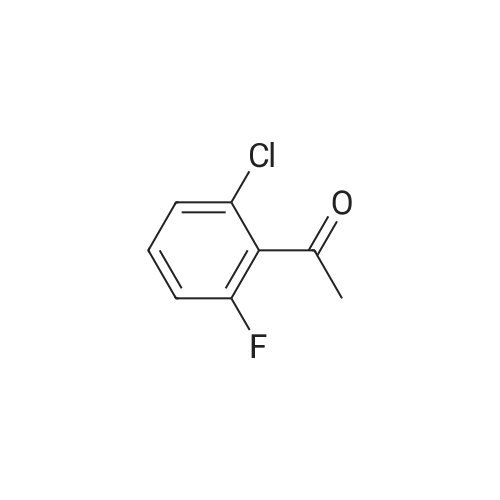
Equipped with a thermometer, constant pressure dropping funnel, drying tube 1000mL flask was added three DCM200mL, VIII (70.62g, 250mmol) ,was added 1 ml of DMF and, at room temperature was slowly added dropwise oxalyl chloride (23.4mL g, 275mmol), was added dropwise after the completion of 20 incubated 2h.After adding concentrated to remove the DCM, oxalyl chloride 200mL DCM dissolved chloride.Equipped with a thermometer, constant pressure dropping funnel, 1000mL three 26mL flask with a drying tube was added fluorobenzene, DCM200mL the system temperature was dropped to -5 deg.] C, graded added to the system aluminum trichloride (36.67g, 275mmol) , dropped acid chloride prepared raw reaction incubated 6h completed.System 40mL conc HCl + 400mL poured into ice water to quench the reaction, the DCM. The combined organic phases were washed until neutral sodium bicarbonate, once with saturated brine separation was washed with water, dried, and concentrated to give VII84.23g, yield 93.4percent.
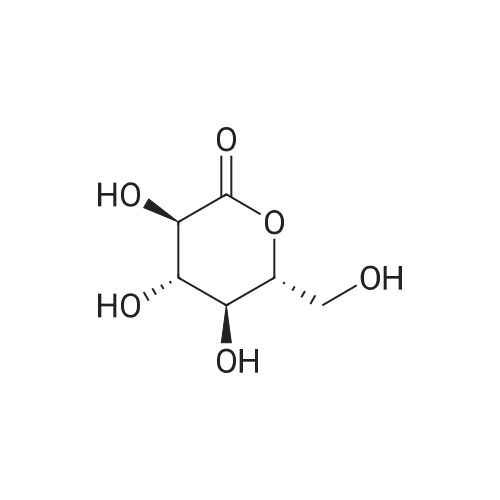
105 g
at 25℃; Inert atmosphere
Oxalyl chloride (72 g, 0.56 moles) was added to a slurry of formula 5a (100 g, 0.35 moles) and a catalytic amount of N, N-dimethylformamide (5mL) in fluorobenzene (250 mL) over about 60 minutes at 15-25 °C under nitrogen atmosphere. The mixture was stirred at 21-25 °C. After completion of the reaction, the reaction mass was concentrated to remove excess oxalyl chloride. The obtained residue was diluted with fluorobenzene (125 mL) and cooled to 15-25 °C. Aluminum chloride (53.5 g, 0.40 moles) was added to this reaction mixture portion- wise while keeping the reaction mass temperature below 25 °C and maintaining it at same temperature to complete the reaction. After completion of reaction, the reaction mass was quenched into precooled dilute hydrochloric acid (5percent, 1700 mL, 0-5 °C) at 5-25 °C. After stirring for 60 minutes at 20-25 °C, the product was extracted with methylene chloride twice (first with 500 mL, then with 250 mL). The combined organic layers were washed with 10percent aqueous sodium hydroxide solution (250mL), water (500mL), and 10percent aqueous sodium chloride solution (250mL), sequentially. Thereafter, the organic layer was concentrated and the obtained residue was diluted with n-heptane (250mL) and stirred to precipitate formula 7a as a white solid (105 g).
42.1 g
With aluminum (III) chloride In dichloromethane at 20℃; for 3 h; Cooling with ice
5-iodo-2-chlorobenzoic acid (33.895 g, 120 mmol) was added DCM (100 mL) DMF (0.175 g, 0.02 eq), Stir, Slowly add oxalyl chloride (12.3 ^, 1.2 called), After the drop is completed at room temperature reaction 21 , The solvent was removed by concentration under reduced pressure, Add DCM (lOOmL), Fluorobenzene (17.298 g, 1.5 eq) was added, Alcia was added to A1C13 (17.6 g, 1 leq) After adding the reaction at room temperature for 3h, After TLC detection reaction was completed, Ice bath slowly add water (100mL), After stirring, The organic phase was washed with 10percent NaCl (50 mL) The organic phase was concentrated under reduced pressure to give a pale yellow oil, Plus isopropyl alcohol (lOOmL), Add water (lOOmL), Ice bath stirring crystallization 2h, Filter, The filter cake was washed with isopropanol / water (1: 1) mixed solution (20 mL) Dried to give 42.1 g of an off-white solid of compound Vb, Yield: 97.3percent Purity: 99.17percent
Reference:
[1] Patent: CN105399735, 2016, A, . Location in patent: Paragraph 0037; 0038; 0039
[2] Patent: US2006/258749, 2006, A1, . Location in patent: Page/Page column 21
[3] Organic Letters, 2014, vol. 16, # 16, p. 4090 - 4093
[4] Patent: WO2015/101916, 2015, A1, . Location in patent: Page/Page column 18; 19
[5] Patent: CN106928040, 2017, A, . Location in patent: Paragraph 0038-0039
2
[ 462-06-6 ]
[ 19094-56-5 ]
[ 915095-86-2 ]
Yield
Reaction Conditions
Operation in experiment
94%
at 25 - 30℃; for 5 h;
Example 1 : Synthesis of the fluoride VIII.1Oxalylchloride (176kg; 1386mol; 1 ,14eq) is added to a mixture of 2-chloro-5-iodo benzoic acid (343kg; 1214mol) (compound IX.1 ), fluorobenzene (858kg) and N,N- dimethylformamide (2kg) within 3 hours at a temperature in the range from about 25 to 30°C (gas formation). After completion of the addition, the reaction mixture is stirred for additional 2 hours at a temperature of about 25 to 30°C. The solvent (291 kg) is distilled off at a temperature between 40 and 45°C (p=200mbar). Then the reaction solution (91 1 kg) is added to aluminiumchloride AICI3 (181 kg) andfluorobenzene (192kg) at a temperature between about 25 and 30°C within 2 hours. The reaction solution is stirred at the same temperature for about an additional hour. Then the reaction mixture is added to an amount of 570 kg of water within about 2 hours at a temperature between about 20 and 30°C and stirred for an additional hour. After phase separation the organic phase (1200kg) is separated into two halves (600kg each). From the first half of the organic phase solvent (172kg) is distilled off at a temperature of about 40 to 50°C (p=200mbar). Then 2-propanole (640kg) is added. The solution is heated to about 50°C and then filtered through a charcoal cartouche (clear filtration). The cartouche may be exchanged during filtration and washed with a fluorobenzene/2-propanole mixture (1 :4; 40kg) after filtration. Solvent (721 kg) is distilled off at a temperature of about 40 to 50°C and p=200mbar. Then 2-propanole (240kg) is added at a temperature in the range between about 40 to 50°C. If the content of fluorobenzene is greater than 1 percent as determined via GC, another 140kg of solvent are distilled off and 2-propanole (140kg) is added. Then the solution is cooled from about 50°C to 40°C within one hour and seeding crystals (50g) are added. The solution is further cooled from about 40°C to 20°C within 2 hours. Water (450kg) is added at about 20°C within 1 hour and the suspension is stirred at about 20°C for an additional hour before the suspension is filtered. The filter cake is washed with 2-propanole/water (1 :1 ; 800kg). The product is dried until a water level of <0.06percentw/w is obtained. The second half of the organic phase is processed identically. A total of 410kg (94percentyield) of product which has a white to off-white crystalline appearance, is obtained. The identity of the product is determined via infrared spectrometry.
94%
Stage #1: With oxalyl dichloride In N,N-dimethyl-formamide at 25 - 30℃; for 5 h; Stage #2: at 20 - 30℃; for 6 h;
Oxalylchloride (176 kg; 1386 mol; 1.14 eq) is added to a mixture of 2-chloro-5-iodo benzoic acid (343 kg; 1214 mol) (compound IX.1), fluorobenzene (858 kg) and N,N-dimethylformamide (2 kg) within 3 hours at a temperature in the range from about 25 to 30° C. (gas formation). After completion of the addition, the reaction mixture is stirred for additional 2 hours at a temperature of about 25 to 30° C. The solvent (291 kg) is distilled off at a temperature between 40 and 45° C. (p=200 mbar). Then the reaction solution (911 kg) is added to aluminiumchloride AlCl3 (181 kg) and fluorobenzene (192 kg) at a temperature between about 25 and 30° C. within 2 hours. The reaction solution is stirred at the same temperature for about an additional hour. Then the reaction mixture is added to an amount of 570 kg of water within about 2 hours at a temperature between about 20 and 30° C. and stirred for an additional hour. After phase separation the organic phase (1200 kg) is separated into two halves (600 kg each). From the first half of the organic phase solvent (172 kg) is distilled off at a temperature of about 40 to 50° C. (p=200 mbar). Then 2-propanole (640 kg) is added. The solution is heated to about 50° C. and then filtered through a charcoal cartouche (clear filtration). The cartouche may be exchanged during filtration and washed with a fluorobenzene/2-propanole mixture (1:4; 40 kg) after filtration. Solvent (721 kg) is distilled off at a temperature of about 40 to 50° C. and p=200 mbar. Then 2-propanole (240 kg) is added at a temperature in the range between about 40 to 50° C. If the content of fluorobenzene is greater than 1percent as determined via GC, another 140 kg of solvent are distilled off and 2-propanole (140 kg) is added. Then the solution is cooled from about 50° C. to 40° C. within one hour and seeding crystals (50 g) are added. The solution is further cooled from about 40° C. to 20° C. within 2 hours. Water (450 kg) is added at about 20° C. within 1 hour and the suspension is stirred at about 20° C. for an additional hour before the suspension is filtered. The filter cake is washed with 2-propanole/water (1:1; 800 kg). The product is dried until a water level of <0.06percent w/w is obtained. The second half of the organic phase is processed identically. A total of 410 kg (94percent yield) of product which has a white to off-white crystalline appearance, is obtained. The identity of the product is determined via infrared spectrometry.
94%
With aluminum (III) chloride; oxalyl dichloride In N,N-dimethyl-formamide at 25 - 45℃; Industry scale
Oxalylchloride (176 kg; 1386 mol; 1.14 eq) is added to a mixture of 2-chloro-5-iodo benzoic acid (343 kg; 1214 mol) (compound IX.1), fluorobenzene (858 kg) and N,N-dimethylformamide (2 kg) within 3 hours at a temperature in the range from about 25 to 30° C. (gas formation). After completion of the addition, the reaction mixture is stirred for additional 2 hours at a temperature of about 25 to 30° C. The solvent (291 kg) is distilled off at a temperature between 40 and 45° C. (p=200 mbar). Then the reaction solution (911 kg) is added to aluminiumchloride AlCl3 (181 kg) and fluorobenzene (192 kg) at a temperature between about 25 and 30° C. within 2 hours. The reaction solution is stirred at the same temperature for about an additional hour. Then the reaction mixture is added to an amount of 570 kg of water within about 2 hours at a temperature between about 20 and 30° C. and stirred for an additional hour. After phase separation the organic phase (1200 kg) is separated into two halves (600 kg each). From the first half of the organic phase solvent (172 kg) is distilled off at a temperature of about 40 to 50° C. (p=200 mbar). Then 2-propanole (640 kg) is added. The solution is heated to about 50° C. and then filtered through a charcoal cartouche (clear filtration). The cartouche may be exchanged during filtration and washed with a fluorobenzene/2-propanole mixture (1:4; 40 kg) after filtration. Solvent (721 kg) is distilled off at a temperature of about 40 to 50° C. and p=200 mbar. Then 2-propanole (240 kg) is added at a temperature in the range between about 40 to 50° C. If the content of fluorobenzene is greater than 1percent as determined via GC, another 140 kg of solvent are distilled off and 2-propanole (140 kg) is added. Then the solution is cooled from about 50° C. to 40° C. within one hour and seeding crystals (50 g) are added. The solution is further cooled from about 40° C. to 20° C. within 2 hours. Water (450 kg) is added at about 20° C. within 1 hour and the suspension is stirred at about 20° C. for an additional hour before the suspension is filtered. The filter cake is washed with 2-propanole/water (1:1; 800 kg). The product is dried until a water level of <0.06percent w/w is obtained. The second half of the organic phase is processed identically. A total of 410 kg (94percent yield) of product which has a white to off-white crystalline appearance, is obtained. The identity of the product is determined via infrared spectrometry.
78.6%
Stage #1: With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane for 3 h; Stage #2: With aluminum (III) chloride In dichloromethane at 0 - 5℃; for 1 h;
5-Iodo-2-chlorobenzoic acid (20.0 g, 0.071 mol) was added to a solution of 2.0 M oxalyl chloride in dichloromethane (39 ml, 0.078 mol), stirred for suspension, and then 8 drops of DMF solution was added dropwise. After the reaction was carried out for 3 hours, the solution was clarified and the reaction was almost complete. The solvent was spin-dried on a rotary evaporator, then 15 ml of methylene chloride was added, and the solvent was dried. After spin-drying, add 30 ml of dichloromethane, stir, cool to 0-5 ° C, add fluorobenzene (7. lg, 0.074 mol), add anhydrous aluminum trichloride (9.9 g, 0.074 mol) in batches, control The temperature is not more than 5 °C, after the completion of the addition, stirring is continued at 4 ° C for 1 h, the reaction is almost complete by TLC, the reaction is quenched on ice-water mixture, the organic phase is separated, and the aqueous phase is extracted with dichloromethane. The phase was washed twice with 1 mol/L hydrochloric acid, washed once with water, and washed twice with 1 mol/L NaOH solution.The mixture was washed twice with saturated sodium chloride and dried over anhydrous sodium sulfate. Drain filtration, spin dry solvent to obtain oilAfter column chromatography, 20. 1g (78.6percent) of a white solid was obtained.
Reference:
[1] Patent: WO2011/39107, 2011, A1, . Location in patent: Page/Page column 18-19
[2] Patent: US2011/237789, 2011, A1, . Location in patent: Page/Page column 11
[3] Patent: US2011/237526, 2011, A1, . Location in patent: Page/Page column 7
[4] Patent: CN108218928, 2018, A, . Location in patent: Paragraph 0196; 0197; 0198
3
[ 19094-56-5 ]
[ 915095-86-2 ]
Reference:
[1] Organic Letters, 2014, vol. 16, # 16, p. 4090 - 4093
[2] Patent: CN105399735, 2016, A,
[3] Patent: CN106928040, 2017, A,
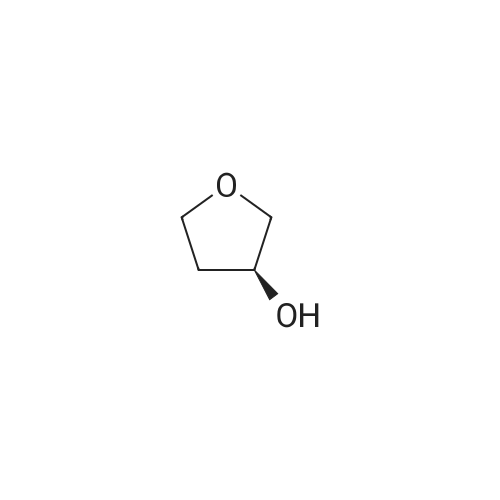
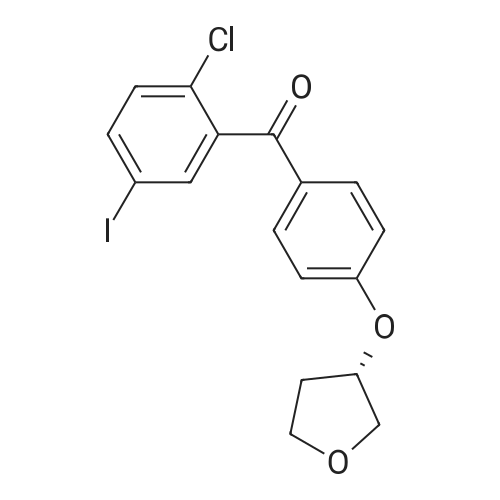
With potassium <i>tert</i>-butylate In tetrahydrofuran at 16 - 25℃; for 3 h;
Example 2: Synthesis of the ketone VII.1To a solution of the fluoride VIII.1 (208kg), tetrahydrofuran (407kg) and (S)-3- hydroxytetrahydrofuran (56kg) is added potassium-terf-butanolate solution (20percent) in tetrahydrofuran (388kg) within 3 hrs at 16 to 25°C temperature. After completion of the addition, the mixture is stirred for 60min at 20°C temperature. Then the conversion is determined via HPLC analysis. Water (355kg) is added within 20 min at a temperature of 21 °C (aqueous quench). The reaction mixture is stirred for 30 min (temperature: 20°C). The stirrer is switched off and the mixture is left stand for 60 min (temperature: 20°C). The phases are separated and solvent is distilled off from the organic phase at 19 to 45°C temperature under reduced pressure. 2- Propanol (703kg) is added to the residue at 40 to 46°C temperature and solvent is distilled off at 41 to 50°C temperature under reduced pressure. 2-Propanol (162kg) is added to the residue at 47°C temperature and solvent is distilled off at 40 to 47°C temperature under reduced pressure. Then the mixture is cooled to 0°C within 1 hr 55 min. The product is collected on a centrifuge, washed with a mixture of 2- propanol (158kg) and subsequently with terf.-butylmethylether (88kg) and dried at 19 to 43°C under reduced pressure. 227kg (91 ,8percent) of product are obtained as colourless solid. The identity of the product is determined via infrared spectrometry.
91.8%
Stage #1: With potassium <i>tert</i>-butylate In tetrahydrofuran at 16 - 25℃; for 4 h; Stage #2: at 20 - 21℃; for 1.83333 h;
To a solution of the fluoride VIII.1 (208 kg), tetrahydrofuran (407 kg) and (S)-3-hydroxytetrahydrofuran (56 kg) is added potassium-tert-butanolate solution (20percent) in tetrahydrofuran (388 kg) within 3 hrs at 16 to 25° C. temperature. After completion of the addition, the mixture is stirred for 60 min at 20° C. temperature. Then the conversion is determined via HPLC analysis. Water (355 kg) is added within 20 min at a temperature of 21° C. (aqueous quench). The reaction mixture is stirred for 30 min (temperature: 20° C.). The stirrer is switched off and the mixture is left stand for 60 min (temperature: 20° C.). The phases are separated and solvent is distilled off from the organic phase at 19 to 45° C. temperature under reduced pressure. 2-Propanol (703 kg) is added to the residue at 40 to 46° C. temperature and solvent is distilled off at 41 to 50° C. temperature under reduced pressure. 2-Propanol (162 kg) is added to the residue at 47° C. temperature and solvent is distilled off at 40 to 47° C. temperature under reduced pressure. Then the mixture is cooled to 0° C. within 1 hr 55 min. The product is collected on a centrifuge, washed with a mixture of 2-propanol (158 kg) and subsequently with tert.-butylmethylether (88 kg) and dried at 19 to 43° C. under reduced pressure. 227 kg (91.8percent) of product are obtained as colourless solid. The identity of the product is determined via infrared spectrometry.
91.8%
With potassium <i>tert</i>-butylate In tetrahydrofuran at 16 - 25℃; for 4 h; Industry scale
To a solution of the fluoride VIII.1 (208 kg), tetrahydrofuran (407 kg) and (S)-3-hydroxytetrahydrofuran (56 kg) is added potassium-tert-butanolate solution (20percent) in tetrahydrofuran (388 kg) within 3 hrs at 16 to 25° C. temperature. After completion of the addition, the mixture is stirred for 60 min at 20° C. temperature. Then the conversion is determined via HPLC analysis. Water (355 kg) is added within 20 min at a temperature of 21° C. (aqueous quench). The reaction mixture is stirred for 30 min (temperature: 20° C.). The stirrer is switched off and the mixture is left stand for 60 min (temperature: 20° C.). The phases are separated and solvent is distilled off from the organic phase at 19 to 45° C. temperature under reduced pressure. 2-Propanol (703 kg) is added to the residue at 40 to 46° C. temperature and solvent is distilled off at 41 to 50° C. temperature under reduced pressure. 2-Propanol (162 kg) is added to the residue at 47° C. temperature and solvent is distilled off at 40 to 47° C. temperature under reduced pressure. Then the mixture is cooled to 0° C. within 1 hr 55 min. The product is collected on a centrifuge, washed with a mixture of 2-propanol (158 kg) and subsequently with tert.-butylmethylether (88 kg) and dried at 19 to 43° C. under reduced pressure. 227 kg (91.8percent) of product are obtained as colourless solid. The identity of the product is determined via infrared spectrometry.
75%
With potassium <i>tert</i>-butylate In tetrahydrofuran at 20℃;
Equipped with a thermometer, pressure-equalizing dropping funnel was added 500mL three bottles VII (84.2g, 234mmol), S -3-hydroxytetrahydrofuran (20.6g, 234mmol), was added THF100mL clear solution after stirring, potassium tert-butoxide (34.1 g, 234mmol) was dissolved 150mLTHF added to the system, stirred at room temperature overnight, TLC monitored the reaction was complete, the reaction was quenched with 230 mL of water, spin-out section THF, EA extracted three times, the combined organic phase was washed with water and saturated brine, dried and concentrated to give 120.5 g crude product, after using 170mL of isopropanol + 20mL water crystallized product 75g, purity 98.2percent, yield 75percent.
64.3%
With potassium <i>tert</i>-butylate In tetrahydrofuran at 16 - 25℃; for 1 h;
5-Iodo-2-chloro-4'-fluorobenzophenone (V-2) (18.5 g, 0.051 mol) was dissolved in 50 ml of tetrahydrofuran solutionThen, (5)-3_light-based tetrahydrofuran (4.9 g, 0.056 mol) was added and stirred, and finally a solution of potassium t-butoxide (6.9 g, 0.061 mol) dissolved in 30 ml of tetrahydrofuran was added dropwise, and the temperature was controlled at 16-25 ° C. After the addition was completed, the reaction was stirred at 20 ° C for 1 hour, and the reaction was almost complete by TLC, and the purified water was slowly added and stirred for 30 min.The solution was separated and the organic phase was dried. After column chromatography, 14. lg (64.3percent) of solid was obtained.
90 g
With potassium <i>tert</i>-butylate In tetrahydrofuran at 2 - 10℃;
A mixture of formula 7a (100 g, 0.277 mole) and (S)-tetrahydrofuran-3-ol (27.4 g, 0.31 mole) in tetrahydrofuran (260 mL) was prepared. Potassium tert-butoxide (46.5 g, 0.414 mole) in tetrahydrofuran (405 mL) was added to this mixture solution slowly over a period of 90 minutes at 2-6 °C, maintaining the reaction mass at 7-10 °C to complete the reaction. Thereafter, precooled water (285 mL) was added to quench the reaction at 5-18 °C and the product was extracted twice with methyl tert- butyl ether (first with 285 mL, then with 145 mL) at 20-25 °C. Thereafter, the combined organic layers were washed with 10percent aqueous sodium chloride (250mL) solution and concentrated under reduced pressure. The obtained residue was crystallized in isopropyl acetate (150 mL) to result in formula 9a as solid (90 g).
Reference:
[1] Patent: WO2011/39107, 2011, A1, . Location in patent: Page/Page column 19-20
[2] Patent: US2011/237789, 2011, A1, . Location in patent: Page/Page column 11
[3] Patent: US2011/237526, 2011, A1, . Location in patent: Page/Page column 7
[4] Organic Letters, 2014, vol. 16, # 16, p. 4090 - 4093
[5] Patent: CN105399735, 2016, A, . Location in patent: Paragraph 0040; 0041; 0042
[6] Patent: CN108218928, 2018, A, . Location in patent: Paragraph 0199; 0200; 0201
[7] Patent: US2006/258749, 2006, A1, . Location in patent: Page/Page column 22
[8] Patent: WO2015/101916, 2015, A1, . Location in patent: Page/Page column 19
9
[ 915095-86-2 ]
[ 141-52-6 ]
[ 1103738-26-6 ]
Yield
Reaction Conditions
Operation in experiment
86.5%
at 20℃; for 1.5 h; Cooling with ice
5-iodo-2-chlorophenyl) (4-fluorophenyl) methanone (18.03 g, 50 mmo 1) was added DMF (90 mL), Ice bath slowly add sodium ethoxide (3.748,1. Eq), After adding the reaction at room temperature for 1.5 h, tlc (after the detection reaction is completed, Ice bath slowly add water (180mL), Stirring crystallization lh, Filter, The filter cake was washed with water (200 mL) Beat with ethanol (50 mL) Filtered and dried to give 16.72 g of the pale yellow solid of compound lib, Yield: 86.5percent Purity: 99.53percent.
Reference:
[1] Patent: CN106928040, 2017, A, . Location in patent: Paragraph 0042-0047
10
[ 915095-86-2 ]
[ 917-58-8 ]
[ 1103738-26-6 ]
Yield
Reaction Conditions
Operation in experiment
87.2%
at 40℃; for 1 h; Cooling with ice
(5-iodo-2-chlorophenyl) (4-fluorophenyl) methanone (18.03 g, 50 mmo 1) Was added DMF (90 mL), Ice bath slowly add potassium ethoxide (5.478,1.3 called), After adding 40 ° (after the next stirring reaction 111,11 (after the reaction is completed, add water (180mL) slowly under ice bath, Stirring crystallization lh, Filter, The filter cake was washed with water (200 mL) Beat with ethanol (50 mL) Filter, Dried to give 16.86 g of the pale yellow solid of compound lib, Yield: 87.2percent Purity: 99.27percent.
Reference:
[1] Patent: CN106928040, 2017, A, . Location in patent: Paragraph 0044-0045
11
[ 915095-86-2 ]
[ 2388-07-0 ]
[ 1103738-26-6 ]
Yield
Reaction Conditions
Operation in experiment
82.7%
at 0℃; for 3 h;
(5-iodo-2-chlorophenyl) (4-fluorophenyl) methanone (18.03 g, 50 mmol) was added DMS0 (80 mL) Ice bath slowly add lithium ethoxide (3.128,1.2 called), After adding 0 ° (below the stirring reaction 311, hole (after the reaction is completed, add water (240mL) slowly under ice bath, Stirring crystallization lh, Filter, The filter cake was washed with water (200 mL) Beat with ethanol (50 mL) Filter, Dried to give 15.99 g of a pale yellow solid of compound lib, yield: 82.7percent, purity: 99.44percent.
Reference:
[1] Patent: CN106928040, 2017, A, . Location in patent: Paragraph 0046-0047
With aluminum (III) chloride; In dichloromethane; at -5℃; for 6h;
Equipped with a thermometer, constant pressure dropping funnel, drying tube 1000mL flask was added three DCM200mL, VIII (70.62g, 250mmol) ,was added 1 ml of DMF and, at room temperature was slowly added dropwise oxalyl chloride (23.4mL g, 275mmol), was added dropwise after the completion of 20 incubated 2h.After adding concentrated to remove the DCM, oxalyl chloride 200mL DCM dissolved chloride.Equipped with a thermometer, constant pressure dropping funnel, 1000mL three 26mL flask with a drying tube was added fluorobenzene, DCM200mL the system temperature was dropped to -5 deg.] C, graded added to the system aluminum trichloride (36.67g, 275mmol) , dropped acid chloride prepared raw reaction incubated 6h completed.System 40mL conc HCl + 400mL poured into ice water to quench the reaction, the DCM. The combined organic phases were washed until neutral sodium bicarbonate, once with saturated brine separation was washed with water, dried, and concentrated to give VII84.23g, yield 93.4percent.
To a solution of 48.94 g 2-chloro-5-iodo-benzoic acid in 180 mL of fluorobenzene and 0.3 mL of N,N-dimethylformamide is added 16.2 mL of oxalyl chloride at 0 to 10° C. The solution is stirred at about 20° C. for 2 hours. The excess amount of oxalyl chloride is evaporated. The residue is diluted in 166 mL of fluorobenzene and 25.93 g of aluminum chloride is added at 0° C. in five portions. The solution is stirred at 75° C. for 1.5 hours and quenched with 300 mL of water at 0 to 25° C. The product is extracted in 300 mL of isopropylacetate and washed with two times of 200 mL brine (3 weight-percent). The residue water and solvent is removed upon evaporation. Yield: 60.56 g (95percent of theory) Mass spectrum (ESI+): m/z=361/363 (Cl)[M+H]+
105 g
With aluminum (III) chloride; at 25℃;Inert atmosphere;
Oxalyl chloride (72 g, 0.56 moles) was added to a slurry of formula 5a (100 g, 0.35 moles) and a catalytic amount of N, N-dimethylformamide (5mL) in fluorobenzene (250 mL) over about 60 minutes at 15-25 °C under nitrogen atmosphere. The mixture was stirred at 21-25 °C. After completion of the reaction, the reaction mass was concentrated to remove excess oxalyl chloride. The obtained residue was diluted with fluorobenzene (125 mL) and cooled to 15-25 °C. Aluminum chloride (53.5 g, 0.40 moles) was added to this reaction mixture portion- wise while keeping the reaction mass temperature below 25 °C and maintaining it at same temperature to complete the reaction. After completion of reaction, the reaction mass was quenched into precooled dilute hydrochloric acid (5percent, 1700 mL, 0-5 °C) at 5-25 °C. After stirring for 60 minutes at 20-25 °C, the product was extracted with methylene chloride twice (first with 500 mL, then with 250 mL). The combined organic layers were washed with 10percent aqueous sodium hydroxide solution (250mL), water (500mL), and 10percent aqueous sodium chloride solution (250mL), sequentially. Thereafter, the organic layer was concentrated and the obtained residue was diluted with n-heptane (250mL) and stirred to precipitate formula 7a as a white solid (105 g).
42.1 g
With aluminum (III) chloride; In dichloromethane; at 20℃; for 3h;Cooling with ice;
5-iodo-2-chlorobenzoic acid (33.895 g, 120 mmol) was added DCM (100 mL) DMF (0.175 g, 0.02 eq), Stir, Slowly add oxalyl chloride (12.3 ^, 1.2 called), After the drop is completed at room temperature reaction 21 , The solvent was removed by concentration under reduced pressure, Add DCM (lOOmL), Fluorobenzene (17.298 g, 1.5 eq) was added, Alcia was added to A1C13 (17.6 g, 1 leq) After adding the reaction at room temperature for 3h, After TLC detection reaction was completed, Ice bath slowly add water (100mL), After stirring, The organic phase was washed with 10percent NaCl (50 mL) The organic phase was concentrated under reduced pressure to give a pale yellow oil, Plus isopropyl alcohol (lOOmL), Add water (lOOmL), Ice bath stirring crystallization 2h, Filter, The filter cake was washed with isopropanol / water (1: 1) mixed solution (20 mL) Dried to give 42.1 g of an off-white solid of compound Vb, Yield: 97.3percent Purity: 99.17percent
With potassium tert-butylate; In tetrahydrofuran; at 16 - 25℃; for 3h;
Example 2: Synthesis of the ketone VII.1To a solution of the fluoride VIII.1 (208kg), tetrahydrofuran (407kg) and (S)-3- hydroxytetrahydrofuran (56kg) is added potassium-terf-butanolate solution (20percent) in tetrahydrofuran (388kg) within 3 hrs at 16 to 25°C temperature. After completion of the addition, the mixture is stirred for 60min at 20°C temperature. Then the conversion is determined via HPLC analysis. Water (355kg) is added within 20 min at a temperature of 21 °C (aqueous quench). The reaction mixture is stirred for 30 min (temperature: 20°C). The stirrer is switched off and the mixture is left stand for 60 min (temperature: 20°C). The phases are separated and solvent is distilled off from the organic phase at 19 to 45°C temperature under reduced pressure. 2- Propanol (703kg) is added to the residue at 40 to 46°C temperature and solvent is distilled off at 41 to 50°C temperature under reduced pressure. 2-Propanol (162kg) is added to the residue at 47°C temperature and solvent is distilled off at 40 to 47°C temperature under reduced pressure. Then the mixture is cooled to 0°C within 1 hr 55 min. The product is collected on a centrifuge, washed with a mixture of 2- propanol (158kg) and subsequently with terf.-butylmethylether (88kg) and dried at 19 to 43°C under reduced pressure. 227kg (91 ,8percent) of product are obtained as colourless solid. The identity of the product is determined via infrared spectrometry.
91.8%
To a solution of the fluoride VIII.1 (208 kg), tetrahydrofuran (407 kg) and (S)-3-hydroxytetrahydrofuran (56 kg) is added potassium-tert-butanolate solution (20percent) in tetrahydrofuran (388 kg) within 3 hrs at 16 to 25° C. temperature. After completion of the addition, the mixture is stirred for 60 min at 20° C. temperature. Then the conversion is determined via HPLC analysis. Water (355 kg) is added within 20 min at a temperature of 21° C. (aqueous quench). The reaction mixture is stirred for 30 min (temperature: 20° C.). The stirrer is switched off and the mixture is left stand for 60 min (temperature: 20° C.). The phases are separated and solvent is distilled off from the organic phase at 19 to 45° C. temperature under reduced pressure. 2-Propanol (703 kg) is added to the residue at 40 to 46° C. temperature and solvent is distilled off at 41 to 50° C. temperature under reduced pressure. 2-Propanol (162 kg) is added to the residue at 47° C. temperature and solvent is distilled off at 40 to 47° C. temperature under reduced pressure. Then the mixture is cooled to 0° C. within 1 hr 55 min. The product is collected on a centrifuge, washed with a mixture of 2-propanol (158 kg) and subsequently with tert.-butylmethylether (88 kg) and dried at 19 to 43° C. under reduced pressure. 227 kg (91.8percent) of product are obtained as colourless solid. The identity of the product is determined via infrared spectrometry.
91.8%
With potassium tert-butylate; In tetrahydrofuran; at 16 - 25℃; for 4h;Industry scale;
To a solution of the fluoride VIII.1 (208 kg), tetrahydrofuran (407 kg) and (S)-3-hydroxytetrahydrofuran (56 kg) is added potassium-tert-butanolate solution (20percent) in tetrahydrofuran (388 kg) within 3 hrs at 16 to 25° C. temperature. After completion of the addition, the mixture is stirred for 60 min at 20° C. temperature. Then the conversion is determined via HPLC analysis. Water (355 kg) is added within 20 min at a temperature of 21° C. (aqueous quench). The reaction mixture is stirred for 30 min (temperature: 20° C.). The stirrer is switched off and the mixture is left stand for 60 min (temperature: 20° C.). The phases are separated and solvent is distilled off from the organic phase at 19 to 45° C. temperature under reduced pressure. 2-Propanol (703 kg) is added to the residue at 40 to 46° C. temperature and solvent is distilled off at 41 to 50° C. temperature under reduced pressure. 2-Propanol (162 kg) is added to the residue at 47° C. temperature and solvent is distilled off at 40 to 47° C. temperature under reduced pressure. Then the mixture is cooled to 0° C. within 1 hr 55 min. The product is collected on a centrifuge, washed with a mixture of 2-propanol (158 kg) and subsequently with tert.-butylmethylether (88 kg) and dried at 19 to 43° C. under reduced pressure. 227 kg (91.8percent) of product are obtained as colourless solid. The identity of the product is determined via infrared spectrometry.
75%
With potassium tert-butylate; In tetrahydrofuran; at 20℃;
Equipped with a thermometer, pressure-equalizing dropping funnel was added 500mL three bottles VII (84.2g, 234mmol), S -3-hydroxytetrahydrofuran (20.6g, 234mmol), was added THF100mL clear solution after stirring, potassium tert-butoxide (34.1 g, 234mmol) was dissolved 150mLTHF added to the system, stirred at room temperature overnight, TLC monitored the reaction was complete, the reaction was quenched with 230 mL of water, spin-out section THF, EA extracted three times, the combined organic phase was washed with water and saturated brine, dried and concentrated to give 120.5 g crude product, after using 170mL of isopropanol + 20mL water crystallized product 75g, purity 98.2percent, yield 75percent.
64.3%
With potassium tert-butylate; In tetrahydrofuran; at 16 - 25℃; for 1h;
5-Iodo-2-chloro-4'-fluorobenzophenone (V-2) (18.5 g, 0.051 mol) was dissolved in 50 ml of tetrahydrofuran solutionThen, (5)-3_light-based tetrahydrofuran (4.9 g, 0.056 mol) was added and stirred, and finally a solution of potassium t-butoxide (6.9 g, 0.061 mol) dissolved in 30 ml of tetrahydrofuran was added dropwise, and the temperature was controlled at 16-25 ° C. After the addition was completed, the reaction was stirred at 20 ° C for 1 hour, and the reaction was almost complete by TLC, and the purified water was slowly added and stirred for 30 min.The solution was separated and the organic phase was dried. After column chromatography, 14. lg (64.3percent) of solid was obtained.
With potassium tert-butylate; In tetrahydrofuran; at 0 - 10℃; for 0.5h;
To a solution of 60.56 g <strong>[915095-86-2](2-chloro-5-iodo-phenyl)-(4-fluoro-phenyl)-methanone</strong> in 170 mL of tetrahydrofuran and 16.46 g of (S)-3-hydroxytetrahydrofuran is added 26 g of potassium tert-butoxide in 250 mL of tetrahydrofuran at 0 to 5° C. The solution is stirred at 10° C. for 0.5 hour. The reaction is quenched with 170 mL of water and 170 mL of methyl tert-butyl ether at 0 to 25° C. The product is washed with 170 mL of brine (3 weight-percent). The solvent is removed upon evaporation and crystallized in 220 mL of iso-propylacetate. Yield: 65.1 g (90percent of theory) Mass spectrum (ESI+): m/z=428/430 (Cl) [M+H]+
90 g
With potassium tert-butylate; In tetrahydrofuran; at 2 - 10℃;
A mixture of formula 7a (100 g, 0.277 mole) and (S)-tetrahydrofuran-3-ol (27.4 g, 0.31 mole) in tetrahydrofuran (260 mL) was prepared. Potassium tert-butoxide (46.5 g, 0.414 mole) in tetrahydrofuran (405 mL) was added to this mixture solution slowly over a period of 90 minutes at 2-6 °C, maintaining the reaction mass at 7-10 °C to complete the reaction. Thereafter, precooled water (285 mL) was added to quench the reaction at 5-18 °C and the product was extracted twice with methyl tert- butyl ether (first with 285 mL, then with 145 mL) at 20-25 °C. Thereafter, the combined organic layers were washed with 10percent aqueous sodium chloride (250mL) solution and concentrated under reduced pressure. The obtained residue was crystallized in isopropyl acetate (150 mL) to result in formula 9a as solid (90 g).
With oxalyl dichloride;N,N-dimethyl-formamide; at 25 - 30℃; for 5h;
Example 1 : Synthesis of the fluoride VIII.1Oxalylchloride (176kg; 1386mol; 1 ,14eq) is added to a mixture of <strong>[19094-56-5]2-chloro-5-iodo benzoic acid</strong> (343kg; 1214mol) (compound IX.1 ), fluorobenzene (858kg) and N,N- dimethylformamide (2kg) within 3 hours at a temperature in the range from about 25 to 30C (gas formation). After completion of the addition, the reaction mixture is stirred for additional 2 hours at a temperature of about 25 to 30C. The solvent (291 kg) is distilled off at a temperature between 40 and 45C (p=200mbar). Then the reaction solution (91 1 kg) is added to aluminiumchloride AICI3 (181 kg) andfluorobenzene (192kg) at a temperature between about 25 and 30C within 2 hours. The reaction solution is stirred at the same temperature for about an additional hour. Then the reaction mixture is added to an amount of 570 kg of water within about 2 hours at a temperature between about 20 and 30C and stirred for an additional hour. After phase separation the organic phase (1200kg) is separated into two halves (600kg each). From the first half of the organic phase solvent (172kg) is distilled off at a temperature of about 40 to 50C (p=200mbar). Then 2-propanole (640kg) is added. The solution is heated to about 50C and then filtered through a charcoal cartouche (clear filtration). The cartouche may be exchanged during filtration and washed with a fluorobenzene/2-propanole mixture (1 :4; 40kg) after filtration. Solvent (721 kg) is distilled off at a temperature of about 40 to 50C and p=200mbar. Then 2-propanole (240kg) is added at a temperature in the range between about 40 to 50C. If the content of fluorobenzene is greater than 1 % as determined via GC, another 140kg of solvent are distilled off and 2-propanole (140kg) is added. Then the solution is cooled from about 50C to 40C within one hour and seeding crystals (50g) are added. The solution is further cooled from about 40C to 20C within 2 hours. Water (450kg) is added at about 20C within 1 hour and the suspension is stirred at about 20C for an additional hour before the suspension is filtered. The filter cake is washed with 2-propanole/water (1 :1 ; 800kg). The product is dried until a water level of <0.06%w/w is obtained. The second half of the organic phase is processed identically. A total of 410kg (94%yield) of product which has a white to off-white crystalline appearance, is obtained. The identity of the product is determined via infrared spectrometry.
94%
Oxalylchloride (176 kg; 1386 mol; 1.14 eq) is added to a mixture of <strong>[19094-56-5]2-chloro-5-iodo benzoic acid</strong> (343 kg; 1214 mol) (compound IX.1), fluorobenzene (858 kg) and N,N-dimethylformamide (2 kg) within 3 hours at a temperature in the range from about 25 to 30 C. (gas formation). After completion of the addition, the reaction mixture is stirred for additional 2 hours at a temperature of about 25 to 30 C. The solvent (291 kg) is distilled off at a temperature between 40 and 45 C. (p=200 mbar). Then the reaction solution (911 kg) is added to aluminiumchloride AlCl3 (181 kg) and fluorobenzene (192 kg) at a temperature between about 25 and 30 C. within 2 hours. The reaction solution is stirred at the same temperature for about an additional hour. Then the reaction mixture is added to an amount of 570 kg of water within about 2 hours at a temperature between about 20 and 30 C. and stirred for an additional hour. After phase separation the organic phase (1200 kg) is separated into two halves (600 kg each). From the first half of the organic phase solvent (172 kg) is distilled off at a temperature of about 40 to 50 C. (p=200 mbar). Then 2-propanole (640 kg) is added. The solution is heated to about 50 C. and then filtered through a charcoal cartouche (clear filtration). The cartouche may be exchanged during filtration and washed with a fluorobenzene/2-propanole mixture (1:4; 40 kg) after filtration. Solvent (721 kg) is distilled off at a temperature of about 40 to 50 C. and p=200 mbar. Then 2-propanole (240 kg) is added at a temperature in the range between about 40 to 50 C. If the content of fluorobenzene is greater than 1% as determined via GC, another 140 kg of solvent are distilled off and 2-propanole (140 kg) is added. Then the solution is cooled from about 50 C. to 40 C. within one hour and seeding crystals (50 g) are added. The solution is further cooled from about 40 C. to 20 C. within 2 hours. Water (450 kg) is added at about 20 C. within 1 hour and the suspension is stirred at about 20 C. for an additional hour before the suspension is filtered. The filter cake is washed with 2-propanole/water (1:1; 800 kg). The product is dried until a water level of <0.06% w/w is obtained. The second half of the organic phase is processed identically. A total of 410 kg (94% yield) of product which has a white to off-white crystalline appearance, is obtained. The identity of the product is determined via infrared spectrometry.
94%
With aluminum (III) chloride; oxalyl dichloride; In N,N-dimethyl-formamide; at 25 - 45℃;Industry scale;
Oxalylchloride (176 kg; 1386 mol; 1.14 eq) is added to a mixture of <strong>[19094-56-5]2-chloro-5-iodo benzoic acid</strong> (343 kg; 1214 mol) (compound IX.1), fluorobenzene (858 kg) and N,N-dimethylformamide (2 kg) within 3 hours at a temperature in the range from about 25 to 30 C. (gas formation). After completion of the addition, the reaction mixture is stirred for additional 2 hours at a temperature of about 25 to 30 C. The solvent (291 kg) is distilled off at a temperature between 40 and 45 C. (p=200 mbar). Then the reaction solution (911 kg) is added to aluminiumchloride AlCl3 (181 kg) and fluorobenzene (192 kg) at a temperature between about 25 and 30 C. within 2 hours. The reaction solution is stirred at the same temperature for about an additional hour. Then the reaction mixture is added to an amount of 570 kg of water within about 2 hours at a temperature between about 20 and 30 C. and stirred for an additional hour. After phase separation the organic phase (1200 kg) is separated into two halves (600 kg each). From the first half of the organic phase solvent (172 kg) is distilled off at a temperature of about 40 to 50 C. (p=200 mbar). Then 2-propanole (640 kg) is added. The solution is heated to about 50 C. and then filtered through a charcoal cartouche (clear filtration). The cartouche may be exchanged during filtration and washed with a fluorobenzene/2-propanole mixture (1:4; 40 kg) after filtration. Solvent (721 kg) is distilled off at a temperature of about 40 to 50 C. and p=200 mbar. Then 2-propanole (240 kg) is added at a temperature in the range between about 40 to 50 C. If the content of fluorobenzene is greater than 1% as determined via GC, another 140 kg of solvent are distilled off and 2-propanole (140 kg) is added. Then the solution is cooled from about 50 C. to 40 C. within one hour and seeding crystals (50 g) are added. The solution is further cooled from about 40 C. to 20 C. within 2 hours. Water (450 kg) is added at about 20 C. within 1 hour and the suspension is stirred at about 20 C. for an additional hour before the suspension is filtered. The filter cake is washed with 2-propanole/water (1:1; 800 kg). The product is dried until a water level of <0.06% w/w is obtained. The second half of the organic phase is processed identically. A total of 410 kg (94% yield) of product which has a white to off-white crystalline appearance, is obtained. The identity of the product is determined via infrared spectrometry.
92%
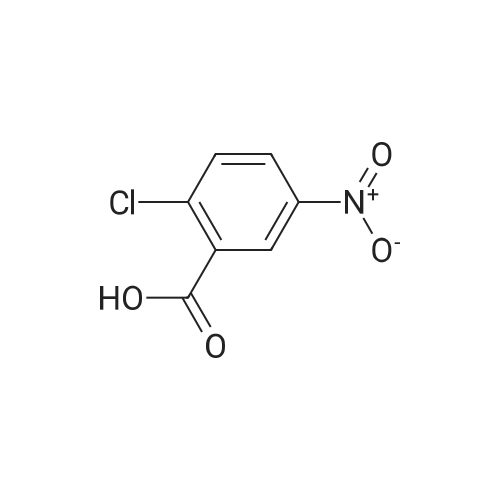
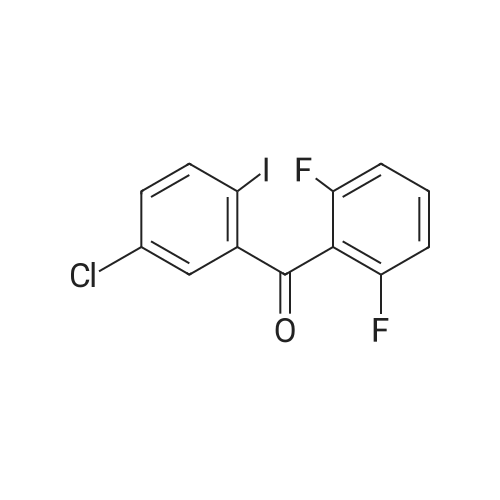
General procedure: To a stirred suspension of <strong>[19094-56-5]2-chloro-5-iodobenzoic acid</strong>(5.65 g, 0.02 mol) in CH2Cl2(20 mL) was added oxalylchloride(1.80 mL, 0.021 mol) and DMF(0.02 mL). Theresulting mixture was stirred for 4 h at room temperature.The mixture was concentrated, and the residual colorlesssolid was dissolved in CH2Cl2(40 mL). To this solutionwere added corresponding substituted benzene(0.02 mol)and then AlCl3(2.75 g, 0.021 mol) portionwise so that thetemperature did not exceed 0 C. After being stirred at 10-20 C for 3 h, the mixture was poured into ice water andextracted with CH2Cl2 three times. The combined organiclayers were washed with 1M HCl, water, and brine, thendried over MgSO4 and concentrated. The residual solid wascrystallized from ethanol to give as white solid.According to this procedure the following compoundswere prepared. (2-chloro-5-iodophenyl)(4-fluorophenyl)methanone (1a)White solid, yield 92%, m.p. 69-70 C. 1H NMR (400MHz,DMSO-d6): delta 7.38-7.40(m, 2H), 7.41(d, J=8.0 Hz, 1H), 7.82(dd, J=2.4 Hz, 8.0 Hz, 2H), 7.90(d, J=2.4 Hz, 1H), 7.92-7.95(m, 1H). 13C NMR (100MHz, DMSO-d6): delta 116.19,129.47, 131.65, 132.1, 132.65, 136.78, 139.69, 140.20,164.54, 166.56, 191.36. HRMS (ESI) m/z (pos):382.91890C13H7ClFIONa [M+Na]+(calcd. 382.91118).
78.6%
5-Iodo-2-chlorobenzoic acid (20.0 g, 0.071 mol) was added to a solution of 2.0 M oxalyl chloride in dichloromethane (39 ml, 0.078 mol), stirred for suspension, and then 8 drops of DMF solution was added dropwise. After the reaction was carried out for 3 hours, the solution was clarified and the reaction was almost complete. The solvent was spin-dried on a rotary evaporator, then 15 ml of methylene chloride was added, and the solvent was dried. After spin-drying, add 30 ml of dichloromethane, stir, cool to 0-5 C, add fluorobenzene (7. lg, 0.074 mol), add anhydrous aluminum trichloride (9.9 g, 0.074 mol) in batches, control The temperature is not more than 5 C, after the completion of the addition, stirring is continued at 4 C for 1 h, the reaction is almost complete by TLC, the reaction is quenched on ice-water mixture, the organic phase is separated, and the aqueous phase is extracted with dichloromethane. The phase was washed twice with 1 mol/L hydrochloric acid, washed once with water, and washed twice with 1 mol/L NaOH solution.The mixture was washed twice with saturated sodium chloride and dried over anhydrous sodium sulfate. Drain filtration, spin dry solvent to obtain oilAfter column chromatography, 20. 1g (78.6%) of a white solid was obtained.
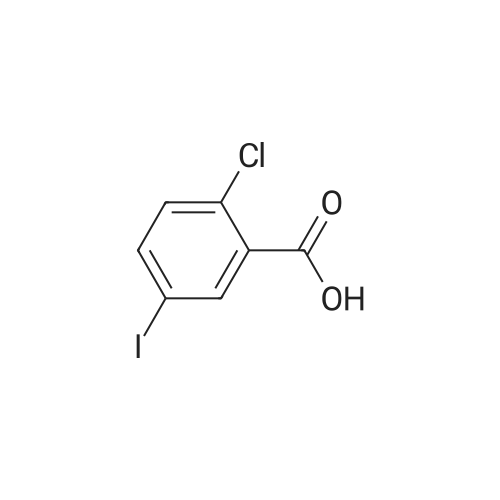
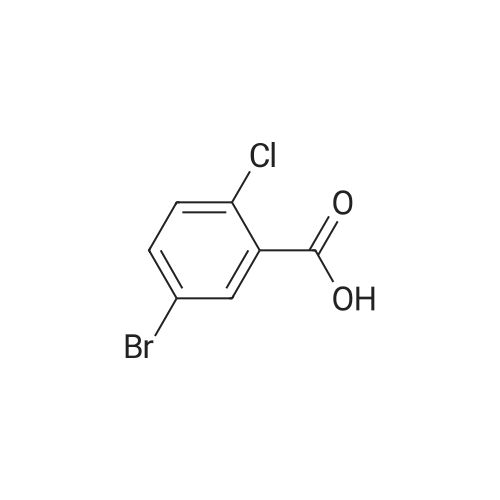
310 g
(282 g, 1 mol) of <strong>[19094-56-5]5-iodo-2-chlorobenzoic acid</strong>, 1.0 L of dichloromethane, (142.8 g, 1.2 mol) of thionyl chloride and a catalytic amount of DMF were added to the reaction flask.The temperature was raised to 39 C - 42 C for 3 hrs - 5 hrs. Cool down to 5-10 C,(160 g, 1.2 mol) of aluminum trichloride and (125 g, 1.3 mol) of fluorobenzene were added.The reaction was heated to reflux overnight. After the reaction, cool down to 0 C,The reaction was quenched by dropwise addition of 100 ml of 6N hydrochloric acid.Add 400ml of water to the organic phase,The organic phase is treated with 400 ml of saturated sodium bicarbonate solution.Wash with 400 ml of saturated sodium chloride solution and dry over anhydrous sodium sulfate.Concentrate to dryness, add 100ml of absolute ethanol to heat and dissolve, and cool down to 5-10 C.Insulation crystallization for 2h, suction filtration,Dry white solid(5-iodo-2-chlorophenyl)(4-fluorophenyl)methanone 310 g.
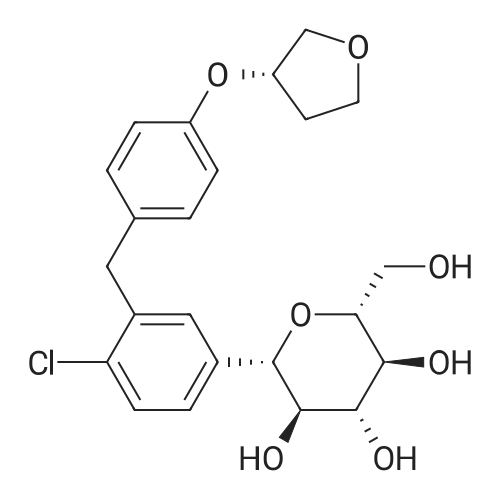
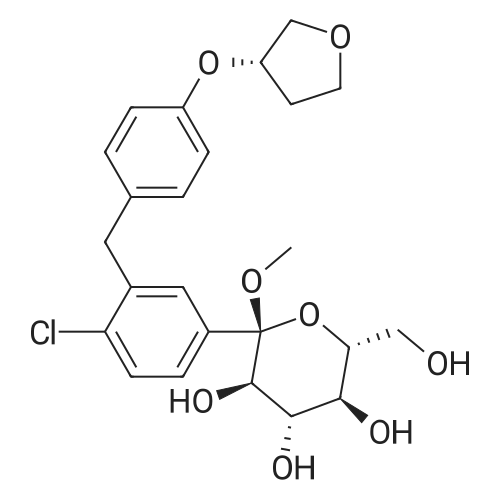
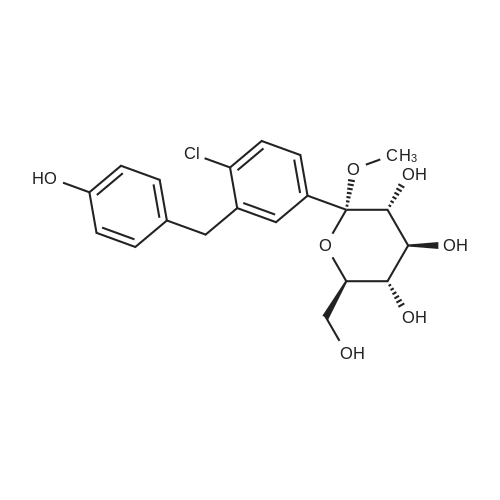
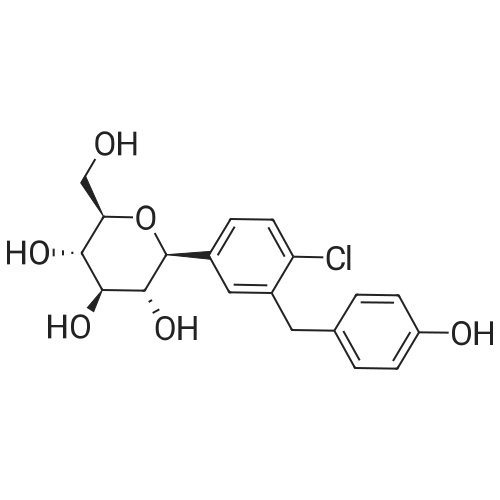
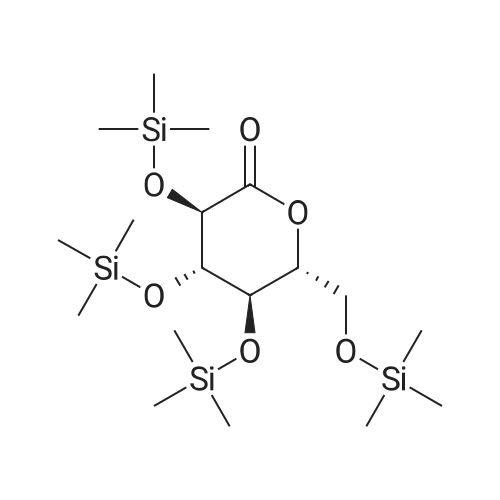
(2S,3R,4S,5R,6R)-6-(acetoxymethyl)-2-(4-chloro-3-(4-(((S)-tetrahydrofuran-3-yl)oxy)benzyl)phenyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate[ No CAS ]
(2S,3R,4S,5R,6R)-3,4,5-tris(benzyloxy)-6-((benzyloxy)methyl)-2-(4-chloro-3-(4-(((S)-tetrahydrofuran-3-yl)oxy)benzyl)phenyl)-2-methoxytetrahydro-2H-pyran[ No CAS ]
(2S,3R,4S,5R,6R)-3,4,5-tris(allyloxy)-6-((allyloxy)methyl)-2-(4-chloro-3-(4-(((S)-tetrahydrofuran-3-yl)oxy)benzyl)phenyl)-2-methoxytetrahydro-2H-pyran[ No CAS ]
(2-chloro-5-aminophenyl)(4-fluorophenyl)methanone[ No CAS ]
[ 915095-86-2 ]
Yield
Reaction Conditions
Operation in experiment
91.6%
1.8 kg of (2-chloro-5-aminophenyl) (4-fluorophenyl) methanone was added to 120 kg of a 20percent aqueous solution of sulfuric acid,Hold the temperature at 0 ~ 10 ° c under stirring, dropping sodium nitrite aqueous solution (0.51kg sodium nitrite dissolved in 2kg7j0, stirring to the reverseTLC (PE / EA: 1/3) After the reaction is complete, add 0.009 kg of urea, stir to cool to 0 ° C, fastAdd potassium iodide solution (1.3kgKI dissolved in 5kg7j0, rose to room temperature, stirring until no bubbles continue to stir for 30min. Filtration, 2kgWashed with water to give a brown solid; the solid was dissolved in 10 kg of ethyl acetate, washed with 7 kg of IN hydrochloric acid, 7 kg of 10percent sodium bisulfate,8kg saturated brine, dried over magnesium sulfate, dried at 50 ° C under reduced pressure to give the crude product. After adding 4 kg of isopropyl alcohol,(2-chloro-5-iodophenyl) (4-fluorophenyl) methanone as a white solid, filtered at 50 ° C under reduced pressure,2.38 kg, purity 99.6percent, yield 91.6percent.
In N,N-dimethyl-formamide; at 20℃; for 1.5h;Cooling with ice;
5-iodo-2-chlorophenyl) (4-fluorophenyl) methanone (18.03 g, 50 mmo 1) was added DMF (90 mL), Ice bath slowly add sodium ethoxide (3.748,1. Eq), After adding the reaction at room temperature for 1.5 h, tlc (after the detection reaction is completed, Ice bath slowly add water (180mL), Stirring crystallization lh, Filter, The filter cake was washed with water (200 mL) Beat with ethanol (50 mL) Filtered and dried to give 16.72 g of the pale yellow solid of compound lib, Yield: 86.5percent Purity: 99.53percent.
In N,N-dimethyl-formamide; at 40℃; for 1h;Cooling with ice;
(5-iodo-2-chlorophenyl) (4-fluorophenyl) methanone (18.03 g, 50 mmo 1) Was added DMF (90 mL), Ice bath slowly add potassium ethoxide (5.478,1.3 called), After adding 40 ° (after the next stirring reaction 111,11 (after the reaction is completed, add water (180mL) slowly under ice bath, Stirring crystallization lh, Filter, The filter cake was washed with water (200 mL) Beat with ethanol (50 mL) Filter, Dried to give 16.86 g of the pale yellow solid of compound lib, Yield: 87.2percent Purity: 99.27percent.
(5-iodo-2-chlorophenyl) (4-fluorophenyl) methanone (18.03 g, 50 mmol) was added DMS0 (80 mL) Ice bath slowly add lithium ethoxide (3.128,1.2 called), After adding 0 ° (below the stirring reaction 311, hole (after the reaction is completed, add water (240mL) slowly under ice bath, Stirring crystallization lh, Filter, The filter cake was washed with water (200 mL) Beat with ethanol (50 mL) Filter, Dried to give 15.99 g of a pale yellow solid of compound lib, yield: 82.7percent, purity: 99.44percent.
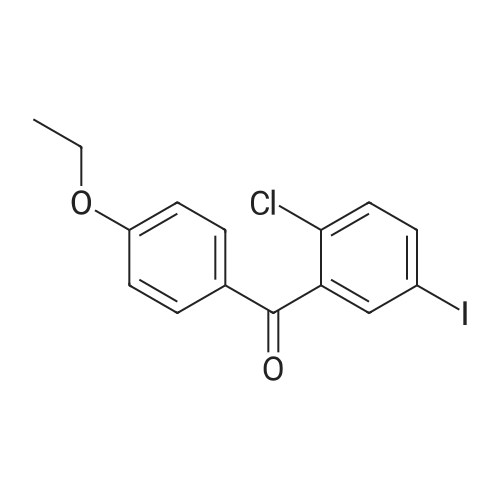
(180 g, 0.5 mol) <strong>[915095-86-2](5-iodo-2-chlorophenyl)(4-fluorophenyl)methanone</strong>, 900 ml of absolute ethanol and (40 g, 1 mol) of NaOH were added to the reaction flask.Raise the temperature to 50 ° C -65 ° C reaction for 5h-8h, after the reaction is over,Distilling 600 ml-700 ml of ethanol under reduced pressure and cooling to 20 ° C to 30 ° C.The reaction solution was poured into 300 g-500 g of ice water and filtered.The filter cake is washed with 300ml of water and dried.The product compound I (5-iodo-2-chlorophenyl) (4-ethoxyphenyl) ketone 182 g,The yield is 94.3percent,
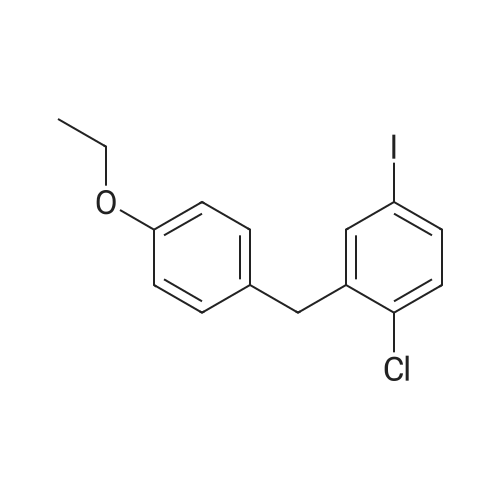
1-chloro-2-(4-fluorobenzyl)-4-iodobenzene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
With triethylsilane; boron trifluoride diethyl etherate; In dichloromethane; acetonitrile; at 0 - 20℃;
General procedure: To a stirred solution of 1 (0.01 mol) in 30 mL of CH2Cl2and 30 mL of CH3CN at 0 °C were added Et3SiH(4.79 mL,0.03 mol), then BF3·Et2O(2.53 mL, 0.02 mol), and theresulting mixture was warmed to room temperature andstirred for 3?8 h. The mixture was poured into saturatedaqueous NaHCO3 and extracted with CH2Cl2 twice. Thecombined organic layers were washed with brine, dried overMgSO4, and concentrated. The residual solid was crystallizedfrom ethanol to give as white solid.According to this procedure the following compoundswere prepared. 1-chloro-2-(4-fluorobenzyl)-4-iodobenzene (2a)White solid, yield 83percent, m.p. 52?54 °C. 1H NMR (400MHz, DMSO-d6): delta 4.02(s, 2H), 7.10?7.13(m, 2H), 7.22?7.25(m, 3H), 7.60(dd, J = 0.8, 6.8 Hz, 1H), 7.72(d, J = 0.8Hz, 1H). 13C NMR (100 MHz, DMSO-d6): delta 36.94, 92.85,115.21(d, J = 68 Hz), 130.30(d, J = 24 Hz), 131.32,133.16, 134.87, 136.82, 139.44, 140.77, 159.80, 161.73.
With acetylhydroxamic acid; potassium carbonate; In dimethyl sulfoxide; at 80℃; for 6h;
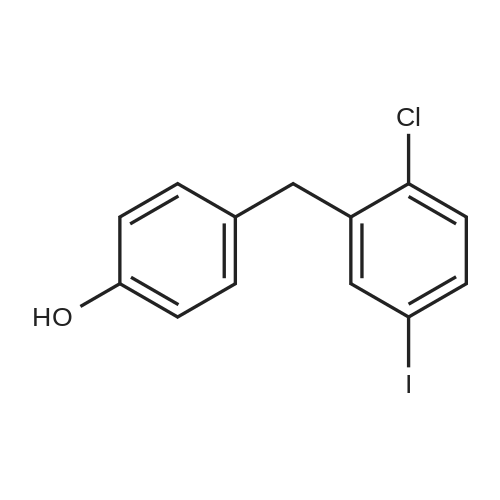
<strong>[915095-86-2](2-chloro-5-iodophenyl)(4-fluorophenyl)methanone</strong> (9.01 g, 25.0 mmol), acetohydroxamic acid (5.63 g, 75.0 mmol, 3.0 eq.) and anhydrous potassium carbonate ( 17.28 g, 125.0 mmol, 5.0 eq.) was dissolved in 50 mL of DMSO after removal of water. After heating to 80 C for 6 h, the TLC monitoring material has been converted. Stop heating, naturally cool to room temperature, and adjust the pH to 3.0 with 1M diluted hydrochloric acid.Extract with ethyl acetate (100 mL × 3), and combine the organic phases. After washing with saturated brine (50 mL×3), dry over anhydrous sodium sulfate, filter, the solvent is removed by rotary evaporation under reduced pressure. A pale yellow oil is obtained which is crystallized from n-hexane. A large amount of white solid precipitated, suction filtration, The filter cake was dried under vacuum to give a white solid product. Namely, (5-chloro-5-iodophenyl)(4-hydroxyphenyl)methanone 8.55 g (yield 95.4%).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping