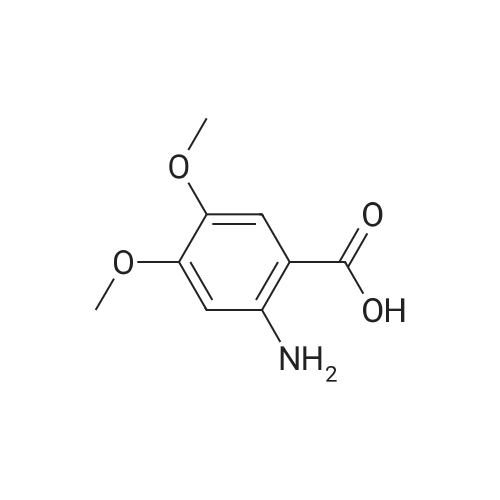
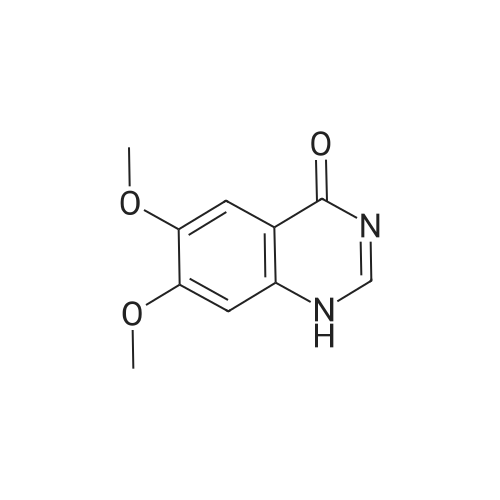
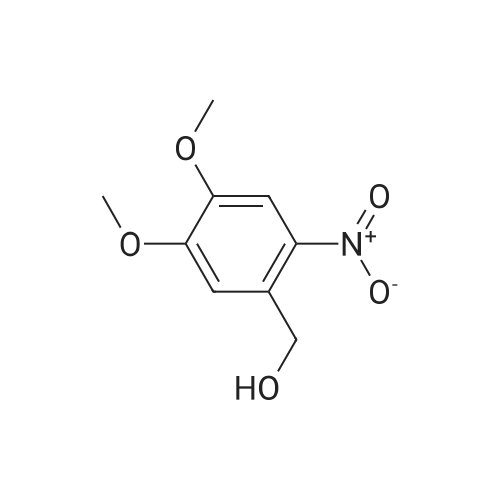
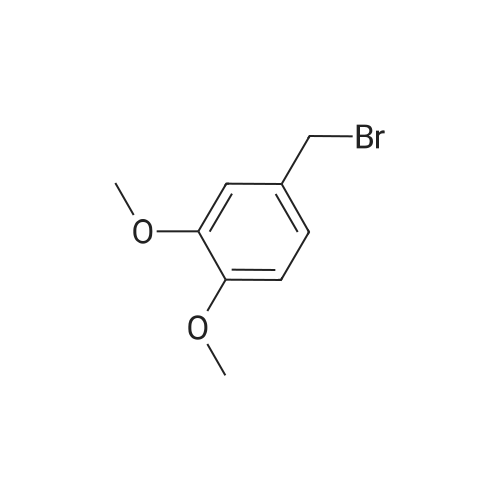
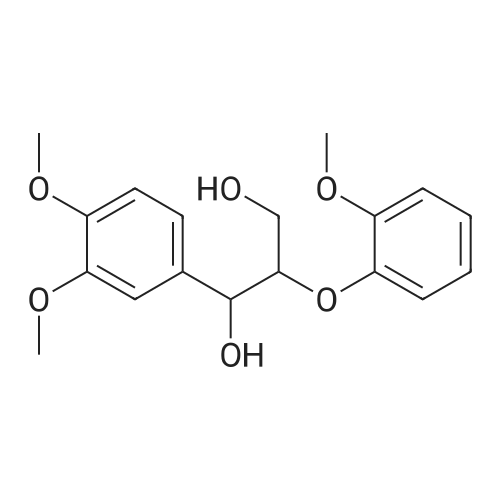
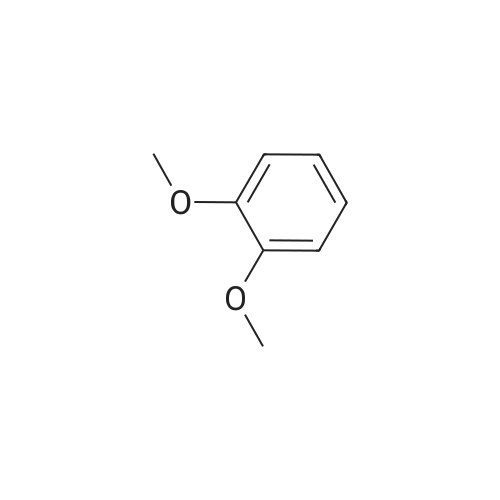
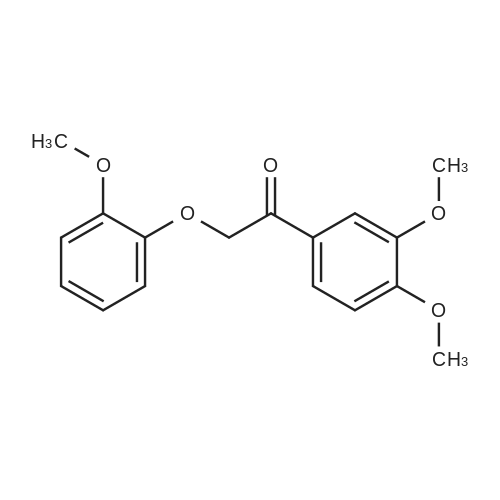
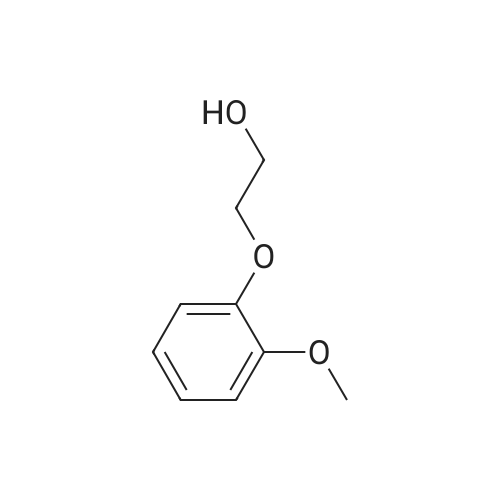
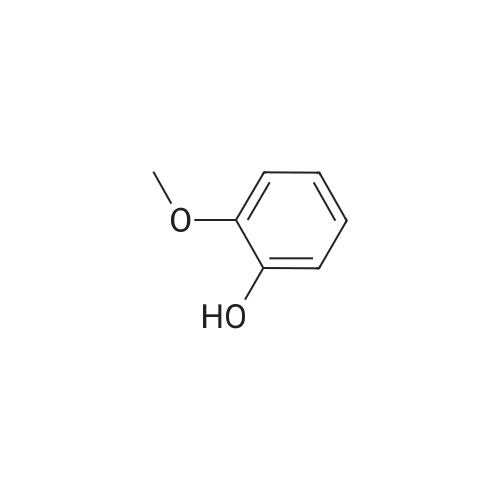
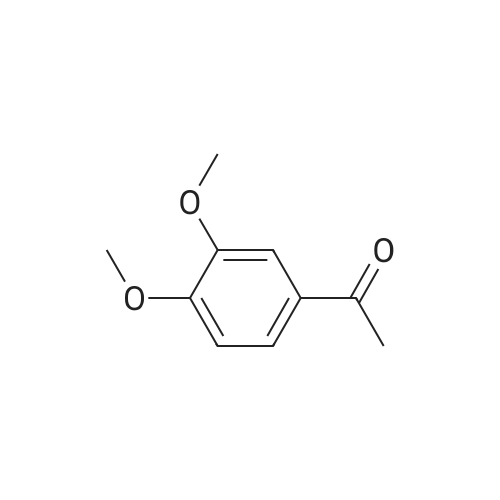
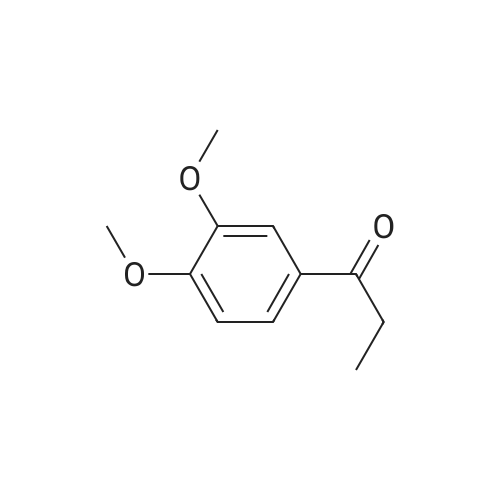
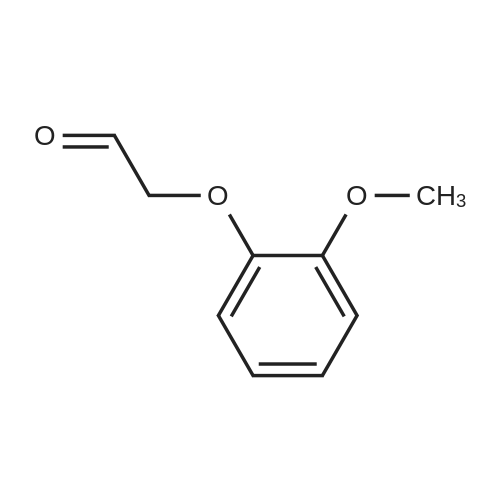
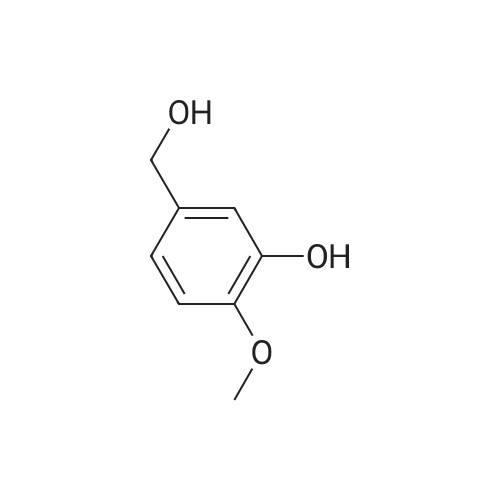
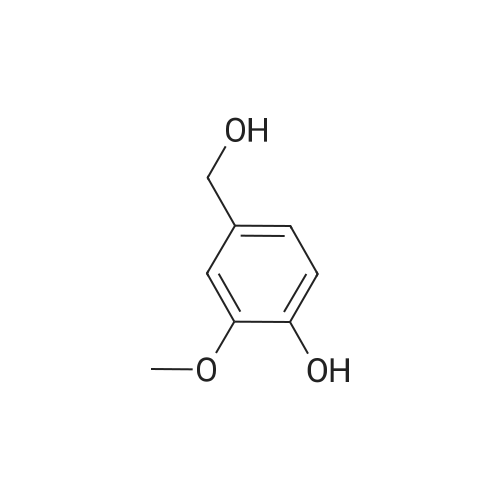
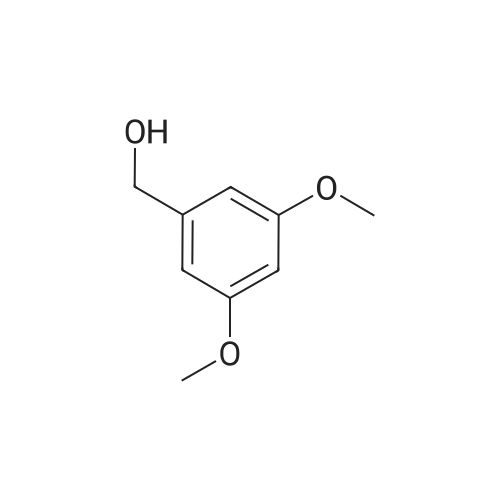
| 100% |
With aluminium chloride anhydrous; 1-butyl-4-aza-1-azoniabicyclo[2.2.2]octane chlorochromate In acetonitrile for 2h; Heating; |
|
| 100% |
With iodosylbenzene; Cl-CH2-PS supported 5-amino-1,10-phenanthroline-Ru In acetonitrile at 60℃; for 1h; |
|
| 100% |
With 1,3,5,7-tetrakis[4-(diacetoxyiodo)phenyl]adamantane; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical In dichloromethane at 20℃; |
|
| 100% |
With K<SUB>2</SUB>OsO<SUB>4</SUB>.2H<SUB>2</SUB>O; potassium carbonate; potassium hexacyanoferrate(III) In water monomer; acetonitrile at 60℃; for 0.5h; chemoselective reaction; |
|
| 100% |
With oxygen In 1,3,5-trimethyl-benzene at 60℃; for 0.5h; |
8.6 Example 8 Application of this method in the reaction of other alcohols to aldehydes and ketones
The typical reaction steps are as follows:1 mmol of the starting alcohol of the reactant column shown in Table 2,OH - Ni3In-LDH 14 mg,Mesitylene 5mL were added to the reactor,Into the oxygen,Atmospheric reaction,The reaction was stirred at 60 for a certain period of time.The solid catalyst was removed by filtration,Using gas chromatography internal standard method (chlorobenzene as internal standard) to analyze the content of liquid products,Calculate yield. |
| 99% |
With dihydrogen peroxide In benzene at 70℃; for 3h; |
|
| 99% |
With laccase; 2,2,6,6-tetramethyl-1-piperidinyloxy free radical; oxygen In water monomer at 20℃; for 24h; |
|
| 99% |
With dihydrogen peroxide In water monomer; acetonitrile at 25℃; for 0.583333h; |
|
| 99% |
With dihydrogen peroxide In water monomer at 20℃; for 0.416667h; |
|
| 99% |
With manganese(IV) oxide; oxygen In toluene at 110℃; for 8h; Green chemistry; |
3.1. Oxidation of benzhydrol 1a in the presence of substoichiometric amounts of activated MnO2: preparation of benzophenone 2 (Table 1, entry 6). General procedure
General procedure: Benzhydrol 1 (0.3831 g, 2.08 mmol) was dissolved in toluene (15 mL) and activated MnO2 (purchased from Aldrich, 0.106 g, 1.92 mmol, 50 mg/mmol) was added to the solution. The reaction mixture was heated at 110 °C under oxygen atmosphere for 4 h. Supernatant of the reaction mixture was scooped by pipet. Additional toluene (5 mL) was added to solid residue and washed the solid then the supernatant was scooped by pipet. This washing procedure was repeated for four times. All of toluene solution was combined and concentrated. Crude product was purified by flash chromatography (silica gel/hexane-EtOAc 3:1) to give 2 in 98% yield (0.373 g, 2.05 mmol). MnO2 residue was examined for the recycling use of the oxidant (Scheme 4, see below). |
| 99% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; oxygen In aq. phosphate buffer; acetonitrile at 20℃; for 24h; Enzymatic reaction; |
|
| 99% |
With titanium(IV) dioxide; oxygen at 29.84℃; for 6h; Sealed tube; Irradiation; |
|
| 99% |
With tert.-butylhydroperoxide; oxygen In decane; toluene at 80℃; Schlenk technique; |
|
| 98% |
With 1-benzyl-1-aza-4-azoniabicyclo<2.2.2>octane periodate In acetonitrile for 0.5h; Heating; |
|
| 98% |
With 1-benzyl-4-aza-1-azoniabiyclo<2.2.2>octane peroxodisulfate In acetonitrile for 0.5h; Heating; |
|
| 98% |
With ammonium hydroxide; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; oxygen In ethanol at 50℃; for 24h; |
5 4.5. General procedure for the aerobic alcohol oxidation
General procedure: Under an air atmosphere, a Schlenk tube was charged with MCM-41-bpy-CuI (40 mg, 0.025 mmol), alcohol (0.5 mmol), TEMPO (4 mg, 0.025 mmol), aqueous ammonia (0.5 mmol, 25e28%, w/w) and EtOH (1.0 mL). The mixture was stirred at 50 °C for 18-48 h. After completion of the reaction, the reaction mixture was cooled to room temperature, diluted with ethyl acetate (10 mL), and filtered. The MCM-41-bpy-CuI complex was washed with EtOH (2*5 mL), and Et2O (5 mL) and reused in the next run. The filtrate was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (petroleum/ethyl acetate=15:1 to 10:1) to provide the desired product. |
| 97% |
With 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane dichromate In dichloromethane for 0.05h; Microwave irradiation; |
|
| 97% |
With methyltriphenylphosphonium peroxydisulfate In acetonitrile for 0.25h; Heating; |
|
| 97% |
With potassium permanganate In various solvent(s) at 20℃; for 1h; |
|
| 97% |
With 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione In neat (no solvent) at 20℃; for 0.75h; Milling; |
|
| 97% |
With tripotassium phosphate tribasic; copper (I) iodide; 1,10-Phenanthroline In 1,4-dioxane at 80℃; Schlenk technique; |
General Procedure-2 [For the open air oxidation of alcohols to carbonyl compounds; GP-2(method B)]:
General procedure: In an oven dried Schlenk tube, were added alcohol 1 (69.0-199.5 mg, 0.5 mmol), CuI (10 mol%)and 1,10-Phenanthroline (20 mol%) and K3PO4 (2 mmol) followed by the addition of dioxane (2mL) at room temperature under open air atmosphere. The stirred reaction mixture was heated inan oil bath at 80 C for 7-48 h. Progress of the reaction was monitored by TLC till the reaction iscompleted. Then, the reaction mixture was cooled to room temperature, quenched with aqueousNH4Cl solution and then extracted with CH2Cl2 (3 10 mL). The organic layer was washed withsaturated NaCl solution, dried (Na2SO4), and filtered. Evaporation of the solvent under reducedpressure and purification of the crude material by silica gel column chromatography (petroleumether/ethyl acetate) furnished the aldehyde/ketone 2 (61-97%). |
| 97% |
With dmap; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; copper trifluoromethanesulfonate; (S)-(-)-5-(2-pyrrolidinyl)-1H-tetrazole In acetonitrile at 25℃; for 1h; chemoselective reaction; |
4 4.1.2. The oxidation of primary alcohols
General procedure: A round-bottom flask was charged with alcohol (2 mmol), CuOTf (0.1 mmol, 0.05 equiv) (S)-5-(pyrrolidin-2-yl)-1H-tetrazole (0.1 mmol, 0.05 equiv), TEMPO (0.1 mmol, 0.05 equiv), DMAP (0.15 mmol, 0.075 equiv) and CH3CN (5 ml). The reaction mixture was stirred at 25 °C open to air until the completion of the reaction, as monitored by TLC. After completion, CH3CN was evaporated under vacuum. The residue was then diluted with CH2Cl2 (5 ml) and filtered through a plug of silica gel to afford the desired product. |
| 96% |
With bismuth(III) chloride; benzyltriphenylphosphonium peroxymonosulfate In acetonitrile for 0.75h; Heating; |
|
| 96% |
With dihydrogen peroxide; 1-n-butyl-3-methylimidazolium tetrafluoroborate for 1.5h; Heating; |
|
| 96% |
With sodium (meta)periodate In dichloromethane; water monomer at 20℃; for 15h; |
|
| 96% |
In butanone Heating; |
|
| 96.5% |
With paraformaldehyd In toluene for 4h; Heating / reflux; |
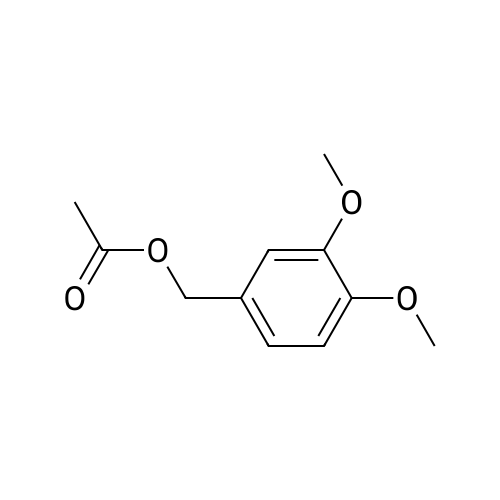
9
Operating as described in example 2, 5.0 g (0.03 mols) of veratric alcohol (3,4- dimethoxy benzyl alcohol) are reacted, in 50 g of toluene, with 2.25 g (0.075 mols of p-formaldehyde in the presence of 1.25 g of zirconia (ZrO2) XZO 632/03 fromMelcat.After filtering off the catalyst and evaporating under vacuum (at 30°C/21 mbar) a crude reaction product containing veratric aldehyde is obtained with a 96.5% GC yield and a conversion of 98.3%. |
| 96% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; H4VPO7; oxygen In water monomer at 80℃; for 4h; Autoclave; |
|
| 96% |
With oxygen; 2,3-dicyano-5,6-dichloro-p-benzoquinone; NaNO2 In dichloromethane; acetic acid at 20℃; for 6h; |
|
| 96% |
With dimanganese decacarbonyl In toluene at 120℃; for 12h; Sealed tube; |
|
| 95% |
With aluminum(III) oxide; potassium permanganate In neat (no solvent) for 0.0333333h; |
|
| 95% |
With benzyltriphenylphosphonium chlorochromate In dichloromethane for 0.0333333h; microwave irradiation; |
|
| 95% |
With benzyltriphenylphosphonium dichromate for 0.166667h; |
|
| 95% |
With potassium dichromate||potassium bichromate||K2Cr2O7||Cr2O7K2; aluminium chloride anhydrous for 0.0333333h; |
|
| 95% |
With aluminium chloride anhydrous; butyltriphenylphosphonium periodate In acetonitrile for 9h; Heating; |
|
| 95% |
With 3,5-dimethylpyrazolium fluorochromate(VI) at 20℃; for 0.0333333h; |
|
| 95% |
With aluminium chloride anhydrous; 1-decyl-4-aza-1-azoniabicyclo[2.2.2]octane chlorochromate In acetonitrile for 1.5h; Heating; |
|
| 95% |
With benzyltriphenylphosphonium dichromate; mesoporous silica at 20℃; for 0.166667h; |
|
| 95% |
With NaNO3 at 40℃; for 0.1h; |
|
| 95% |
With 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione In water monomer at 25℃; for 24h; Micellar solution; |
General procedure for the oxidation of alcohols
General procedure: A vial was charged with alcohol (1 mmol), IBX (1.2 mmol, 1.2 equiv) and 2 wt % GMPGS-2000/H2O solution (5 mL). The mixture was stirred for 24 h at 25 °C and filtered. The solid was washed with CH2Cl2 and the filtrate was extracted with CH2Cl2 (3×10 mL). Then, the organic phase was combined and dried with anhydrous Na2SO4, evaporated to dryness. The crude product was purified was purified by column chromatography on silica gel eluted with (petroleum ether/EtOAc) to afford the desired product. |
| 95% |
With Trametes villosa (viz. Poliporus pinsitus) laccase; oxygen; 6-methyl-1-hydroxybenzotriazole In 1,4-dioxane at 30℃; for 5h; Enzymatic reaction; |
4.4 Enzymatic oxidations
General procedure: The mediators 6-X-HBTs (10μmol), laccase from Trametes villosa (7-40 units) and the substrate (30 μmol) were added to a 5.0 mL of a buffered water solution (0.1M sodium citrate, pH 5.0) with 25% dioxane as cosolvent, purged with O2 for 30min before the addition of the reagents. The mixture was magnetically stirred at 30°C for 5-24 h under oxygen (filled balloon). Reaction products were extracted with ethyl acetate, characterized by GC-MS and 1H NMR and quantified by GC and 1H NMR analysis. A good material balance (>95%) was observed in all the experiments. In the absence of the mediator or the enzyme no formation of oxidation products was observed in significant amounts (<0.1%). |
| 95% |
With tert.-butylnitrite; oxygen; 2,3-dicyano-5,6-dichloro-p-benzoquinone In 2-methoxy-ethanol at 80℃; for 14h; chemoselective reaction; |
|
| 95% |
With dmap; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; copper chloride (I) In water monomer at 55℃; for 10h; Green chemistry; |
|
| 95% |
With 9-fluorenone In dimethyl sulfoxide at 20℃; Irradiation; |
|
| 94% |
With vanadyl(IV) acetylacetonate; oxygen; 1-n-butyl-3-methylimidazolium tetrafluoroborate at 80℃; for 12h; |
|
| 94% |
With nitric acid; diphosphorus pentoxide; mesoporous silica for 0.0333333h; |
|
| 94% |
With triphenylmethylphosphonium dichromate at 20℃; for 0.0666667h; Neat (no solvent); chemoselective reaction; |
|
| 94% |
With hydrogenchloride; 4-acetylamino-2,2,6,6-tetramethyl-1-piperidinoxy; oxygen; nitric acid In water monomer; acetonitrile at 45℃; for 24h; High pressure; Sealed tube; chemoselective reaction; |
|
| 94% |
With oxygen In N,N-dimethyl-formamide at 20℃; for 6h; UV-irradiation; |
Photocatalytic oxidation of benzyl alcohols by 3D-RGO/ZnO photocatalyst
General procedure: A 25 mL round-bottomed flask was charged with alcohol (1 mmol),3D-RGO/ZnO (40 mg) and N,N-dimethyl formamide (5 mL). The resultant mixture was stirred under O2 with two white LED lamps (12W). After completion of the reaction, the 3D-RGO/ZnO catalyst was recycled by filtration and the organic phase of the filtrate was extracted with EtOAc, washed three times with water and dried over Na2SO4.The pure product was then isolated by silica chromatography using petroleum ether/EtOAc mixtures as the eluent. |
| 94% |
With 2,4,6-trimethyl-pyridine; 4-acetylamino-2,2,6,6-tetramethyl-1-piperidinoxy; iodine; Sodium hydrogenocarbonate In dichloromethane; water monomer at 20 - 22℃; for 1h; |
1.10 Preparative synthesis of compounds 2a,b,d-al (general procedure)
General procedure: A solution of corresponding alcohol 1a,b,d-al (8 mmol), nitroxide 4a (0.085 g, 0.4 mmol) and compound 6d (0.097 g, 0.8 mmol) in CH2Cl2 (10 mL) was added to a vigorously stirred solution of NaHCO3 (2.016 g, 24 mmol) in water (10 mL) at 20 °C. Then I2 (4.06 g, 16 mmol) powder was added in one portion to the formed reaction mixture at vigorous stirring and temperature 20-22 °C. The reaction mixture was stirred at 20-22 °C for appropriate time (see Table 1 in the article). Then, a saturated solution of sodium thiosulfate was added to the stirred reaction mixture for discoloration. Organic and aqueous phases were separated and the aqueous phase was then extracted with CH2Cl2 (3×5 mL). Organic phase and the extracts were combined and washed subsequently with saturated aqueous solution of NaCl (5 mL), aqueous solutionof HCl (1%) saturated with NaCl (3 mL), and then with water (5 mL). The washed extract was dried with anhydrous Na2SO4 and evaporated to dryness to give crude product, which was then purified by vacuum distillation under argon atmosphere or by recrystallization. |
| 93% |
With aluminium chloride anhydrous; 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane dichromate for 0.0111111h; |
|
| 93% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; copper (II) acetate In water monomer; acetonitrile at 20℃; for 2h; Green chemistry; |
General procedure: A mixture of alcohol (5.0 mmol), Cu(OAc)2 (9.1 mg, 0.05 mmol), and TEMPO (7.8 mg, 0.05 mmol) in CH3CN/H2O (5/10 mL) was stirred at room temperature for specified time. After completion of the reaction (monitored by TLC, eluents: petroleum ether/ethyl acetate = 4/1), dichloromethane (10 mL) was added to the resulting mixture. The dichloromethane phase was separated, and the aqueous phase was further extracted with dichloromethane (10 mL × 2). The combined organic layers were dried over anhydrous sodium sulfate and concentrated to give a residue, which was purified by column chromatography (eluents: petroleum ether/ethyl acetate = 10/1) to provide the desired product. |
| 93% |
With potassium carbonate In water monomer at 60℃; for 24h; |
4.1. General Procedure for Heck Coupling and the Oxidationof Alcohols
General procedure: A typical reaction was carried out as follows: bromobenzene(1a) (1.0 mmol), alkene (1.5 mmol), HCOONa (1.5mmol), 50 mg of wool-Pd complex catalyst (Pd 11.74 %),were added to 15 mL aqueous media (PEG-400 = 33 mg) ina 25 mL beaker, and stirred at 80 °C under ambient conditions.After the completion of the reaction, the catalyst was filtrated, washed and dried. Subsequently, 40 mg of the solid catalyst, benzyl alcohol (0.2 mmol), K2CO3 (0.2 mmol) were stirred in 15 mL aqueous media (PEG-400 = 33 mg), atmosphericair was used as the source of molecular oxygen at 60°C for 24 h, and the oxidation monitored by TLC. After completion, an extraction with ethyl acetate was performed.The organic layer was dried under reduced pressure to givethe desired crude product. Analytically pure products were obtained by column chromatography using petroleum ether and ethyl acetate as eluent. Formation of products and consumptionof substrates were monitored by GC. The identity of products was determined either by comparison with authentic samples using GC or by NMR analysis. The conversion and product selectivity were determined using GC analysis. |
| 93% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; C110H202N8O47; oxygen; copper(II) bis(trifluoromethanesulfonate); potassium carbonate In water monomer at 20℃; for 6h; Green chemistry; |
General procedure for aerobic oxidation of alcohols in water
General procedure: To a 48 mL tube, were added Cu(II) or Cu(I) salt (0.05 mmol), PEG-PyTa (0.025 mmol) and H2O (3.0 mL). The mixture was stirred for 30 min at room temperature and a clear dark-blue solution was observed. Then alcohols (1.0 mmol), TEMPO (0.05 mmol), and K2CO3 (0.2 mmol) were sequentially added, followed by connecting a balloon of oxygen. The reaction mixture was stirred at room temperature until the reaction completed based on GC analysis. After that, the reaction mixture was extracted with MTBE (3 mL×3) and the extracts were combined, dried over anhydrous Na2SO4 and concentrated under vacuum. Finally, the residue was purified by flash chromatography on silica to afford the desired aldehydes. |
| 93% |
With sulfuric acid In dimethyl sulfoxide for 0.666667h; Reflux; |
Protocol A:
General procedure: A mixture of the benzylic alcohol 1 (1 mmol) and 98% H2SO4 (1 mmol) in DMSO (3 mL) was stirred for the appropriate time under reflux conditions. The mixture was then cooled to r.t., and brine (4 mL) was added. The organic phase was extracted with CH2Cl2 (6 mL), and the organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. In all cases, the reaction products were obtained with high purity, and did not require further purification by distillation or column chromatography. |
| 92% |
With mesoporous silica; pyridinium chlorochromate In dichloromethane at 18℃; for 0.333333h; |
|
| 92% |
With laccase; oxygen; benzotriazol-1-ol In various solvent(s) at 20℃; for 24h; |
|
| 92% |
With hydrogenchloride; 4-acetylamino-2,2,6,6-tetramethyl-1-piperidinoxy; oxygen; nitric acid In water monomer; acetonitrile at 45℃; for 24h; Sealed tube; |
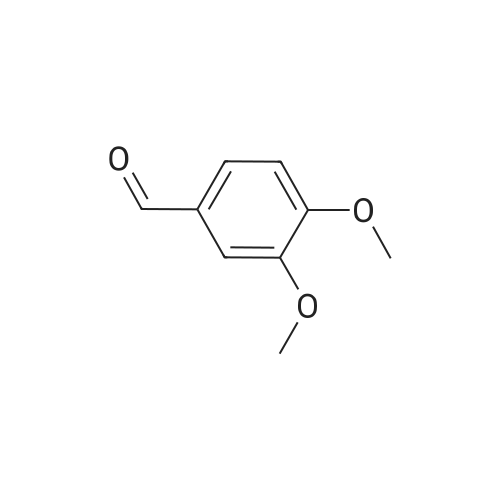
3,4-Dimethoxy-benzaldehyde (3a)
General procedure: To a 25 mL high pressure tube with a stir bar was added lignin model compound (1 mmol) and 5 mol % of NHAc-TEMPO (0.05 mmol, 10.6 mg). The tube was sealed, evacuated and filled to 1.1 atm with oxygen gas. Nitric acid (67%; 9.4 μL) (0.1 mmol, 6.3 mg) in 1 mL acetonitrile and 9.9 μL of hydrochloric acid (37%) (0.1 mmol, 3.65 mg) in 1 mL of acetonitrile was injected through the septum individually. Additional 3 mL of acetonitrile and 260 μL of water were injected. The reaction mixture was stirred for 24 h at 45° C. The solvent was evaporated and the residue was subjected to column chromatography to ascertain the amount of the corresponding carbonyl compound. The reaction of 1 with optimized catalyst system was repeated several times. The results are shown in FIG. 5. As is clearly depicted in the figure, the conversion and yield results for this catalytic system (HNO3/HCl/NHAc-TEMPO) are highly consistent. The six identical runs yielded statistically identical conversions and yields. Spectral data were consistent with those reported in the literature. (Jeena, V.; Robinson, R. S. Chem. Commun. 2012, 48, 299.) 1H NMR (400 MHz, CDCl3) δ 9.86 (s, 1H), 7.47 (dd, J=8.2, 2.0 Hz, 1H), 7.41 (d, J=1.8 Hz, 1H), 6.97 (d, J=8.2 Hz, 1H), 3.97 (s, 3H), 3.95 (s, 3H). HRMS (EI) calculated for C9H10O3 [M]+ 166.0630. found 166.0626. |
| 92% |
With hydrogenchloride; 4-acetylamino-2,2,6,6-tetramethyl-1-piperidinoxy; oxygen; nitric acid In water monomer; acetonitrile at 45℃; for 24h; Sealed tube; |
3a General Procedure for Oxidation of Lignin Model Compounds
General Procedure for Oxidation of Lignin Model Compounds: (0198) To a 25 mL high pressure tube with a stir bar was added lignin model compound (1 mmol) and 5 mol of NHAc-TEMPO (0.05 mmol, 10.6 mg). The tube was sealed, evacuated and filled to 1.1 atm with oxygen gas. Nitric acid (67%; 9.4 μΕ) (0.1 mmol, 6.3 mg) in 1 mL acetonitrile and 9.9 μΕ of hydrochloric acid (37%) (0.1 mmol, 3.65 mg) in 1 mL of acetonitrile was injected through the septum individually. Additional 3 mL of acetonitrile and 260 μL· of water were injected. The reaction mixture was stirred for 24 h at 45°C. The solvent was evaporated and the residue was subjected to column (0199) chromatography to ascertain the amount of the corresponding carbonyl compound. The reaction of 1 with optimized catalyst system was repeated several times. The results are shown in Fig. 5. As is clearly depicted in the figure, the conversion and yield results for this catalytic system (HNO3/HCI/NHAC-TEMPO) are highly consistent. The six identical runs yielded statistically identical conversions and yields. |
| 91% |
With 4-(N,N-dimethylamino)pyridinium chlorochromate In dichloromethane for 14h; |
|
| 91% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; oxygen; copper (II) acetate; diethylamine In acetonitrile at 50℃; for 3h; chemoselective reaction; |
General procedure for synthesis of carbonyl compounds
General procedure: A mixture of an alcohol (5.0 mmol), Cu(OAc)2*H2O (50 mg, 5 mol%), TEMPO (39 mg,5 mol%) and diethylamine (25 μL, 5 mol%) in CH3CN (10 mL) was stirred at 50°C under oxygen balloon for a specified time as noted in the Table I. For the analysis of the products, gas chromatography (equipped with a Capillary Column of VF-1 ms, 15 m,0.25 mm, 0.25 μm) was employed. Time-to-time quantification of the reactants consumed and products generated was examined by GC by comparing the peak area with the standard starting alcohol and the product. After a specified time, the reaction mixture was passed through a celite pad and washed with acetonitrile, and concentrated to get the crude, which was purified by column chromatography (hexane/ethyl acetate = 4.75/0.25) to obtain the desired product. |
| 90% |
With n-butyltriphenylphosphonium permanganate In acetonitrile at 20℃; for 0.333333h; |
|
| 90% |
With iodine; potassium carbonate; potassium iodide at 90℃; for 0.416667h; |
|
| 90% |
With potassium peroxodisulfate at 40℃; for 0.0833333h; Ionic liquid; |
|
| 90% |
With ferric(III) chloride; nitric acid In acetone at 20℃; for 0.416667h; Sonication; |
|
| 90% |
With ammonium cerium (IV) nitrate; 1-hexyl-3-methylimidazolium hydrogen sulfate at 80℃; for 0.333333h; neat (no solvent); chemoselective reaction; |
|
| 90% |
With C18H26Cl2Cu3N4O10; dihydrogen peroxide In neat (no solvent) at 100℃; for 5h; |
|
| 90% |
With tripotassium phosphate tribasic; carbon dioxide In dimethyl sulfoxide at 90℃; for 48h; |
|
| 89% |
With dmap; oxygen In various solvent(s) at 20℃; for 5h; |
|
| 89% |
With dihydrogen peroxide; manganese(II) acetate In n-heptane; water monomer at 25℃; for 0.5h; Ionic liquid; chemoselective reaction; |
2.3 General procedures for the oxidation of alcohols
General procedure: To a 25 mL round-bottom flask, ionic liquid (5.6 mmol), Mn(OAc)2 (0.025 mol%), alcohol (3 mmol), and n-heptane (6 mL) were added. De-ionized water (0.5 mL) was added to lower the viscosity of the mixture. The resulting solution was then kept stirring vigorously. Under ambient conditions, aqueous hydrogen peroxide (6× 60 μL, 35 wt.% in water) was injected into the reaction mixture in 3 min intervals over 20 min. After the reaction had been completed, the n-heptane solution (upper layer) was carefully decanted from the ionic liquid medium. In order to completely extract the aldehyde from the ionic liquid with residual n-heptane, additional n-heptane (2× 5 mL) was added and the combined organic solution was then dried over anhydrous sodium sulfate. After the removal of solvent under vacuum, the aldehyde was isolated as a colourless liquid and characterized by GC-MS and 1H NMR. The n-heptane collected was reused in catalysis and extraction process. It was found that the use of n-heptane, n-hexane, n-pentane, or petroleum ether (40-60 °C) as the extraction phase in the catalysis showed no observable difference as these solvents are immiscible with the ionic liquid. However, heptane was chosen because it is considered as a greener alternative solvent for chemical processes [20]. |
| 89% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; anhydrous sodium carbonate; N-Phenylglycine; copper (II) bromide In water monomer for 1h; Reflux; Schlenk technique; |
General Procedures for the Copper-Catalyzed Primary BenzylicAlcohol Oxidation under Air in Water (p-MethylbenzylAlcohol)
General procedure: A mixture of p-methylbenzyl alcohol (1.0 mmol), N-phenylglycine(0.0076 g, 0.05 mmol), CuBr2 (0.0112 g, 0.05 mmol),Na2CO3 (0.1060 g, 1.0 mmol), TEMPO (0.0078 g, 0.05 mmol),H2O (3.0 mL) were added to a 100 mL Schlenk tube, which wasvigorously stirred in air under reflux for 0.5 h. After the reaction,the product was extracted with CH2Cl2 (3 × 2.0 mL). Thecombined organic phase was washed with H2O (3.0 mL) anddried over anhydrous MgSO4. After concentration undervacuum, the residue was purified by column chromatography toafford p-methylbenzaldehyde.Isolated yield: 0.1080 g (90%). |
| 89% |
With iodine In water monomer; dimethyl sulfoxide at 100℃; for 3h; chemoselective reaction; |
Procedure
General procedure: A mixture of benzyl alcohol (0.108 g, 1 mmol), I2 (0.279 g, 1.1 mmol) in DMSO (1 mL)and water (2 mL) was stirred at 100 C for 3 h. Then, the reaction mixture was cooledto room temperature. To the reaction mixture was added 2mL of aqueous sodium thiosulfatesolution and the organic phase was extracted with 5mL of dichloromethane.Finally, the organic layer was dried over anhydrous Na2SO4, filtered and the solventwas removed under reduced pressure. In all cases, the reaction products were obtainedin high purity, and did not require further purification by distillation or columnchromatography |
| 88% |
With ammonium nitrate In dichloromethane at 20℃; for 0.416667h; |
|
| 88% |
With 1-methyl-1H-imidazole; copper (I) iodide; C20H25N4O2 In acetonitrile at 25℃; for 1.5h; |
|
| 88% |
With 2C36H60N9(3+)*3O4W(2-); dihydrogen peroxide In water monomer at 20℃; for 0.166667h; Green chemistry; |
|
| 88% |
With C6H4MoNO7(1-)*C19H42N(1+); oxygen In water monomer at 100℃; for 18h; Green chemistry; chemoselective reaction; |
2.3. General procedure for the catalytic oxidation of alcohols toaldehydes
General procedure: A mixture of alcohol (0.75 mmol), and catalyst Mo1 (13 mg,3.0 mol%) taken in 0.5 mL of water was stirred at 100 ° C under oxygenatmosphere (O2 bladder) and the stirring was continued for16-24 h as per requirement. The progress of reaction was monitoredby TLC. After completion of the reaction, ethyl acetate was added to the mixture. The aqueous phase was extracted with ethyl acetate 2-3 times. Then the combined organic extracts were driedover anhydrous sodium sulfate and the solvent was removed under reduced pressure. The crude product so obtained was purified by column chromatography using hexane-ethyl acetate as eluent. While the known products were characterized by spectroscopic techniques and compared with reported data and the new products 22b and 36b were characterized completely. The characterization detail is provided in supporting information section. |
| 88% |
With NiCl2/γ-Al2O3; potassium-t-butoxide In toluene at 80℃; for 7h; chemoselective reaction; |
|
| 88% |
With potassium peroxodisulfate; V2O5/TiO2 In water monomer; acetonitrile at 80℃; Sealed tube; Green chemistry; |
|
| 87% |
With 2,2,6,6-tetramethyl-1-piperidinyloxy free radical; silver(I) nitrate In water monomer for 10h; UV-irradiation; |
|
| 87% |
With bis(diphenylphosphonium)ferrocene perruthenate grafted on SiO2 coated Fe3O4 magnetic nanoparticles In tetrahydrofuran for 4h; Reflux; |
General Procedure for Oxidation of Alcohol
General procedure: A primary alcohol (1 mmol) was added to a suspension of 50 mg of [DppfSilNMag](RuO4)2 8 in 5mL of THF and refluxed. TLC was used to monitor the reaction progress. Compound 8 was isolated magnetically after completion of the reaction. Column chromatography (ethyl acetate/petroleum ether) wasused to purify the reaction mixture to afford pure products. |
| 86% |
With aluminium chloride anhydrous; benzyltriphenylphosphonium periodate In acetonitrile for 3.33333h; Heating; |
|
| 86% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; dimethyl 3-methyl-9-oxo-7-(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-heptadecafluorodecyl)-2,4-di(pyridin-2-yl)-3,7-diazabicyclo-[3.3.1]nonane-1,5-dicarboxylate; copper(II) bromide In water monomer at 20℃; for 7h; Green chemistry; |
|
| 86% |
With ferrocene-labeled Merrifield resin-supported ionic liquid ([FemDMMerA]RuO4) In tetrahydrofuran for 5h; Reflux; Green chemistry; |
General procedure for the oxidation of alcohols
General procedure: A mixture of aryl alcohol (1 mmol) and [FemDMMerA]Y (100 mg) in solvent(5 mL) was refluxed in oil bath. After completion of the reaction as monitored byTLC, the reaction mixture was filtered to remove insoluble SILP catalyst.Evaporation of solvent in vacuuo followed by column chromatography over silicagel using petroleum ether/ethyl acetate (95:5 v/v) afforded pure aldehydes. |
| 85% |
With oxygen; potassium carbonate In toluene at 90℃; for 10h; |
|
| 84% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; C20H36N5(3+)*3Br(1-); oxygen; copper atom In water monomer at 35℃; for 12h; Schlenk technique; |
Cu/L2/TEMPO-Catalyzed Aerobic Oxidation of Benzylic Alcohols; General Procedure
General procedure: A 30-mL Schlenk tube was evacuated and filled with O2, the benzyl alcohol (1 mmol), Cu powder (5% mol), imidazolium (5% mol), TEMPO (5% mol), and H2O (3 mL) was added and stirred at 35 °C under an O2 atmosphere by connecting an O2 balloon. When the reaction was complete, the mixture was extracted with CH2Cl2 (3 × 10 mL). The combined extracts were dried (MgSO4). Then the solvent was removed and the mixture was purified by column chromatography (silica gel) to give the product. |
| 83% |
With tripotassium phosphate tribasic; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; Cu2(phenanthroline)2(μ-Cl)2Cl2; oxygen In acetonitrile at 20℃; for 2h; |
|
| 83% |
With palladium; oxygen; Sodium hydrogenocarbonate In water monomer at 80℃; for 2h; |
2.7 Vanillic Alcohol, Veratryl AlcoholandHydrobenzoin Oxidations
The reactions were performed in a 100mL round-bottomedflask. A colloidal suspension of PVP stabilized Pd NPs(20mL, 7.05 × 10-5mol of Pd), or a dispersion of Pd/TiO2(277mg, 7.05 × 10-5mol Pd) in 20mL of HPLC-gradewater, was introduced in the flask. NaHCO3(400mg) wasadded and the mixture heated at 80°C under vigorous stirring.The reactant (7.05 × 10-3mol for vanillic and veratrylalcohol, 100 equiv./Pd or 7.05 × 10-4mol for hydrobenzoin,10 equiv./Pd) was then added. The mixture was heated atthe desired temperature stirred under air or O2flow, for adetermined time with regular sampling for analysis. |
| 82% |
With acetic anhydride; NaNO2 at 25℃; |
|
| 82% |
With copper chloride (II) In tetrahydrofuran at 80℃; for 1.5h; chemoselective reaction; |
|
| 82% |
With [RuCl2(p-cymene)(1,4-dibutyl-3-methyl-1,2,3-triazolylidene)] In toluene at 110℃; for 16h; |
|
| 82% |
With dmap; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; copper (II) acetate In neat (no solvent) at 25℃; for 36h; Green chemistry; |
|
| 82% |
With chlorocyclopentadienylbis(triphenylphosphine)ruthenium(II); oxygen In tetrahydrofuran for 0.48h; Reflux; |
|
| 81% |
With C23H35N3O3(1+)*Br(1-); copper atom In chlorobenzene at 80℃; for 15h; |
|
| 80% |
With 3 A molecular sieve; pyridinium chlorochromate In dichloromethane at 25℃; for 0.166667h; |
|
| 80% |
With 3 Angstroem MS; pyridinium chlorochromate In dichloromethane for 0.166667h; |
|
| 80% |
With [Cu3(1,3,5-benzenetricarboxylate)2]; 9-azabicyclo<3.3.1>nonane-N-oxyl; oxygen In 1,2-dichloro-ethane at 70℃; for 15h; Green chemistry; |
5. General procedures of the aerobic oxidation catalyzed by HKUST-1/ABNO
General procedure: A 15 mm flame-dried test tube, which was equipped with a magnetic stir bar and charged with alcohol (0.5 mmol, in case of solid), HKUST-1 (10 mol %, 0.05 mmol), ABNO (5 mol %, 0.025 mmol), and Cs2CO3 (1.0 equiv, 0.5 mmol), was evacuated and backfilled with oxygen (this process was repeated 3 times). After 0.5 mL of DCE was added, alcohol (0.5 mmol, in case of liquid), and DCE (0.5 mL) were added in sequence. The reaction mixture was stirred for 15 h at room temperature under O2 balloon. The reaction was diluted by adding CH2Cl2 and filtered through celite. The solvent was removed under vacuo. The residue was purified by column chromatography to give the desired product |
| 80% |
With C15H25Cl2N3NiO3; potassium-t-butoxide In toluene at 110℃; for 12h; Schlenk technique; Inert atmosphere; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping