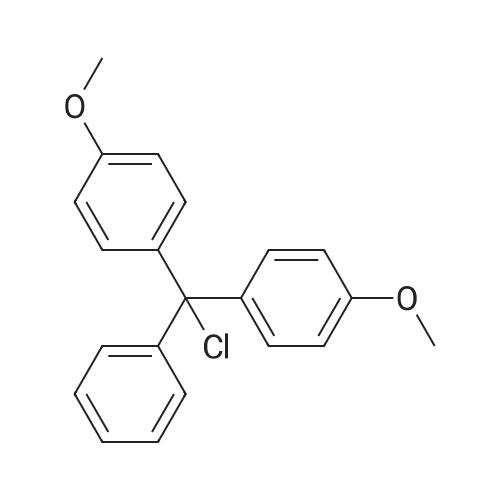
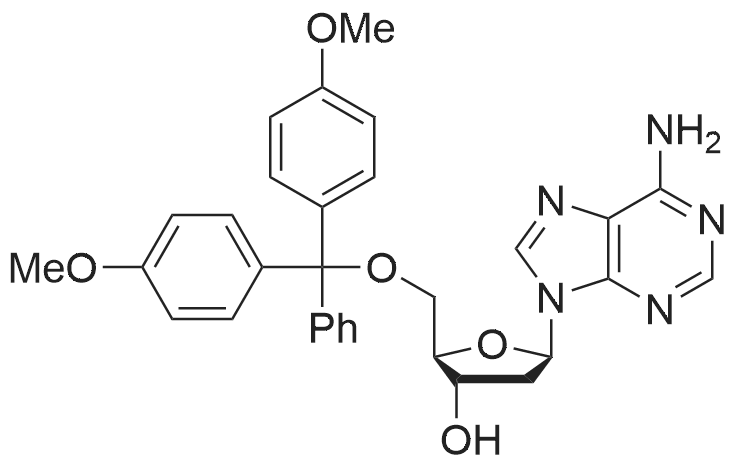
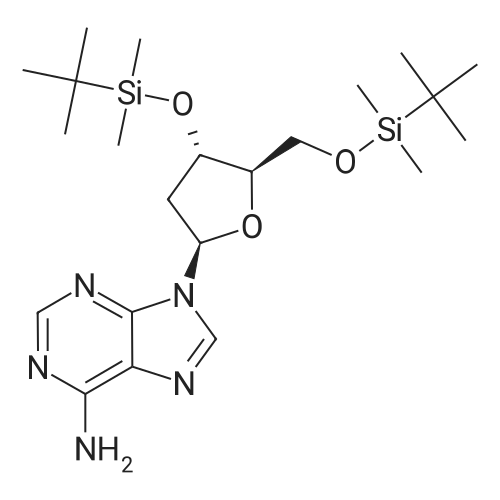
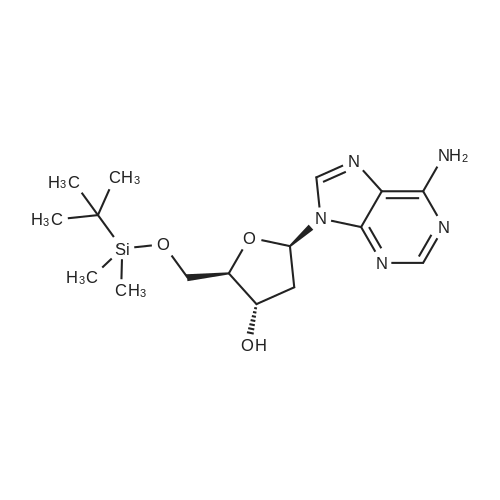
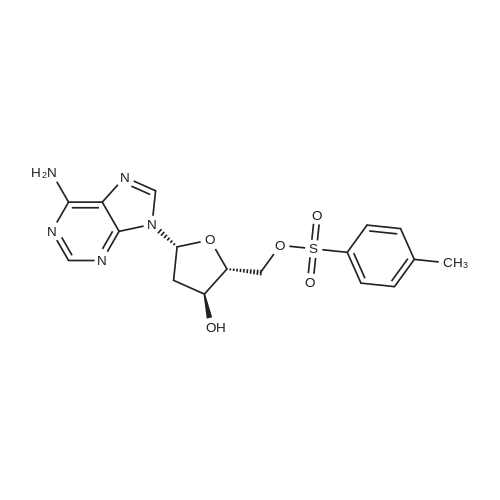
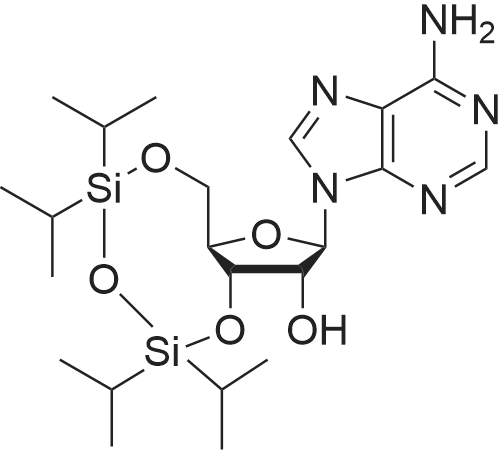
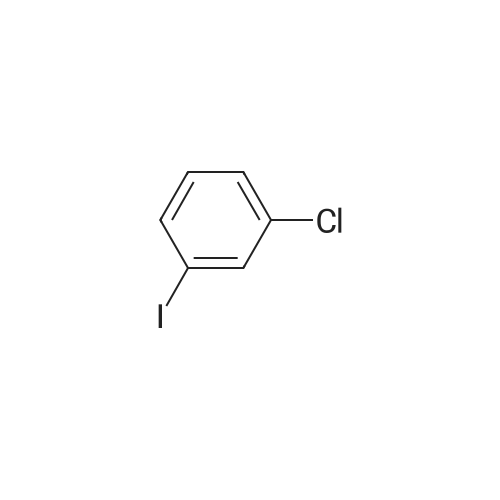
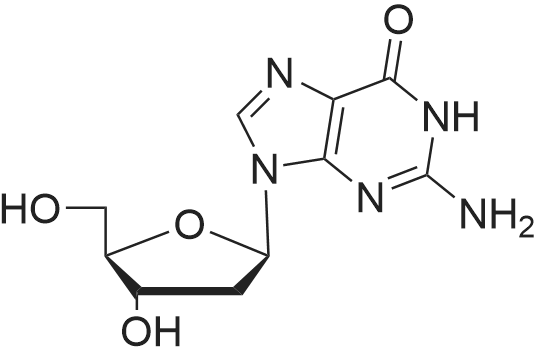
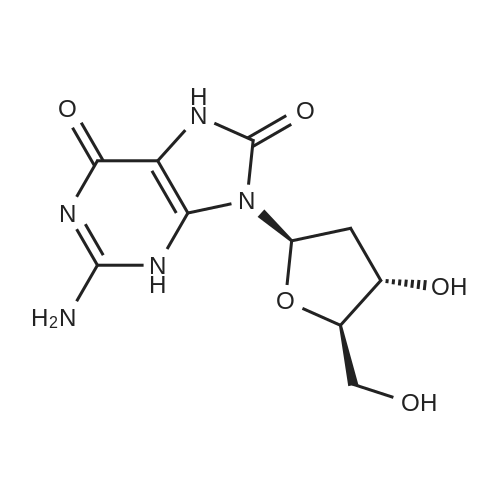
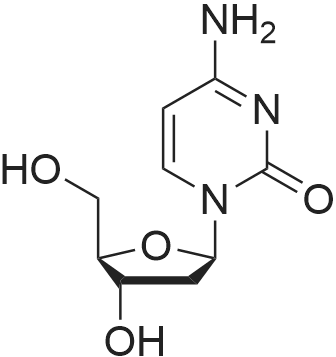
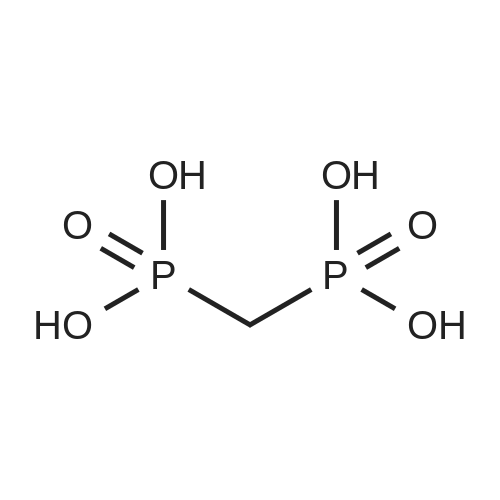
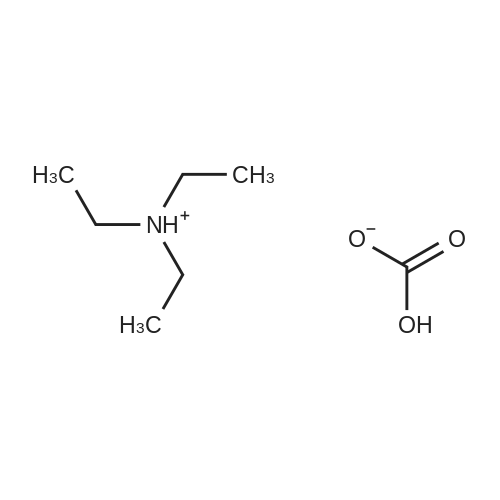
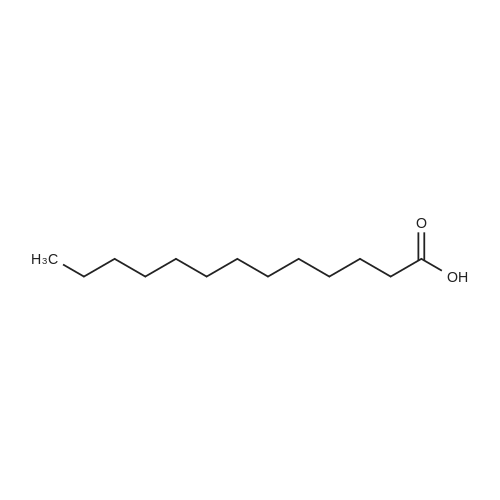
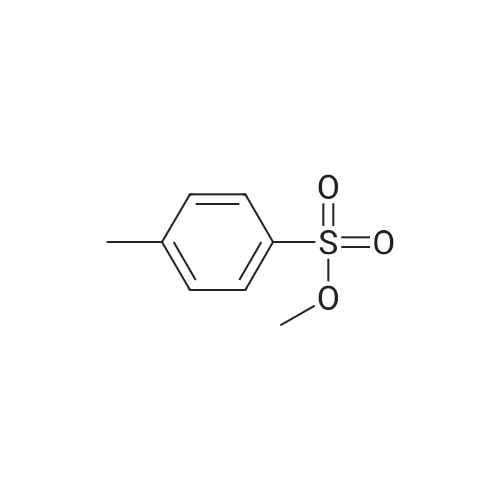
| 79% |
With pyridine In dichloromethane at 20℃; for 1.5h; Cooling with ice; |
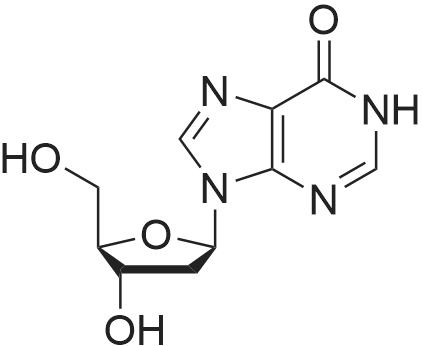
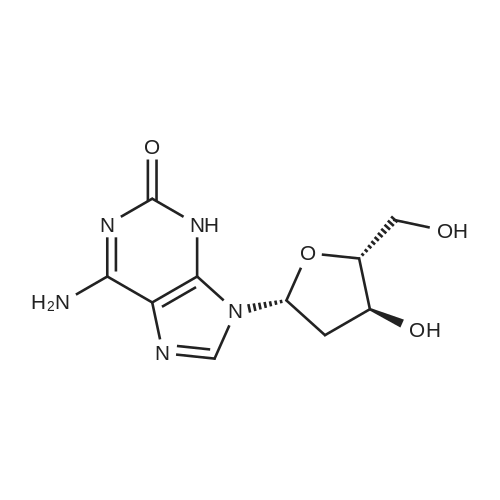
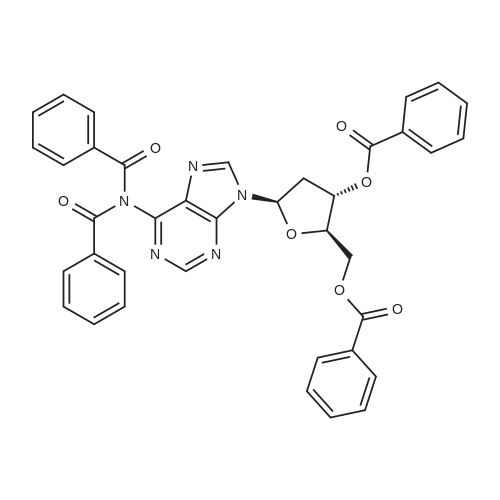
Synthesis of 3′,5′-di-O-acetyl-2′-deoxyadenosine (b)
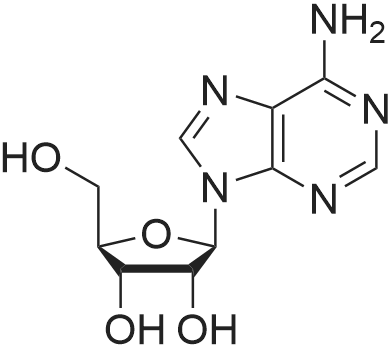
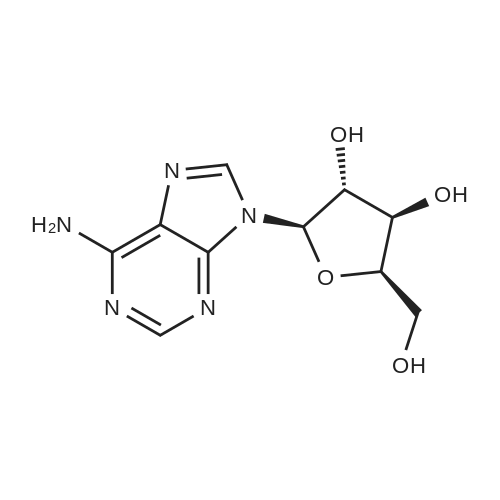
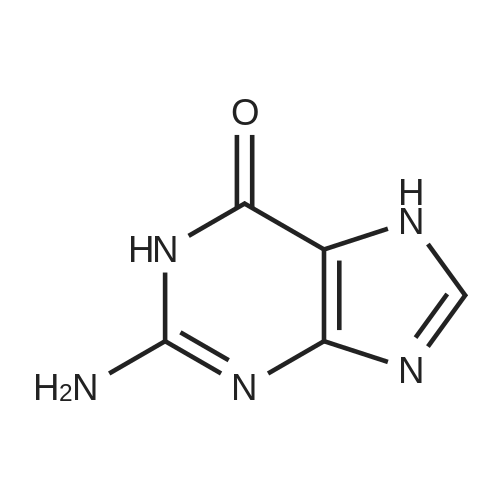
To the solution of 2′-deoxyadenosine a (3 g, 11.95 mmol)in 40 mL of anhydrous pyridine 12 mL of acetic anhydride(119.5 mmol) were slowly dropped and the reactionmixture was stirred for 1.5 h at room temperature.Then, after cooling in the ice bath, 10 mL of CH2Cl2were added and the solution was washed with 10 % aq.NaHCO3 (50 mL). The aqueous layer was extracted twicewith 30 mL of chloroform. The combined organic layerswere dried with MgSO4, the drying agent was filtered offand chloroform evaporated under reduced pressure. Theresulting precipitate was three times evaporated with toluene(3 × 15 mL) and purified by crystallization fromethanol to give 3.18 g (9.5 mmol) of acetylated 2′-deoxyadenosineb (yield 79 %), TLC: Rf = 0.66, (CHCl3/MeOH 80:20 v/v). 1H NMR (250 MHz, CDCl3) δ 2.09(s, 3H, CH3COO), 2.14 (s, 3H, CH3COO), 2.62 (ddd,1H, JH2′,H3 = 2.5 Hz, JH2′,H1′ = 5.9 Hz, Jgem = 14.1 Hz,H2′), 2.96 (ddd, 1H, JH2″,H3′ = 6.3 Hz, JH2″,H1′ = 8.1 Hz,Jgem = 14.1 Hz, H2″), 4.32-4.46 (m, 3H, H4′, H5′,H5″), 5.43 (dt, 1H, JH3′,H2′ = JH3′,H4′ = 2.5 Hz,JH3′,H2″ = 6.3 Hz, H3′) 5.85 (bs, 2H, NH-6), 6.46 (dd, JH3′,H2″ = 6.3 Hz, H3′) 5.85 (bs, 2H, NH-6), 6.46 (dd,1H, JH1′,H2′ = 5.9 Hz, JH1′,H2″ = 8.1 Hz, H1), 7.99 (s, 1H,H2), 8.36 (s, 1H, H8). |
|
With dmap; trimethylamine In acetonitrile for 8h; Reflux; |
148
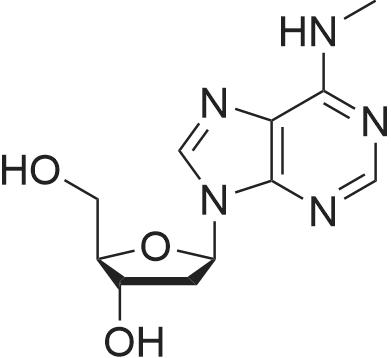
First step, a mixture of 2-deoxy-adenosine(3.6 g), acetic anhydride (5.47 g), trimethyl (4.07 g) and DMAP (0.16 g) in anhydrous acetonitrile (40 ml) was refluxed for 8 h. Acetonitrile was removed under reduced pressure. H2O (40 ml) was added to the residue, and the resulting solution was extracted with EtOAc (3 × 40 ml). The EtOAc of the combined organic layer was dried with sodium acetate and The EtOAc layer was filtered and the filtrate was concentrated. The residue was separated by column chromatography over silica gel and eluted with CHCl3-CH3OH (100 : 1) to yield 3',5'-diacetyl-2'-deoxy-adenosine (4.0 g). Second step, a mixture of 3', 5'- diacetyl-2'-deoxy-adenosine(1.2 g), tert-butyl nitrite (7.42 g), and tribromomethane (20 ml) was refluxed for 2 h. The excess tert-butyl nitrite was removed under reduced pressure. The residue was separated by column chromatography over silica gel and eluted with CHCl3-CH3OH (80 : 1) to yield 3',5'-diacetyl-2'-deoxy-6-bromo-adenosine (595 mg). Third step, a mixture of 3', 5'- diacetyl-2'-deoxy-6-bromo-adenosine (398 mg), 3-methoxy-4-hydroxybenzylamine (379.3 mg, the hydrochloride) and triethylamine (253 mg) in anhydrous EtOH (20 ml) was refluxed for 5 h. After evaporation, the residue was separated by column chromatography over silica gel and eluted with CHCl3-CH3OH (50 : 1) to yield N6-(3-methoxy-4-hydroxy-benzyl)-2-deoxy-3, 5'-diacetyl adenosine (340 mg): 1H NMR (300 MHz, acetone-d6):the 2-deoxy- adenosine moiety δ 8.29 (1H, s, H-8), 8.16 (1H, s, H-2), 7.33 (1H, t, J= 6.0 Hz, NH), 6.44 (1H, dd, J= 7.8, 6.3 Hz, H-1), 5.48 (1H, m, H-3'), 4.36 (1H, dd, J= 6.3, 12.9 Hz, H-5'a), 4.30 (1H, dd, J= 6.0, 12.9 Hz, H-5'b), 4.29 (1H, m, H-4'), 3.21 (1H, ddd, J= 7.5, 7.8, 15.0 Hz, H-2'a), 2.59 (1H, ddd, J = 2.4, 6.0, 15.0, H-2'b); the 3-methoxy-4-hydroxybenzyl moiety δ 7.63 (1H, brs, OH), 7.05 (1H, d, J= 1.5 Hz, H-2"), 6.87 (1H, dd, J= 7.8, 1.5 Hz, H-6"), 6.74 (1H, d, J= 7.8 Hz, H-5"), 4.76 (2H, brs, H-7"), 3.75 (3H, s, OMe); the acetyl δ2.09 (3H, s, CH3CO), 2.01 (3H, s, CH3CO); 13C NMR (300 MHz, acetone-d6):the 2-deoxy- adenosine moiety δ 155.9 (C-6), 153.6 (C-2), 149.1 (C-4), 139.9 (C-8), 121.2 (C-5), 85.2 (C-1), 83.1 (C-4'), 75.6 (C-3'), 64.5 (C-5'), 37.0 (C-2'); the 3-methoxy-4-hydroxy moiety δ 148.2 (C-3"), 146.5 (C-4"), 132.0 (C-1), 121.2 (C-2"), 115.6 (C-6"), 112.3 (C-5"), 56.1 (OMe), 44.2 (C-7"); the acetyl δ170.8, 170.7, 20.9, 20.6ο |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping