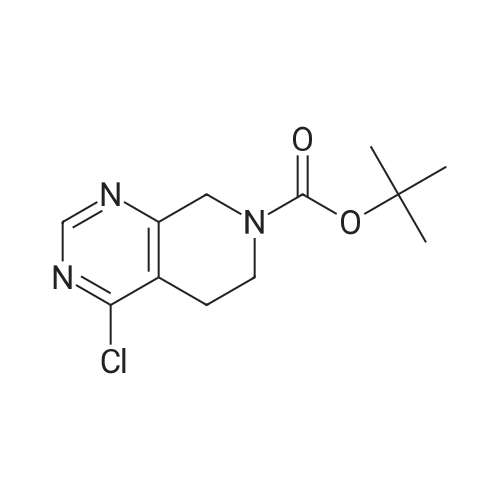
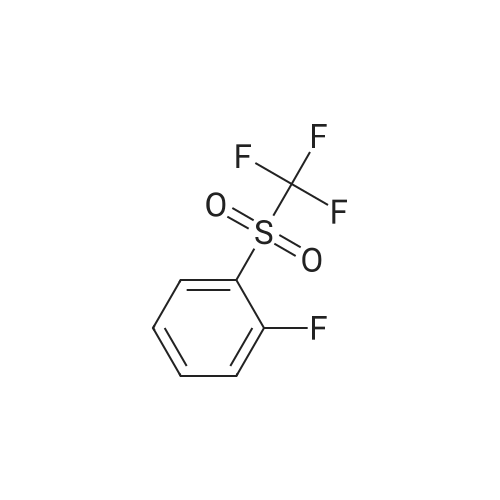
|
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 30℃; |
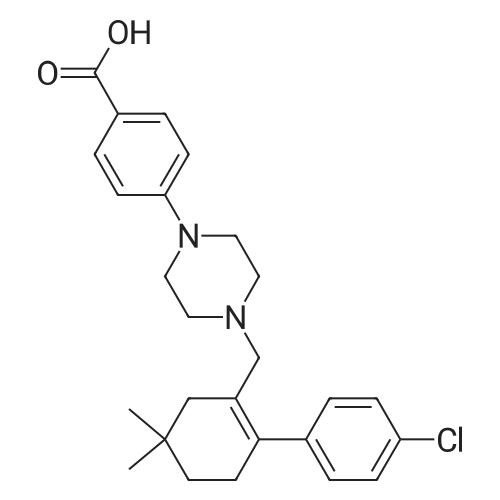
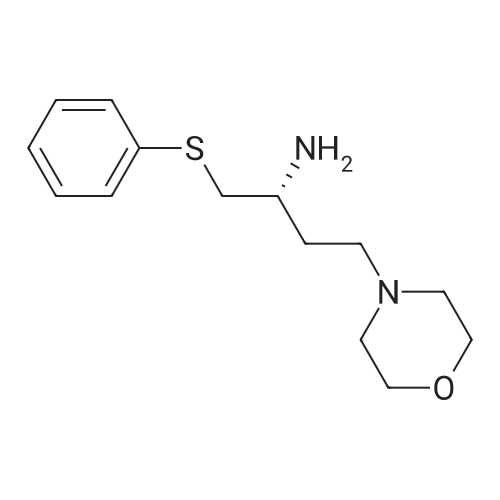
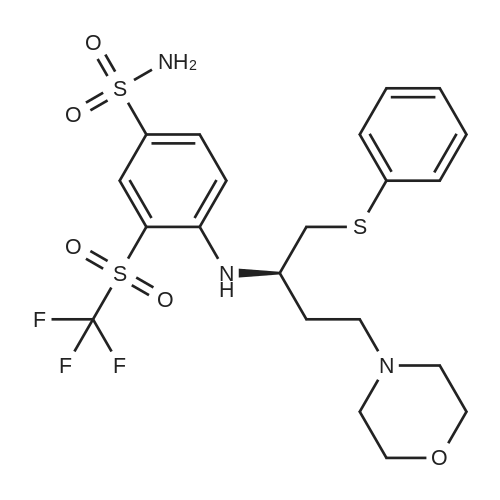
EXAMPLE 7N-(4-(4-((2-(4-chlorophenyl)-5,5-dimethyl-1-cyclohex-1-en-1-yl)methyl)piperazin-1-yl)benzoyl)-4-(((1R)-3-(morpholin-4-yl)-1-((phenylsulfanyl)methyl)propyl)amino)-3-((trifluoromethyl)sulfonyl)benzenesulfonamide; A mixture of 4-(((1R)-3-morpholin-4-yl-1-((phenylthio)methyl)propyl)amino)-3-((trifluoromethyl)sulfonyl)benzenesulfonamide (8.575 g) (EXAMPLE 17), EXAMPLE 6 (6.4 g), 4-dimethylaminopyridine (4.4 g), 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride (4.1 g) and dichloromethane (80 g) was heated at 30 C., cooled to room temperature, quenched with N,N-dimethylethylenediamine (1.1 g), distilled to 30 ml, treated with water (5.4 g) and ethyl acetate (97 g), distilled to 65 ml, treated with water (5.4 g) and ethyl acetate (97 g), distilled to 80 ml and treated with 10% acetic acid/0.75% brine (182 g) and ethyl acetate (65 g). The layers were separated, and the extract was washed with 10% aqueous acetic acid/0.75% brine (182 g), 25% aqueous K2HPO4 (180 g) and pH 7 buffer solution (163 g), concentrated to 35 ml and chase distilled with ethyl acetate (120 g, 120 g and 60 g) with concentration to 35 ml after each addition. The extract was then treated with ethyl acetate (60 g), and the solution was diluted with ethanol (71 g) and polish-filtered through a polypropylene 0.5 mum filter into a reactor with an ethyl acetate (20 g) rinse. In a separate reactor, HCl (2.8 g) in ethanol (80 g) was prepared and polish-filtered through a separate filter and housing into the reactor. The polish filtration of the solution removed residual phosphate salts from the final extraction. The solution was concentrated to about 150 ml and maintained at that level while an additional chase of ethanol (160 g) was conducted, heated at 45 C., treated with seeds (90 mg) in ethanol (1 g), stirred for 12 hours, cooled to 20 C. and stirred for another 4 hours. Analysis of the filtrates indicated the crystallization was complete. The slurry was filtered, and the solids were rinsed with ethanol (2×57 g). The rinses were applied in a slurry fashion with no vacuum, (contact time 15-25 minutes for each) then removed by vacuum filtration. The wet cake was sampled for impurities to determine if a recrystallization would be necessary. The solids were dried under vacuum and nitrogen at 50 C. for 3 days. Analysis of the dryer sample (GPAS residual solvents method) showed that drying was complete. 1H NMR (400 MHz, methanol-d4) delta ppm 1.08 (s, 5H) 1.57 (t, J=6.38 Hz, 2H) 2.11 (s, 2H) 2.26-2.34 (m, 1H) 2.40 (t, J=5.69 Hz, 2H) 3.18-3.27 (m, 4H) 3.42 (dd, J=14.48, 4.87 Hz, 2H) 3.69 (s, 2H) 3.87 (s, 2H) 4.14 (s, 1H) 6.99 (td, J=6.07, 2.54 Hz, 3H) 7.11-7.19 (m, 5H) 7.29-7.32 (m, 2H) 7.37-7.41 (m, 2H) 7.76 (d, J=9.06 Hz, 2H) 8.07 (dd, J=9.19, 2.33 Hz, 1H) 8.29 (d, J=2.20 Hz, 1H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping