| 45.7% |
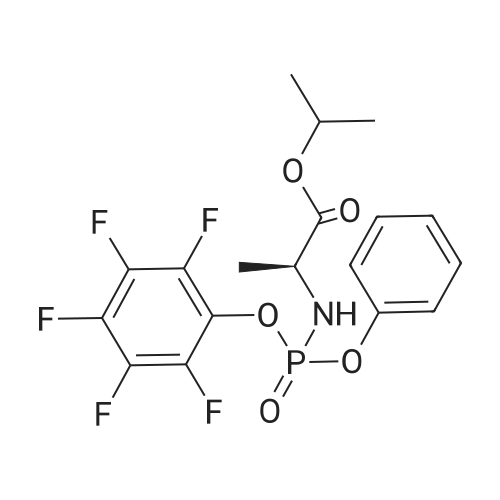
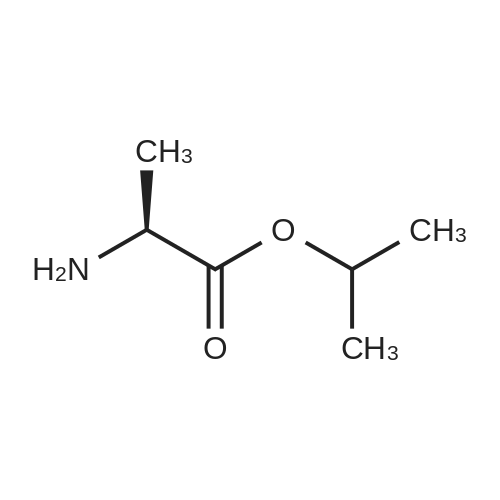
Stage #1: O-phenyl phosphorodichloridate; alanine isopropyl ester hydrochloride With 1-methyl-1H-imidazole In dichloromethane at -5 - 5℃; for 1.5h;
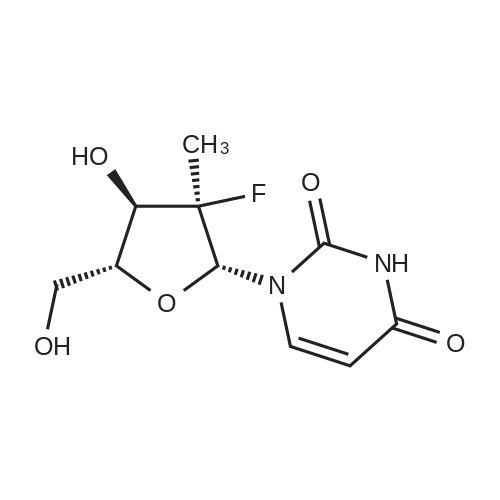
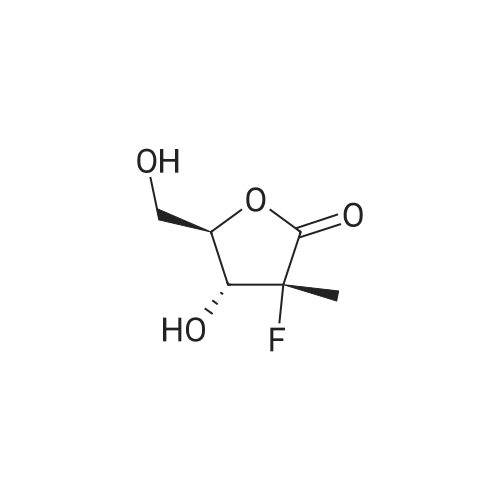
Stage #2: 2'-deoxy-2'-fluoro-2'-methyluridine In dichloromethane at 0 - 20℃; for 5h; |
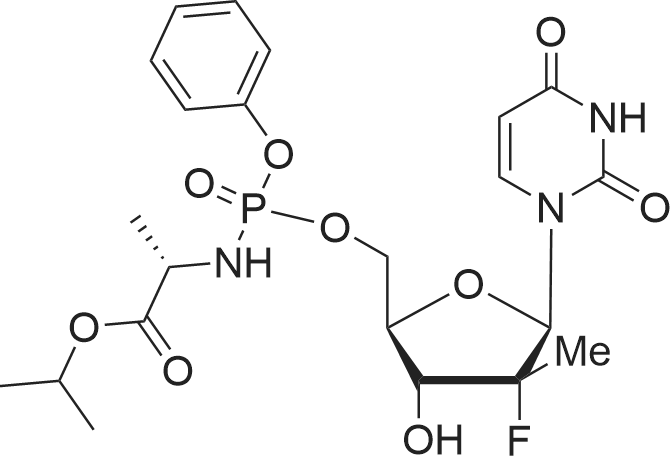
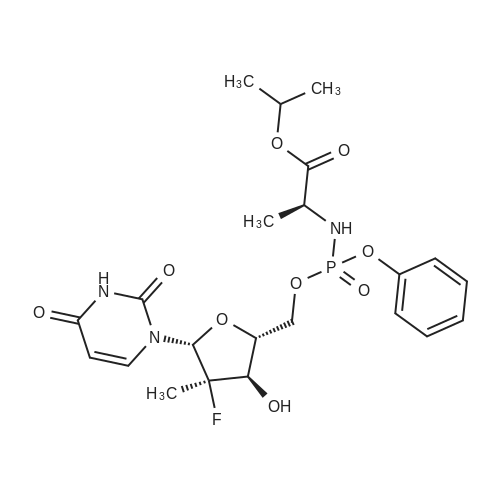
9 Preparation of (S)-2-[(1 R,4R,5R)-5-(2,4-Dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-4-(R)-fluoro-3- hydroxy-4-methyl-tetrahydro-furan-2-yl-methoxy]-phenoxyphosphorylamino}-propionic acid isopropyl ester (from US2010/0298257 Example 2)
A 5 L 3-necked flask was fitted with a mechanical stirrer, brine ice bath, internal thermometer, and a nitrogen atmosphere. The flask was charged with L-alanine isopropyl ester hydrochloride (82.0 g, 0.490 moles) and anhydrous dichloromethane (0.80 L). While this was stirring, phenyl dichlorophosphate (85.0 g, 0.40 moles) was added in one lot and stirred. While maintaining the internal temperature between -5 to 5 °C, a solution of N-methylimidazole (N I, 250 g, 3.07 moles) in dichloromethane (250 mL) was added over a period of a half hour. The solution was allowed to stir for 1 h in this temperature range. 2'-Deoxy-2'-fluoro-2'-C-methyl-uridine (3,80.0 g, 0.307 moles) was added at 0° C. in one portion and then the reaction flask was allowed to warm up slowly in the brine bath. At 1 h, the internal temperature was up to -2° C. TLC (5% Methanol in HCL) at 1 h showed that more than 50% of nucleoside was consumed. The bath was removed and the reaction flask reached ambient temperature over 1 h more. TLC after 3 h and at 5 h total showed 95% of the starting nucleoside was consumed. The reaction mixture was quenched by adding methanol (100 mL) and stirring the reaction for 5 minutes. The reaction mixture was washed with 1 HCI (2x500 mL) followed by saturated sodium bicarbonate solution (2x500 mL). The separated organic layer was dried over anhydrous sodium sulfate (50 g) and filtered. The solution was evaporated under reduced pressure and then under high vacuum to dryness to give the crude product as a viscous oil (170 g). NMRs of the crude product (31P and 1H) were taken. The 31P-NMR indicated about 1% of the total phosphorus integration was due to the presence of the 3' isomer 5. To the crude product was added anhydrous pyridine (1700 mL). The solvent was evaporated under reduced pressure and then under high vacuum in order to reduce the water content of the crude mixture through co-evaporation. The resulting oil was re-dissolved in anhydrous pyridine (500 ml) and then was added excess t-butyldimethylsilyl chloride (9.0 g, 60 m ). The reaction was stirred at ambient temperature. Reaction progress was monitored by UPLC/MS. After 3 hours, the 3' impurity 5 could no longer be detected and the reaction was quenched by the addition of methanol (50 mL). The reaction was evaporated under reduced pressure to an oil. The residue was dissolved in ethyl acetate (1.5 L) and washed with 1 N HCI (2x500 mL), followed by saturated sodium bicarbonate solution (2x500 mL). The organic layer was dried over anhydrous sodium sulfate (50 g), filtered and evaporated under reduced pressure to give the crude product as a pale yellow oil. The crude oil was diluted with the same volume of dichloromethane and loaded onto a 2.5 Kg silica gel cartridge n a radial compression module at 100 psi of air pressure. Using a gradient pump at 60 psi and a flow rate of 400 ml/min, the cartridge was washed with methylene chloride (4L) followed by a gradient 1-4% methanol in methylene chloride (48 L). Most of the major impurities (di-(isopropylalanyl) phenyl phosphate, 3',5'-bis phosphoramidate, 3'- phosphoramidate-5'-TBDMS adduct (7)) eluted with ~3% gradient. The desired product eluted between 3 and 4% methanol. The product containing fractions were sorted into two lots. The first contained small amounts of upper impurities and the latter was pure product. The first set of fractions contained small amounts of less polar impurities (upper impurities) such as the 3', 5'- bis phosphoramidate and the di-alanylphenyl phosphate and a mostly the Rp diastereomer and required a second column purification. (The relative terminology, upper vs. lower refers to the elution on normal phase silica-gel chromatography, where the "upper isomer" means the first eluting isomer.) The second set of fractions did not have a significant amount of impurities-just the remaining Rp and mostly the Sp diasterereomers. It was later recombined with the twice- columned fractions. The solvent was evaporated under reduced pressure and the resulting white foam was further dried (0.20mmHg) for 1 h to give 42 g of the impure lot (4:1 upper vs lower isomer based on 31P-NMR) and 38 g of the pure lot (1 :3 upper vs lower isomer). The impure lot was recolumned in a similar manner to give 3.8 g of 97% pure upper isomer (fraction set aside) and 36 g ofpure product in a 4: 1 ratio. The two main lots were dissolved in HCL, combined, evaporated under reduced pressure and dried (50° C, 0.2 mmHg, 24 h) to get 74 g (45.7%) of pure product (Compound 9) with a diastereomeric ratio of 48:51 , as a white foam, mp about 75-85° C. In order to produce an amorphous solid of the diastereomeric mixture, 74 g of the white foam was stirred in with t-butyl methyl ether (750 mL) resulting in a partial solution and a gummy solid residue. While stirring, heptanes (750 mL) was added slowly and the suspension was mechanically stirred for 1 hour until most of the gum was converted to a white solid. The solid was scraped up with a spatula and the resulting slurry was filtered. The solid was washed with heptanes (4x50mL) and dried under vacuum (50° C, 0.2mmHg, 24 h) to give a white, amorphous powder (64 g) with a broad melting range of ca 70-80° C. H and 31P NMR conformed to structure and HPLC showed a purity of 99.8% with a diastereomeric ratio of 46:54 (also confirmed by 3 P NMR). Alternative method to make a solid mixture of Compound 9. After chromatography, the residue was co-evaporated with dichloromethane twice (5 mlJg) and dried for 24 h at 35-40° C. at 35- 45 mTorr. The foam residue was sieved through a 250 micron screen and further dried under the same conditions until the residual dichloromethane fell below 400 ppm as measured by headspace GC. The resulting fine off-white to white amorphous powder has a glass transition temperature range of 53.7 to 63.5° C. Characterization of Compound 9 (mixture of isomers): 1H-NMR (CDCI3) 010.05 (brs, 1 H, NH, Sp), 10.00 (brs, 1 H, NH, Rp), 7.49 (d, 1 H, C6-H, Sp), 7.36 (m, 5H, C6-H, Rp, aromatic), 7.23-7.14 (m, 6H, Rp/Sp, aromatic), 6.18 (br d, 2H, Cl'-H, Rp/Sp), 5.63 (d, 1 H, C5-H, Sp), 5.58 (d, 1 H, C5-H, Rp), 5.01 (m, 2H, CH-(CH3)2 Rp/Sp), 4.46- 4.33 (m, 8H, C-5'-H2 , ala-NH, C3'-OH, Rp/Sp), 4.12 (m, 2H, ala-CHCH3, Rp/Sp), 4.01-3.85 (m, 4H, C3'-H, C4'-H, Rp/Sp), 1391.22 (m, 12H, all CH3, Rp/Sp). 31P-NMR (CDCI3) 03.60 (Rp ), 3.20 Sp relative to triphenylphosphate at -17.80 ppm. ES-MS M+1 530.2. Elemental Analysis: Calculated % (including 0.29% water as found by Karl Fisher analysis) C, 49.75; H, 5.54; N, 7.90, F, 3.58, P, 5.84. Found %: C, 49.50; H, 5.44; N, 7.85; F, 3.62; P, 6.05. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping