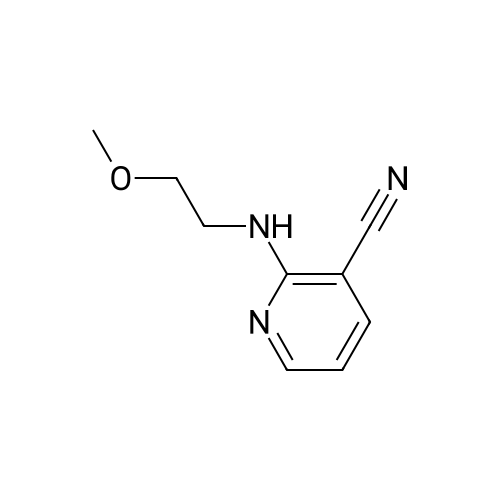
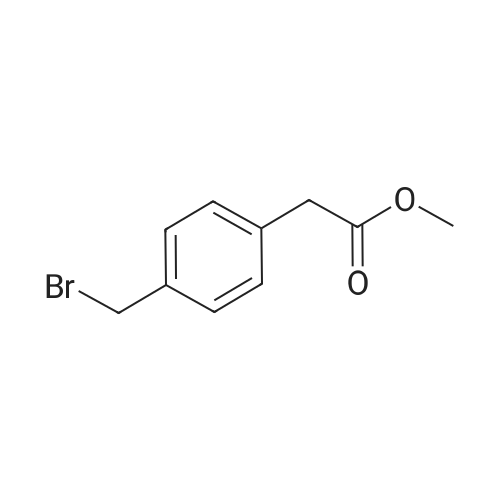
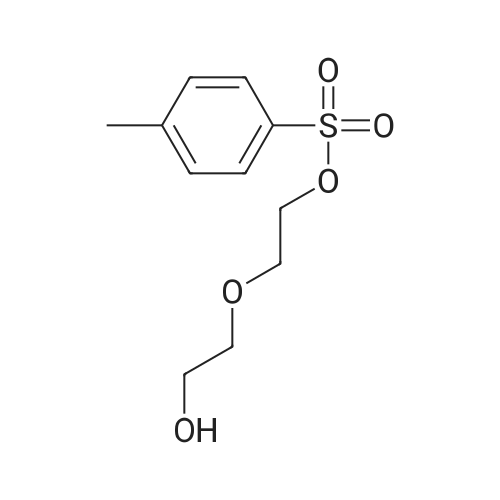
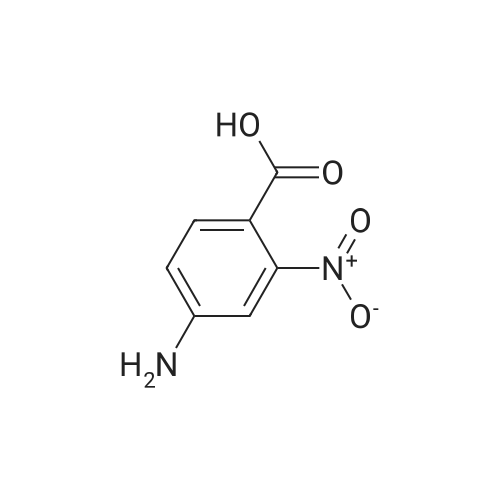
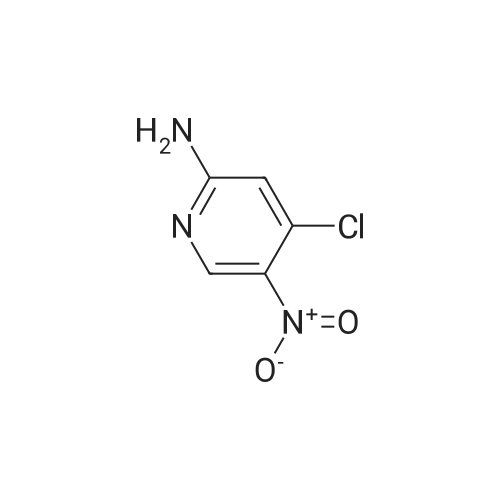
| 86% |
Stage #1: With sodium hydroxide In 2-methyltetrahydrofuran at 20 - 25℃; for 1 h; Large scale
Stage #2: With triethylamine In 2-methyltetrahydrofuran at 0 - 25℃; for 6 h; Large scale
Stage #3: at 50 - 55℃; for 13 h; Large scale |
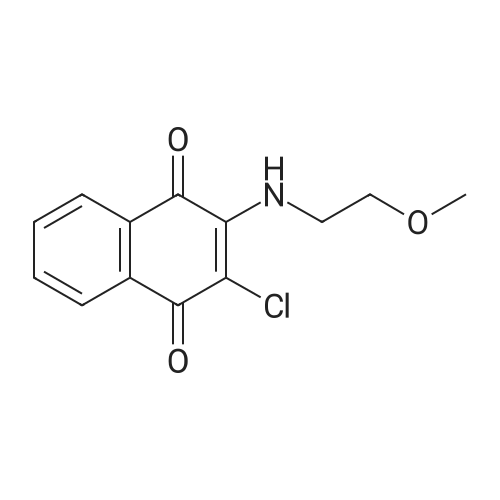
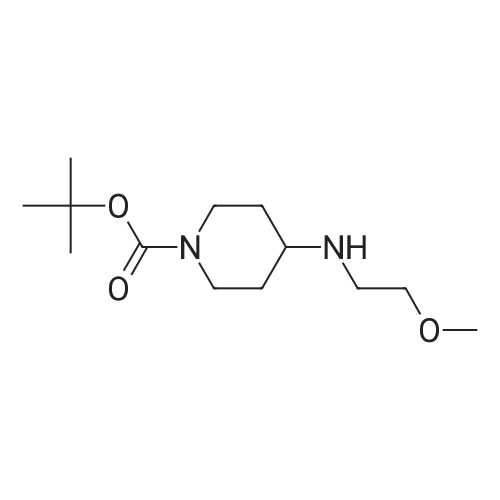
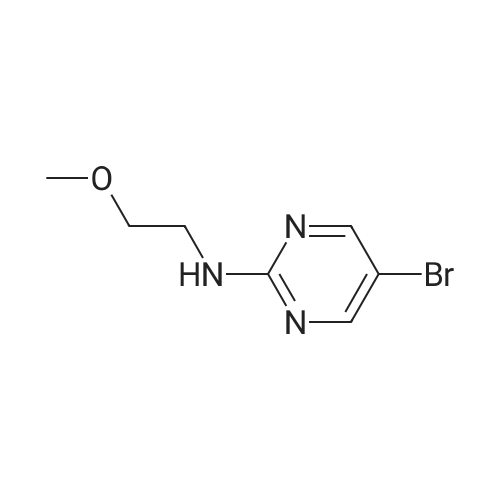
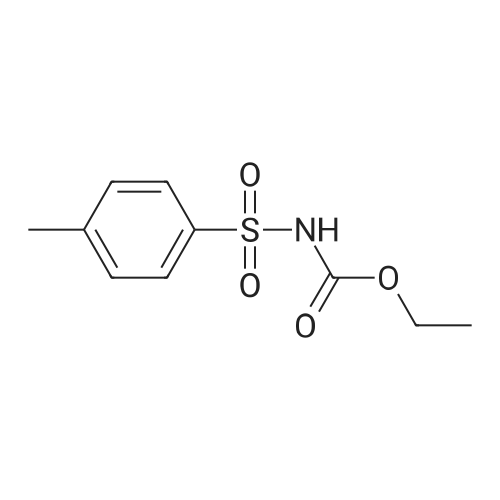
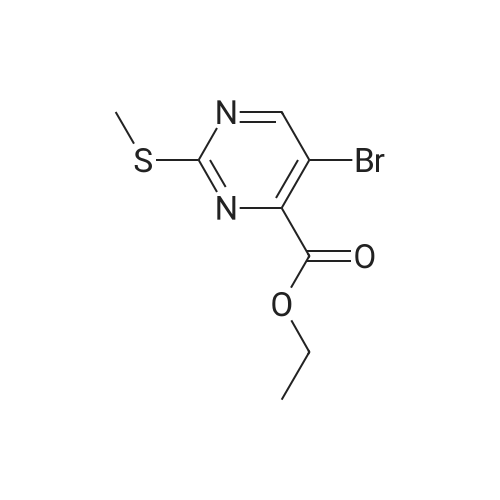
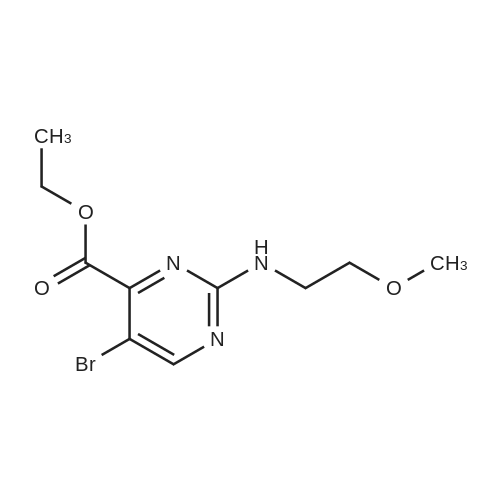
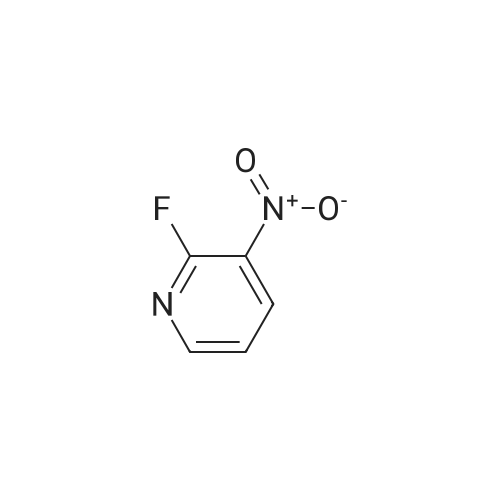
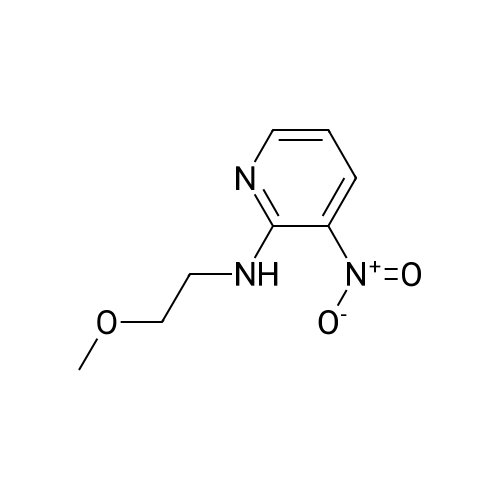
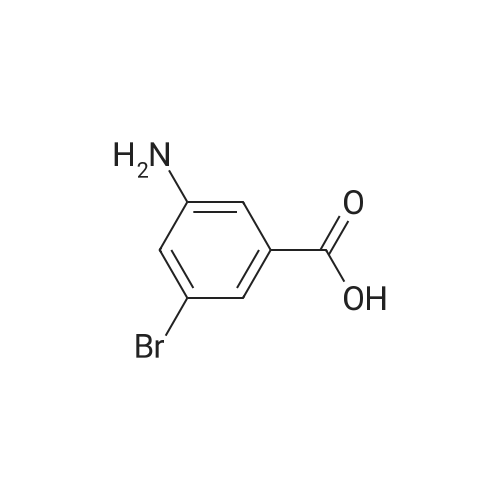
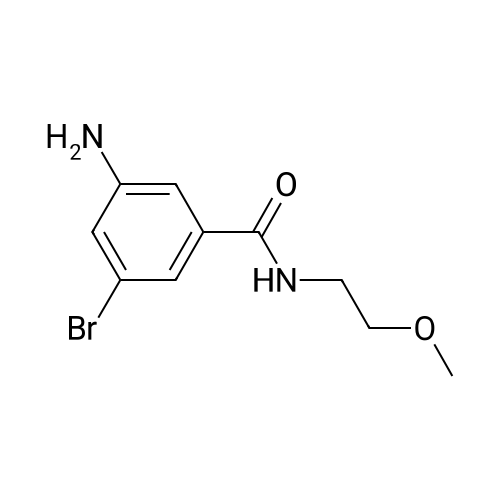
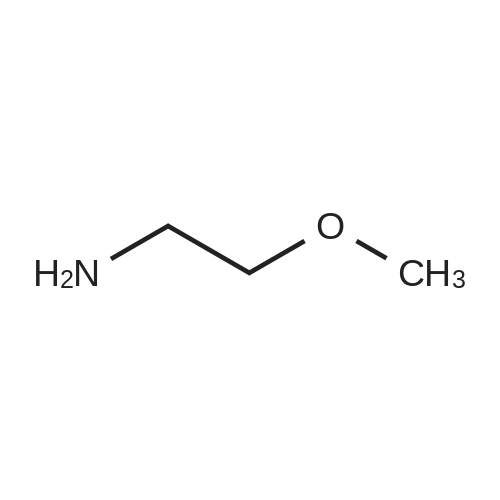
Starting material: Compound 4 Methanesulfonyl chloride (MsCl) [CAS 124-63-0] 2-Methoxyethylamine [CAS 109-85-3] Reagents: Sodium Chloride (NaCl) 50percent Sodium Hydroxide (NaOH) Triethylamine (Et3N) (0258) Solvents: n-Heptane Isopropyl Acetate (zPrOAc) 2-Methyl Tetrahydrofuran (2-MeTHF) A 100-gallon reactor was charged with Compound 4 (10.4 kg, 23.2 mol, 1.0 eq) and 2- methyltetrahydrofuran (2-MeTHF, 132.6 kg, 155.2 L, 15 vol). A solution of 1.0 M NaOH (48.5 L, 48.5 mol, 2.1 eq) was added in one portion to the slurry and the resulting biphasic mixture was allowed to stir at 20-25 °C for 1.0 h. The phases were allowed to settle, the lower aqueous layer was removed and the organic layer was washed with 2.5percent NaCl (52 L, 5 vol). The organic layer was concentrated down to 104 L (10 vol) and chased with 2-MeTHF (44.0 kg, 51.5 L, 5 vol) a total of five times to achieve the desired water content of <0.1percent (0.08percent). After pohsh-filtering the 2-MeTHF solution into a clean 100-gallon reactor, triethylamine (Et3N, 3.5 kg, 4.9 L, 34.8 mol, 1.5 eq) was added and the mixture was cooled to 0-5 °C. Methanesulfonyl chloride (MsCL 4.0 kg, 2.7 L, 34.8 mol, 1.5 eq) was added over a period of 1 h while keeping the internal temperature < 20 °C. Once the addition of MsCl was complete, the reaction temperature was adjusted to 20-25 ''C and the mixture was stirred for 2 h. Analysis by HPLC indicated the presence of 3.7percent Compound 4. Additional Et3N (0.4 kg, mL, 0.55 L, 4.0 mol, 0.2 eq) and MsCl (0.4 kg, 0.27 L, 3.5 mol, 0.15 eq) were charged and the mixture was stirred at 20-25 °C for 1.5 h. At this point, 0.57percent Compound 4 was detected by HPLC. Additional Et3N (0.1 kg, mL, 0.14 L, 1.0 mol, 0.05 eq) and MsCl (0.1 kg, 0.07 L, 1.0 mol, 0.05 eq) were charged and the mixture was stirred at 20-25 °C for 1.5 h. Water (93.5 kg, 9 vol) was added and the biphasic mixture was stirred for 2.5 h. The phases were allowed to settle for 1 h and the aqueous layer was then transferred to a clean 200-gallon reactor. The aqueous layer was back-extracted with 2-MeTHF (44.6 kg, 52.2 L, 5 vol) and the upper layer was transferred to the 100-gallon reactor to combine organic layers before being washed with 5percent NaCl (51.6 kg, 5 vol). The resulting 2-MeTHF solution was concentrated down to -104 L (10 vol) and then chased with 2-MeTHF (44.0 kg, 51.5 L, 5 vol) a total of five times to achieve the desired water content of <0.1percent (0.02percent). After polish-filtering the 2-MeTHF solution into a clean 100-gallon reactor, the solution containing Compound 5 was concentrated down to 52 L (5 vol). 2-methoxyethylamine (35.8 kg, 41.4 L, 4 vol) was added, and the resulting reaction mixture was heated to 50-55 °C. The reaction mixture was allowed to stir at temperature for 13 h and HPLC analysis indicated complete conversion. Once the transformation was deemed complete, isopropylacetate (iPrOAc, 117.8 kg, 135L, 13 vol) and water (104 kg, 10 vol) were charged to the reactor while maintaining a temperature of 50-55 °C. After stirring for 1.5 h, the water layer was transferred to a clean 200-gallon reactor and extracted with iPrOAc (61.8 kg, 70.9 L, 7 vol). The upper layer was transferred to the 100- gallon reactor to combine organic layers and then re-equilibrated at 50-55 °C. The combined organic layer was washed with water (4x20.8 kg, 4x2 vol) before being vacuum distilled down to 63L (6 vol). The resulting slurry was chased with w-heptane (3x85.0 kg, 3x124 L, 3x12 vol) down to ~6 vol to achieve < 8.5 wtpercent of residual iPrOAc (1.1 wtpercent). The slurry was diluted with n-heptane (42.7 kg, 62.4 L, 6 vol) and stirred at 20-25 °C for 16.0 h before being filtered. The filter cake was washed with heptane (2x28.4 kg, 2x41.5 L, 2x4 vol) and then dried at 40-45 °C for 30 h. Compound A was obtained (9.4 kg, 86percent yield, 96.6percent AUC by HPLC) as a cream colored solid. Example 4. Preparation of ARQ 087-2 HC1 Crystalline Form D |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping