Alternatived Products of [ 166734-83-4 ]
Product Details of [ 166734-83-4 ]
| CAS No. : | 166734-83-4 |
MDL No. : | N/A |
| Formula : |
C7H10ClN3O3
|
Boiling Point : |
- |
| Linear Structure Formula : | - |
InChI Key : | - |
| M.W : |
219.63
|
Pubchem ID : | - |
| Synonyms : |
Levornidazole;(S)-Ornidazole
|
Chemical Name : | (S)-1-Chloro-3-(2-methyl-5-nitro-1H-imidazol-1-yl)propan-2-ol |
Safety of [ 166734-83-4 ]
Application In Synthesis of [ 166734-83-4 ]
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
- Downstream synthetic route of [ 166734-83-4 ]
- 1
-
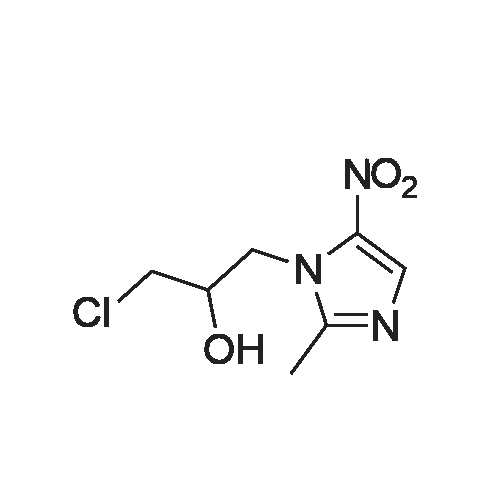 [ 16773-42-5 ]
[ 16773-42-5 ]

-
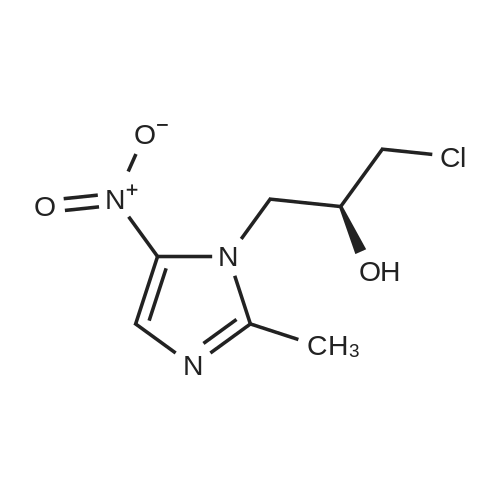 [ 166734-83-4 ]
[ 166734-83-4 ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 2 steps
1: lipase Amano PS / 384 h / Ambient temperature
2: 60 percent / conc. HCl / 5 h |
|
|
Stage #1: ornidazole In ethanol; water at 70℃; for 0.5h;
Stage #2: With hydrazine In ethanol; water at 3℃; for 3h; |
2 example 2
Crystalline α L-ornidazole preparation method is as follows: the 5.0g dissolved in L-ornidazole 15ml75% aqueous solution of ethanol, water bath heated to 50 ±3 °C, stirring to complete dissolution, the solution is transferred to the temperature in the constant temperature water bath 20 ±2 °C 30 minutes, then agitating leng Jing devitrify 3 ±2 °C 3 hours. After filtration, the filtration cake at the hot air drying under 45 ±3 °C 8 hours to obtain 4.0g crystalline α crystals of L-ornidazole, detection HPLC purity of 99.96%. |
Reference:
[1]Skupin, Rolf; Cooper, Trevor G.; Froehlich, Roland; Prigge, Joerg; Haufe, Guenter
[Tetrahedron Asymmetry, 1997, vol. 8, # 14, p. 2453 - 2464]
[2]Current Patent Assignee: SHANDONG QIDU PHARMACEUTICAL - CN105777648, 2016, A
Location in patent: Paragraph 0030; 0031; 0032; 0033
- 2
-
[ 166734-83-4 ]

-
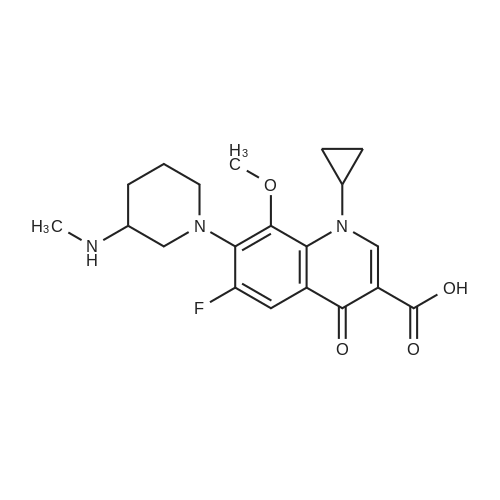 [ 127294-70-6 ]
[ 127294-70-6 ]
-
 [ 1643323-96-9 ]
[ 1643323-96-9 ]
| Yield | Reaction Conditions | Operation in experiment |
| 68% |
With potassium hydroxide In ethanol; water for 12h; Reflux; |
|
Reference:
[1]Li, Qing; Xing, Junhao; Cheng, Haibo; Wang, Hui; Wang, Jing; Wang, Shuai; Zhou, Jinpei; Zhang, Huibin
[Chemical Biology and Drug Design, 2015, vol. 85, # 1, p. 79 - 90]
- 3
-
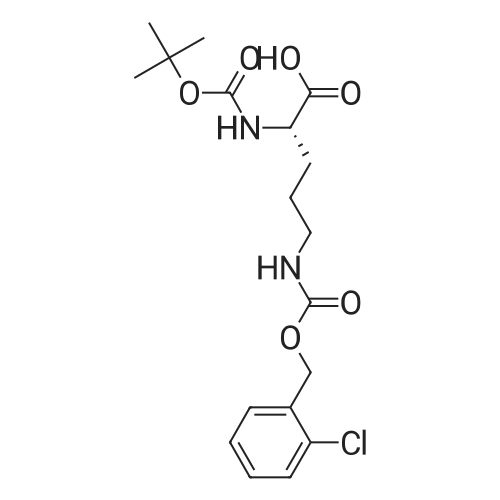 [ 118554-00-0 ]
[ 118554-00-0 ]
-
[ 166734-83-4 ]
-
[ 2088486-60-4 ]
| Yield | Reaction Conditions | Operation in experiment |
| 63.1% |
Stage #1: N-tert-butoxycarbonyl-N'-(2-chlorobenzyloxycarbonyl)-L-ornithine; (S)-(-)-1-(3-chloro-2-hydroxypropyl)-2-methyl-5-nitroimidazole With dmap; dicyclohexyl-carbodiimide In dichloromethane at 20℃;
Stage #2: With hydrogenchloride In 1,4-dioxane at 20℃; for 1h; |
9 Synthesis of (S)-3-chloro-1-(2-methyl-5-nitro-1H-imidazol-1-yl)propane-2-yl-2,5-diaminovaleric acid hydrochloride (Compound 4)
In the water bath, N-tert-butoxycarbonyl-N'-(2-chlorobenzyloxycarbonyl)-L-ornithine (22 mmol, 7.5 g) N, N'-dicyclohexylcarbodiimide (4. 4 mmol, 0.91 g), 4-dimethylaminopyridine (2. 2 mmol, 0.3 g) And stirred with anhydrous dichloromethane (30 ml). Followed by addition of L-ornidazole (26 mmol, 5.7 g) and stirred overnight at room temperature. The insoluble matter was removed by filtration, and the filtrate was evaporated to dryness. The ether was added to the residue and the insoluble impurities in the solution were removed by filtration. The filtrate was concentrated and the column was rapidly passed through ether and the product fractions were collected. The product was concentrated under reduced pressure.The obtained product was directly added with 4M HCl / dioxane solution (20 ml) and stirred at room temperature for 1 h. The solvent was distilled off under reduced pressure and concentrated to dryness. The residue was isolated by HPLC (TFA) system to give product 3. 6 g; compound 9 yield 63. 1%. |
- 4
-
 [ 123-41-1 ]
[ 123-41-1 ]
-
[ 166734-83-4 ]
-
[ CAS Unavailable ]
| Yield | Reaction Conditions | Operation in experiment |
| 79% |
Stage #1: (S)-(-)-1-(3-chloro-2-hydroxypropyl)-2-methyl-5-nitroimidazole With trichlorophosphate In acetonitrile at 45 - 55℃; for 48h;
Stage #2: cholin hydroxide In ethanol |
6 Example 6: Preparation of Nitroimidazole Compound II Choline Salt
Add 200ml of acetonitrile, 17g of phosphorus oxychloride and 50g of levornidazole to the reaction flask,Turn on the stirring, heat up to 45-55°C and react for 48 hours. After the reaction is over,Add 50g purified water, raise the temperature to 60-70°C, hydrolyze for 24 hours,Concentrate under reduced pressure to dryness, add 1000ml ethanol to the residue to dissolve, filter, and adjust the pH of the filtrate to 7-8 with 30% choline hydroxide.After filtration, the filtrate was concentrated to dryness under reduced pressure, and the residue was heated to 50-60°C with 150ml ethanol/water (2/1) to dissolve by heating.After dissolving, add 1.5g activated carbon, stir and decolorize for 10 minutes, filter, and cool the filtrate to -5-0°C, stir and crystallize for 24 hours.After filtration, the filter cake was dried at 20°C to 30°C for 2 to 4 hours to obtain 39.5 g of nitroimidazole compound II choline salt, with a yield of 79.0%. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping


