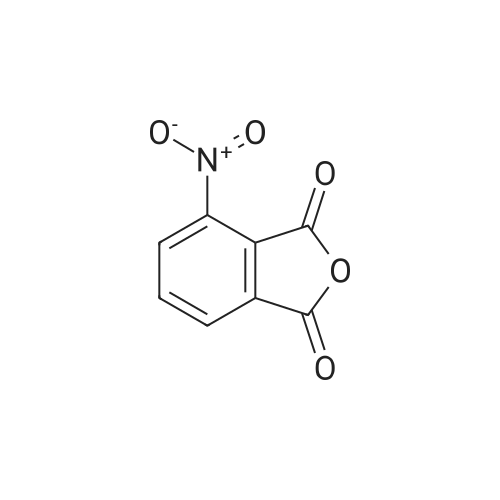
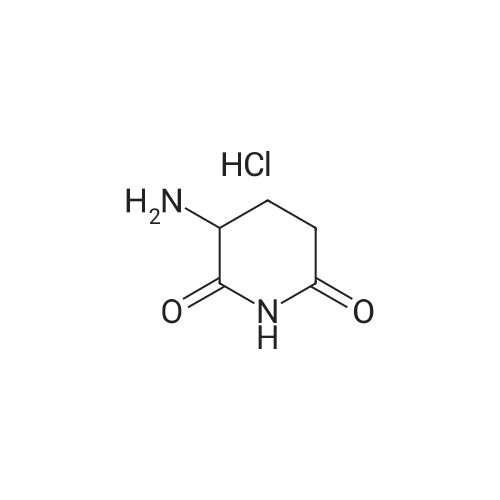
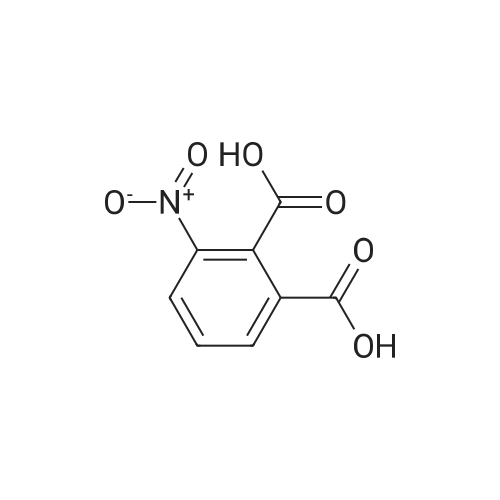
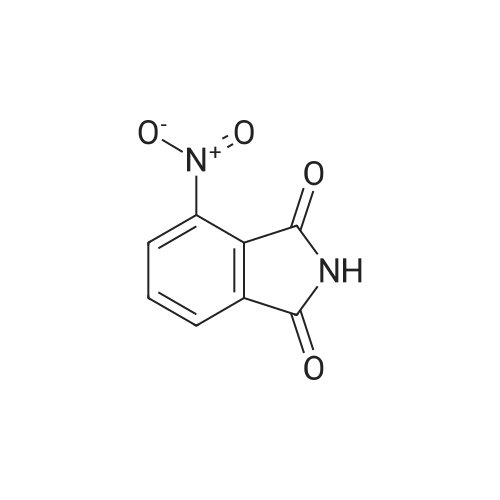
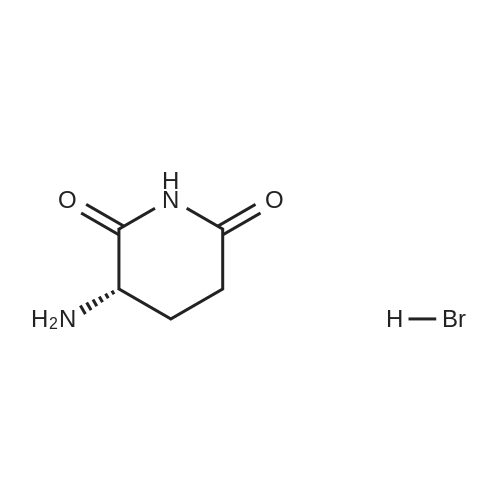
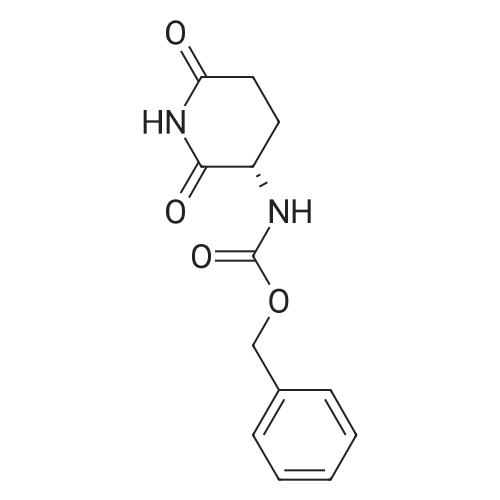
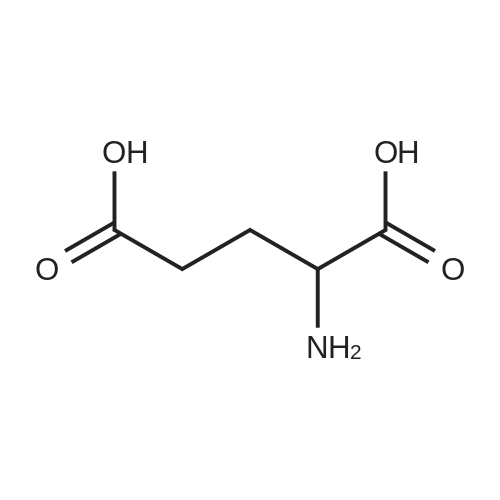
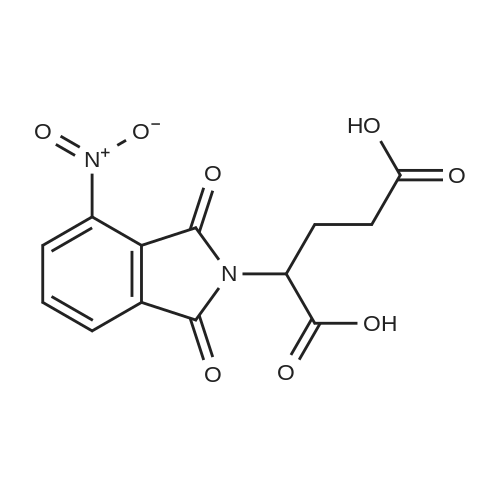
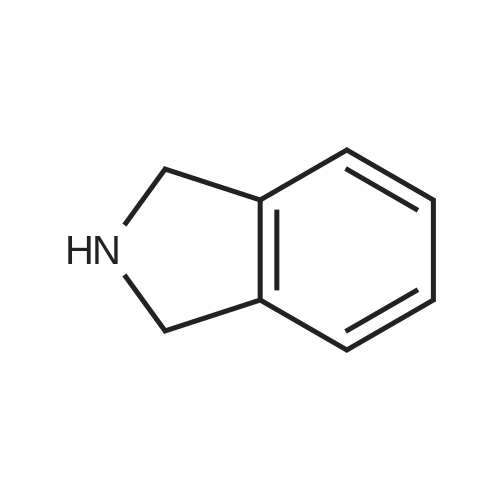
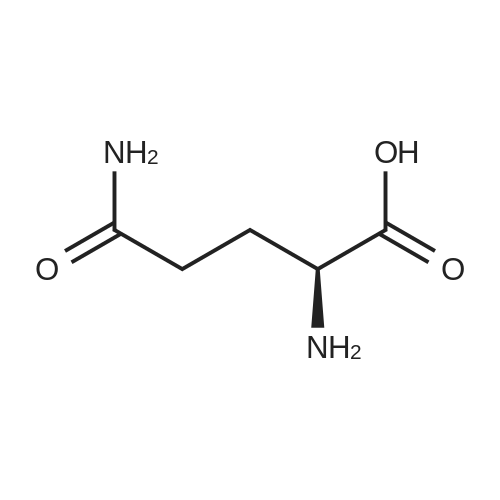
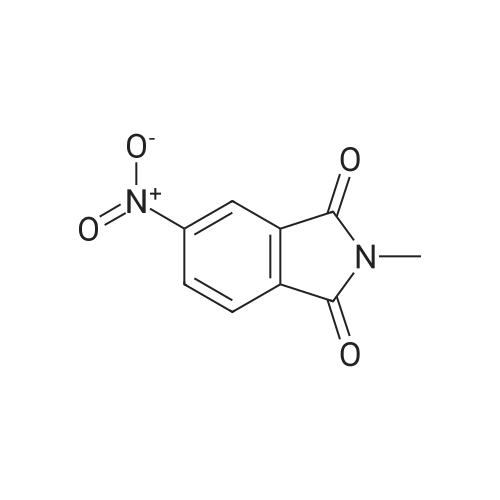
| 91% |
With palladium on activated charcoal; hydrogen; In 1-methyl-pyrrolidin-2-one; at 45℃; under 2250.23 Torr; |
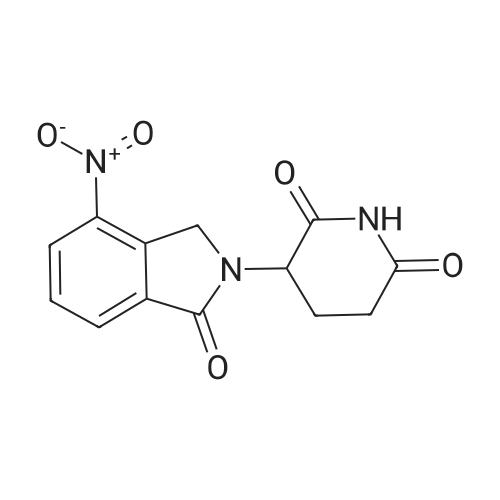
(1) 100g 3-nitro-N-(2,6-dioxo-3-piperidinyl)phthalimide, 10g of the palladium-carbon and 1000 ml N-methylpyrrolidone were added into a 2000 ml hydrogenation reactor. Vacuum. After nitrogen replacement 3 times, place hydrogen gas, regulating the reactor pressure to 0.3 MPa, control temperature at 45 C. After the reaction is complete, filtering, the filtrate by adding 30g charcoal, heating up to 70 C, stirring 3 hours, filtered;(2) to the step (1) and the finally obtained by adding sodium carbonate aqueous solution in the filtrate (2g sodium carbonate dissolved in 10 ml purified water to prepare water), stirring at room temperature for 0.5 hours, then slowly dropping 2000 ml purified water, lowering the temperature to 10 C crystallization, filtered, the filter cake is added to 500 ml of purified water, stirring at room temperature for 0.5 hours, filtered, washed with water purification cake, solid 60 C obtained by vacuum drying the yellow powdery solid 82g, yield 91%. HPLC detection the purity is 99.88%. |
| 80% |
With palladium 10% on activated carbon; hydrogen; In N,N-dimethyl acetamide; at 30℃; for 7h; |
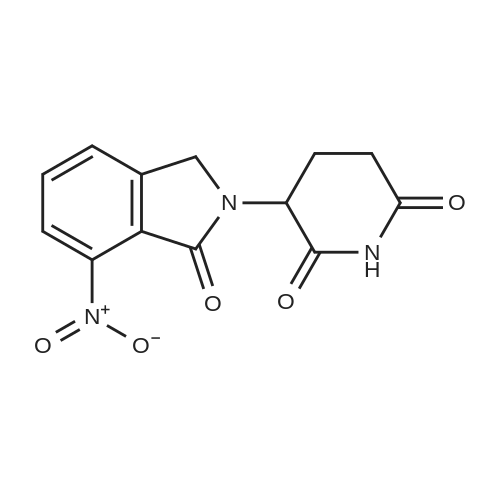
Add 5 g of 1,3-dioxo-2-(2,6-dioxopiperidin-3-yl)-5-nitroisoindoline and 0.4 g of 10% palladium carbon to 50 mL of N,N - in dimethylacetamide,The hydrogenation reaction is then carried out at 0.45 MPa at 30 C.Lasted 7 hours,Filter to remove the catalyst,The filtrate was concentrated under vacuum.The concentrate was beaten with water overnight.Obtained 3.6 g of a yellow solid.That is, the target compound.Yield 80%,The purity measured by HPLC was 99.4%. |
| 79.5% |
With palladium 10% on activated carbon; hydrogen; In 1,4-dioxane; at 60 - 65℃; for 6h; |
In a mixer equipped with a mechanical stirrer, A thermometer and a ventilator was charged with 31.7 g (wet product 48.2 g, 0.105 mol, 1.0 eq) 3-nitro-N-(2,6-dioxo-3-piperidinyl) phthalimide,1.6 g of 10% palladium on carbon and 395 mL of solvent 1,4-dioxane,Stirring to form a suspension of hydrogen gas bubbling, bubbling speed 1 times / second, the temperature of the reaction solution to 60 ~ 65 C, continue stirring for 6 hours, HPLC control: to be raw materials and intermediate content <0.5% The filtrate was concentrated under reduced pressure and 158 mL of ethyl acetate was added to the residue. The mixture was stirred for 2 hours and then filtered. The cake was transferred to another 500 mL reaction flask and 158 mL of N , N-dimethylformamide, heating to 55 ~ 60 C stirring, 55 ~ 60 C drop 316 mL of purified water, heating to 70 ~ 75 C for 2 hours, cooling to a temperature of 30 ~ 35 C, filter, wet goods to vacuum drying oven 50 ~ 55 C drying, 3-amino-N - (2,6-dioxo-3-piperidyl) phthalimide 22.7 G, pale yellow powdery solid, yield 79.5%, m.p. 315.0~318.0 C (DSC). The purity of the product was 99.73% by HPLC, with a single impurity <0.10%, of which 13% UV retention time = 11.5 min.), And using 0.1% (v (v / v)) as the mobile phase, / Nu) phosphoric acid aqueous solution and acetonitrile as a mobile phase at a flow rate of 1.0 ml / min. |
| 77.7% |
With hydrogen; In 1,4-dioxane; water; at 20 - 25℃; under 3620.13 Torr; for 60h; |
A round bottomed flask was charged with 1,4-dioxane (600 ml) and 2-(2,6-dioxopiperidin-3-yl)-4-nitroisoindoline-1,3-dione (10 g). The mixture was stirred at 25 C. to 30 C. for 15 minutes, the temperature was raised up to 80 C. and the reaction mass stirred to obtain a clear solution. To the clear solution activated charcoal (5 g) was added at 80 C., the mixture stirred and the solution cooled to 25 C. to 30 C. The reaction mass was further stirred for 20 minutes at this temperature and filtered. To the obtained solution 1,4-dioxane (300 ml), water (180 ml) and Raney nickel (0.5 g) were added. The reaction mixture was stirred at 20 C. to 25 C. under 70 psi hydrogen pressure for 12 hrs. Raney nickel (0.5 g) was added to the reaction mixture at 20 C. to 25 C. and the reaction continued at the same temperature and pressure for 12 hrs. Another portion of Raney nickel (0.5 g) was added to the reaction mixture and the reaction continued at the same temperature and pressure for 12 hr. Another portion of Raney nickel (0.5 g) was added to the mixture and the reaction continued at the same temperature and pressure for 12 hr. Another portion of Raney nickel (0.5 g) was added to the reaction mixture and the reaction continued at the same temperature and pressure for 12 hr. The reaction mixture was then filtered through a celite bed and the solvent was distilled off from the reaction mixture under vacuum at 50 C. to obtain a solid. To the solid obtained 1,4-dioxane (50 ml) and ethyl acetate (5 ml) were added and the mixture was heated to 80 C. with stirring for 1 hr. The reaction mass was filtered at 80 C. and the obtained cake washed with 1,4-dioxane (10 ml) at 80 C. To the wet cake ethyl acetate (100 ml) was added and the mixture was refluxed with stirring for 1 hr. Then the reaction mass was cooled to 25 C. to 30 C., the solid filtered, washed with ethyl acetate (20 ml) and dried under vacuum to get 4-amino-2-(2,6-dioxopiperidin-3-yl) isoindoline-1,3-dione (7 g). Yield: 77.7% Purity: 99%. |
| 64.5% |
With palladium 10% on activated carbon; hydrogen; In N,N-dimethyl acetamide; at 40℃; for 16h; |
To a solution of 2-(2,6-dioxo-3-piperidyl)-4-nitro-isoindoline-1,3-dione (43.0 g, 142 mmol) in DMA (1.5 L) was added Pd/C (15.0 g, 14.1 mmol, 10% Pd) under a blanket of N2. The mixture was degassed under vacuum and purged with H2 (3*). The purged mixture was stirred at 40 C. under an atmosphere of H2 (40 psi) for 16 hours. The reaction mixture was purged with N2, filtered and the filtrate was concentrated under vacuum. The residual solid was washed with ethyl acetate (200 mL) and dried under vacuum to give 4-amino-2-(2,6-dioxo-3-piperidyl)isoindoline-1,3-dione as a yellow solid (75 g, 64.5% yield). 1H NMR (400 MHz DMSO-d6) delta ppm 11.07 (s, 1H), 7.46 (d, J=6.4 Hz, 1H), 7.00 (t, J=7.0 Hz, 2H), 6.51 (br s, 2H), 5.06-5.01 (m, 1H), 2.93-2.89 (m, 1H), 2.60-2.49 (m, 2H), 2.03-2.00 (m, 1H). |
| 46% |
With triethylsilane; potassium fluoride; palladium diacetate; In tetrahydrofuran; water; at 20℃; for 1h; |
A solution of 2-(2,6-dioxopiperidin-3 -yl)-4-nitroisoindoline- 1,3 -dione (173 mg, 0.854 mmol), Pd(OAc)2 (12.8 mg, 0.0854 mmol, 10 mol%) and potassium fluoride (66 mg, 1.71 mmol, 2 equiv) in THF:water (8:1) (5.7 mL, 0.1 M) was stirred at room temperature. Triethylsilane (3651iL, 3.41 mmol, 4 equiv) was added slowly, and the resulting black solution was stirred at room temperature for 1 hour. The reaction mixture was filtered through a pad of celite, which was washed excessively with ethyl acetate. The filtrate was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel (CH2C12:MeOH (7:1)) to afford the titlecompound as a yellow powder (72 mg, 46%). 1H NMR (500 IVIHz, DMSO-d6) 11.08 (s, 1H),7.47 (dd, J 8.5, 7.0 Hz, 1H), 7.06 - 6.95 (m, 1H), 6.59 -6.44 (m, 1H), 5.04 (dd, J= 12.7, 5.4Hz, 1H), 2.93 - 2.82 (m, 1H), 2.64 - 2.45 (m, 2H), 2.05 - 1.98 (m, 1H); MS (ESI) calcd forC13H11N304 [M+H] 274.08, found 274.23. |
| 46% |
With triethylsilane; potassium fluoride; palladium diacetate; In tetrahydrofuran; water; at 20℃; for 1h; |
A solution of 2-(2,6-dioxopiperidin-3-yl)-4-nitroisoindoline-1,3-dione (173 mg, 0.854 mmol), Pd(OAc)2 (12.8 mg, 0.0854 mmol, 10 mol%) and potassium fluoride (66 mg, 1.71 mmol, 2 equiv) in THF:water (8:1) (5.7 mL, 0.1 M) was stirred at room temperature. Triethylsilane (365 muL, 3.41 mmol, 4 equiv) was added slowly, and the resulting black solution was stirred at room temperature for 1 hour. The reaction mixture was filtered through a pad of celite, which was washed excessively with ethyl acetate. The filtrate was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel (CH2Cl2:MeOH (7:1)) to afford the title compound as a yellow powder (72 mg, 46%). 1H NMR (500 MHz, DMSO-d6) delta 11.08 (s, 1H), 7.47 (dd, J = 8.5, 7.0 Hz, 1H), 7.06- 6.95 (m, 1H), 6.59- 6.44 (m, 1H), 5.04 (dd, J = 12.7, 5.4 Hz, 1H), 2.93- 2.82 (m, 1H), 2.64- 2.45 (m, 2H), 2.05- 1.98 (m, 1H); MS (ESI) calcd for C13H11N3O4 [M+H]+ 274.08, found 274.23. |
| 44.4% |
With iron; ammonium chloride; In ethanol; water; at 20℃; |
9 (410.0 mg, 1.4 mmol), NH4Cl(360.0 mg, 7.0 mmol) and iron powders (380.0 mg, 7.0 mmol) weremixed in a EtOH/H2O (v:v=7:3) solution (40 mL). The resultingmixture was kept stirring at room temperature overnight. Then saturatedNaHCO3 solution was added to the reaction mixture which wasthen extracted with ethyl acetate. The organic layer was dried oversodium sulfate, filtered and concentrated under reduced pressure togive compound 10 as a yellow powder (Yield: 163.0 mg, 44.4%). ESIMS:calcd m/z=273.07, found [M+H]+=274.07, [M+Na]+=296.05 and [2 M+Na]+=569.12 . 1H NMR (ppm,400 MHz, DMSO-d6): delta 11.11 (s, 1H), delta 7.47 (q, J=6.0 Hz, 1H), delta 7.01(q, J=6.0 Hz, 2H), delta 6.54 (s, 2H), delta 5.05 (q, J=10.0 Hz, 1H), delta 2.89(m, 1H), delta 2.57 (m, 2H), delta 2.02 (m, 1H). 13C NMR (ppm, 100 MHz,DMSO-d6): delta 173.32, 170.63, 169.04, 167.85, 147.19, 135.94, 132.47,122.18, 111.46, 108.97, 48.95, 31.46, 22.62 |
|
With palladium 10% on activated carbon; hydrogen; In methanol; dichloromethane; at 20℃; for 7h; |
3. Pomalidomide (9) A mixture of 2-(2,6-dioxopiperidine-3-yl)-4-nitrophthalimide (7) (66.0 mg, 0.2 mmol) and palladium on activated carbon (10 wt. %, 45.0 mg) in methanol (80.0 mL) was shaken under an atmosphere of hydrogen (44 lbs) at room temperature for 7.0 h. Thereafter, the catalyst was filtered through Celite and the filtrate was concentrated by rotary evaporator. The residue was purified by chromatography using CH2Cl2:methanol as the eluent to afford compound (9). |
| 136 g |
With palladium 10% on activated carbon; hydrogen; In N,N-dimethyl-formamide; at 20℃; for 3h; |
Dimethylformamide (1500 ml) was added to 3-(4-nitro-l-oxoisoindolin-2- yl)piperidine-2,6-dione (150 gm) and then stirred for 30 minutes at room temperature to obtain a clear solution. To the solution was added 10% palladium carbon and then applied 4 Kg of hydrogen pressure at room temperature. The reaction mass was stirred for 3 hours at room temperature and temperature of the reaction mass was raised to 60 to 65C. The reaction mass was filtered through hi-flow bed and then concentrated to obtain a residual solid. To the residual solid was added ethyl acetate (600 ml) and then heated to reflux. The reaction mass was maintained for 30 minutes at reflux and then cooled to room temperature. The contents were stirred for 1 hour at room temperature and filtered. The solid obtained was then dried to obtain 136 gm of pomalidomide. Chromatographic purity: 97.5%. |
| 8.2 g |
With palladium 10% on activated carbon; hydrogen; In N,N-dimethyl-formamide; under 760.051 Torr;Inert atmosphere; |
Example 3: Synthesis of 4-amino-2-(2,6-dioxopiperidin-3-yl)-isoindoIine-l,3-(lione (Pomalidomide) Catalyst palladium (10%) on carbon (0.5 g. or 5% w/w of substrate ) was added to a stirred solution of 3-nitrophthalidomide (10 g or 0.033 mole) (Example 1 ) in N,N- dimethyl formamide (100 ml) maintained under nitrogen. Hydrogen gas was bubbled through the reaction mixture at atmospheric pressure. After completion of hydrogenation reaction, as monitored by TLC, hydrogen bubbling was stopped and the catalyst was separated by filtration of the reaction mixture on celite bed. The filtrate was distilled at 60 to 65 C under reduced pressure until half of the solvent was removed. The solution was then cooled at ambient temperature and methanol (50 ml) was added while stirring. The solid obtained was filtered, washed with methanol (50 ml), and suck dried to obtain pomalidomide 8.2 g (90% molar yield) having HPLC purity > 99.0%. |
| 216.7 g |
With iron; at 105℃; for 5h; |
Add 240g iron powder in batches,Control reaction temperature does not exceed 105 stirring reaction 5 hours,Cool to room temperature.filter,Filter cake washed with water,And then washed with sodium bicarbonate solution,After filtration, the crude product.The crude product was dissolved in 2400 ml of dimethylsulfoxide,filter.Add water to the filtrate,The precipitated solid was collected by filtration,Dry at 60 for 4 h.216.7 g of a yellow solid, yield: 79.3%, purity: 99.93%. |
| 3.7 g |
With palladium 10% on activated carbon; hydrogen; In N,N-dimethyl acetamide; at 30℃; under 3000.3 Torr; for 6h; |
5 g of 1,3-dioxo-2- (2,6-dioxopiperidin-3-yl) -5-nitroisoindoline 10% palladium carbon 0.4g mixed into 70 mL of N, N-dimethylacetamide, The hydrogenation reaction was then carried out at 0.4 MPa, The reaction temperature was 30 C, Lasted 6 hours, Filter out the catalyst, The filtrate was concentrated in vacuo, The residue was beaten overnight at 40 C with 40 mL of water, Get 3.7g Yellow-green solid of thalidomide crude. HPLC with a purity of 99.0%. |
|
With palladium 10% on activated carbon; hydrogen; In 1,4-dioxane; methanol; at 35℃;Industrial scale; |
N,N-Dimethylformamide (50kg) was drawn into the reactor.Glutamic acid (32 kg), 3-nitrophthalic anhydride (40 kg) was added under stirring, followed by heating and reaction at 95-100C for 3 hours.Then, N,N-dimethylformamide was evaporated under reduced pressure, acetic anhydride (50 kg) was added with cooling, and the mixture was heated to 100-110 C. for 1 hour. The excess anhydride was distilled off under reduced pressure. DMSO (400 L) was added and the reaction was heated to 200C,Ammonia was slowly introduced under stirring. After the reaction was complete, methanol (800 L) was added to cool to room temperature, the crystals were centrifuged, and dried to obtain 3-nitrophoramide (62.76 kg) in a yield of 71%. Purity 99.99%.3-nitrophoramide (30kg) was dissolved in 1,4-dioxanemethanol=300L300L, 10% palladium on carbon, 35C, 0.4M,Hydrogenation to the completion of the reaction, pressure filter, concentrated under reduced pressure, add DMF (300L) dissolved, isopropyl ether (600L) crystallization, centrifugedThe yellow product was recrystallized from ethanol to obtain high-purity pomametide. HPLC: 99.84%. |
| 85 g |
With palladium 10% on activated carbon; hydrogen; In N,N-dimethyl-formamide; at 15 - 20℃; |
In to a well cleaned and oven dried 3 .OL 4neck RB flask, to a mechanical stirrer and equipped with thermometer socket, condenser (fitted with gas outlet dipped in to the water), gas-inlet(connected with single trap filled with dimethylfonnamide up to bubbling nossile), 100.Og of 2-(2, 6-dioxo-3 -piperidyl)-4-nitro-isoindoline- 1, 3 -dione, 1 550.Oml of dimethylformamide were charged. Reaction mass was stirred for 10-15mm at 25-35C, clear solution was observed. 13.0g of 10% Pd/c was charged in to the reaction mass, in presence of N2 gas atmosphere, washing was given with 50.0 ml of dimethylfonnamide. H2 gas was bubbled in to the reaction mass, for 6.5-7.0 hours, at 15-20C. After completion of reaction, eaction mass is filtered under vacuum, through hyflow, in presence of N2 gas atmosphere, washing is given with 300.Oml of dimethylfonnamide. Filtrate is transferred in to a 5.OL 4 neck RB flask. Slowly added DM Water (1900.0m1) to the reaction mass over a period of 45-60mm at 15-20C, Stirred the reaction mass at 15-20C for 60-90mm. Solid reaction mass was filtered under vacuum, washing was given with 200.Oml DM Water, suck dried for 45-60mm. wet compound is dried in a vacuum oven at 60-650C, under vacuum (600-65OmmJHg) for 2-3hours and characterized as form A of pomalidomide.Yield: 85.0 g |
|
With palladium 10% on activated carbon; hydrogen; In N,N-dimethyl-formamide; at 25 - 30℃; under 2250.23 - 2625.26 Torr; |
10% Palladium on carbon (50% wet; 5 g) was added to a solution of 2-(2,6- dioxopiperidin-3-yl)-4-nitro-lH-isoindole-l,3(2H)-dione (100 g; Formula II; obtained in Example 2) in N, N-dimethylformamide (1000 mL). The solution was hydrogenated with 3.0 kg/cm2 to 3.5 kg/cm2 hydrogen pressure for 2 hours to 3 hours at 25C to 30C. The catalyst was removed by filtration and the filtrate was treated with activated carbon. The filtrate was heated to 50C to 55C and then water was added slowly at 50C to 55C to obtain a slurry. The slurry was slowly cooled to 25 C to 30C and then stirred for 30 minutes at 25C to 30C. The slurry was filtered to obtain a wet solid. The wet solid was dried at 50C to 55C under reduced pressure to obtain the title compound. (0079) Yield: 85 g (94%) |
|
With palladium 10% on activated carbon; hydrogen; In water; N,N-dimethyl-formamide; at 20℃; under 3102.97 Torr; for 18h; |
To a stirred solution of 2-(2,6-dioxopiperidin-3-yl)-4-nitroisoindoline-1,3-dione (29a, 10.0 g, 33.00 mmol) in DMF:H20 (9: 1, 75 mL) was added 10 % Pd/C (2.0 g). The reaction was performed in par shaker at 60 psi under hydrogen atmosphere at RT for 18 h. After completion of the reaction (monitored by TLC) the reaction mixture was filtered though celite. Then cold water was added to the filtrate to get the solid which was filtered off and dried under vacuum to afford the title compound (5.50 g, 61.1 %) which was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) d 11.07 (bs, 1H), 7.46 (t, / = 6.8 Hz, 1H), 7.00 (t, / = 8.0 Hz, 2H), 6.51 (bs, 2H), 5.06-5.01 (m, 1H), 2.89-2.84 (m, 1H), 2,60-2.54 (m, 2H), 2.03-2.0 (m, 1H); LC-MS: m/z 274.1 (M+l)+. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping