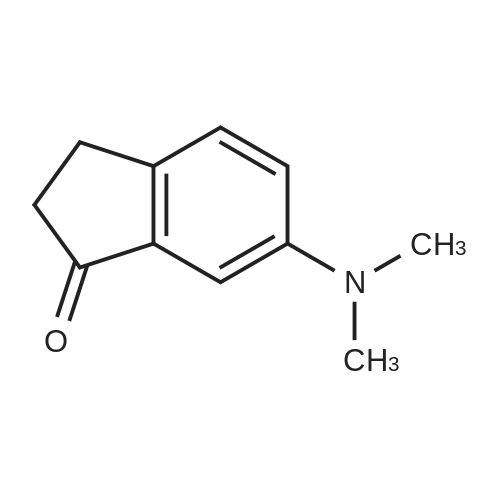
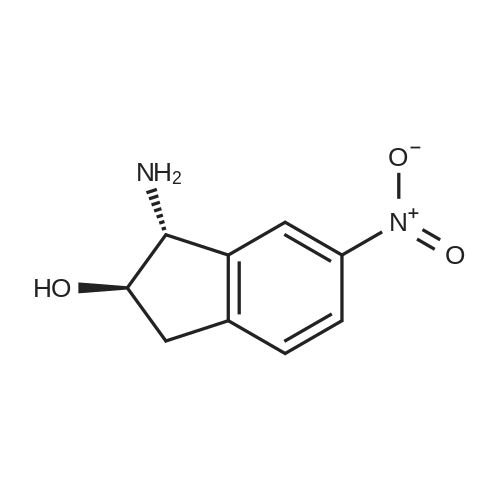
| 64% |
With sulfuric acid; potassium nitrate at 0℃; for 0.5h; |
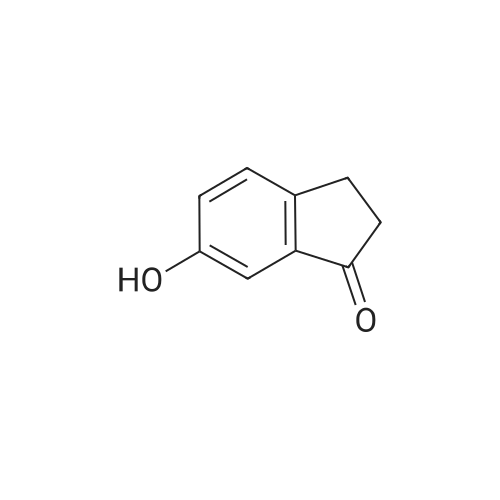
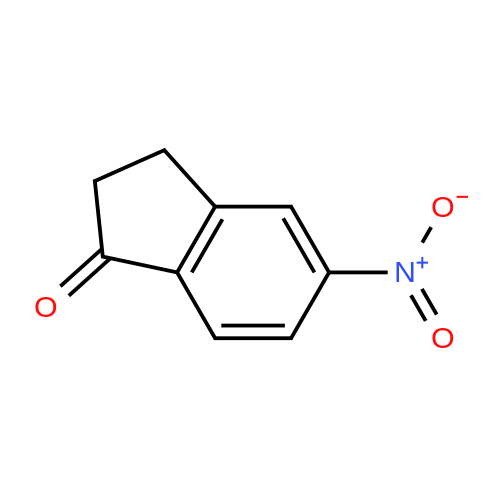
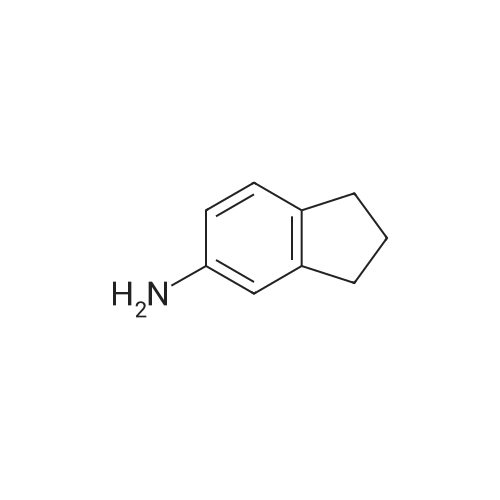
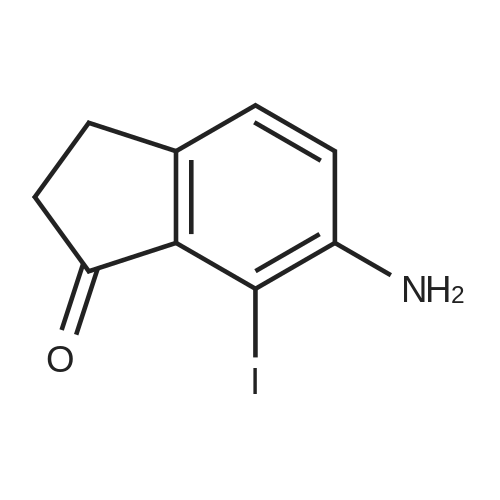
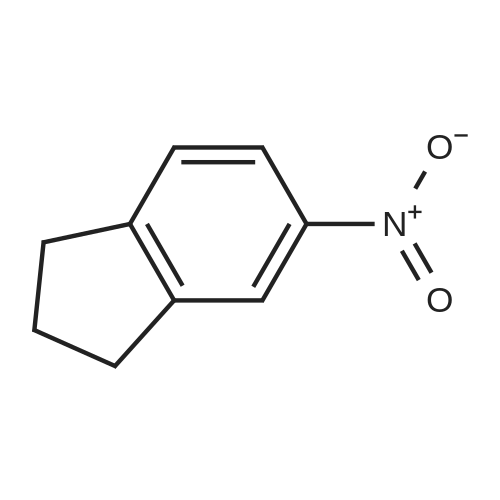
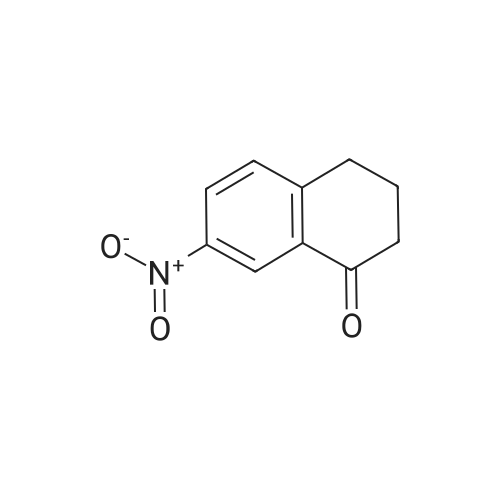
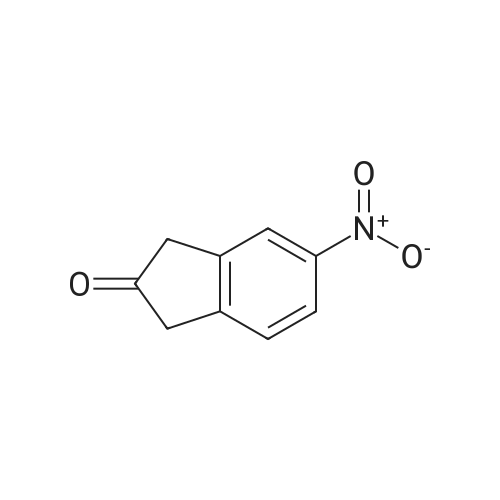
6-Nitro-2,3-dihydro-lH-inden-l-one (SR5-120)
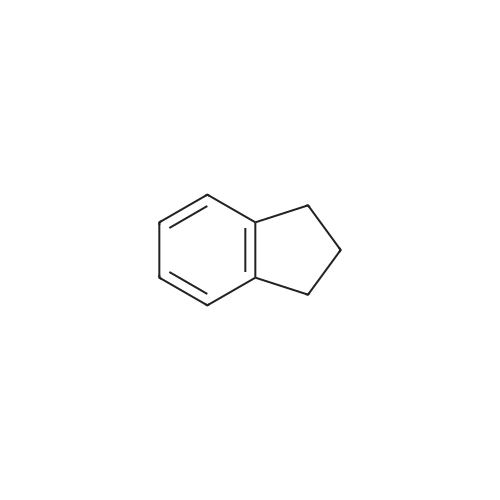
To l-indanone (4.00 g, 30.266 mmol) in cone. H2S04 (29 mL) at 0 °C was added solution of KNO3 (3.060 g, 30.266 mmol) in cone. H2SC (9 mL) over 30 min. The mixture was stirred at 0 °C for 1 h and poured into ice/water mixture. The mixture was extracted with EtOAc (3 x 100 mL), dried (Na2S04) and concentrated under reduced pressure. Purification by flash column chromatography using EtO Ac/hexane (20:80-100:0) as eluent afforded SR5-120 as a pale yellow solid (3.410 g, 64 %). 1 H NMR (500 MHz, DMSO-de) d 8.49 (dd, 7 = 8.4, 2.3 Hz, 1H), 8.29 (dd, 7 = 2.3, 0.7 Hz, 1H), 7.87 (dd, 7 = 8.4, 0.8 Hz, 1H), 3.27- 3.21 (m, 2H), 2.81-2.76 (m, 2H); |
| 62% |
With sulfuric acid; potassium nitrate at -5℃; for 4h; |
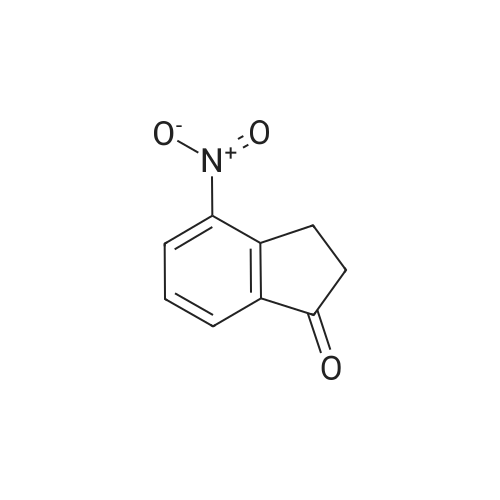
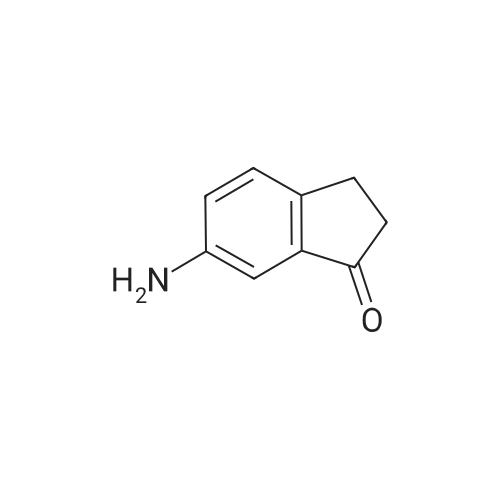
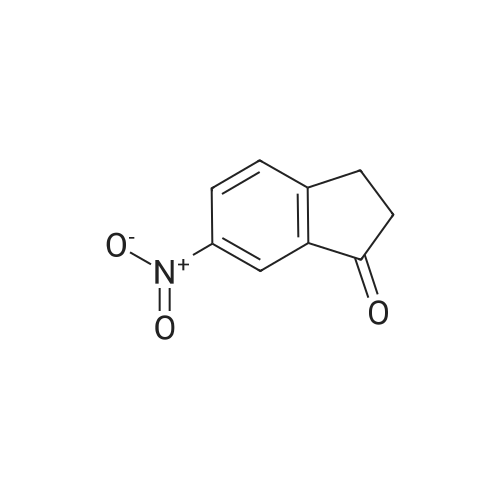
6-Nitro-2,3-dihydro-1H-inden-1-one (10)
To a solution of 2,3-dihydro-1H-inden-1-one (9, 1.32 g,10.0 mmol) in concentrated sulfuric acid (10 mL), KNO3 (1.21 g, 12.0 mmol) in concentrated sulfuricacid (10 mL) was added dropwise at 5°C in 30 min. The mixture was stirred at 5 C for 4 h.After adding ice water slowly, the mixture was partitioned between water and CH2Cl2. The organiclayer was washed with a saturated aqueous solution of NaHCO3 and brine, dried over anhydrousNa2SO4, and concentrated under vacuum. The residue was purified by column chromatographyon silica gel (eluent: hexane/EtOAc = 7/1) to give 10 (1.10 g, 62%) as a beige solid. m.p. 71~74°C;1H-NMR: δ 8.59 (d, J = 1.9 Hz, 1H), 8.47 (dd, J = 8.4, 2.2 Hz, 1H), 7.68 (t, J = 8.4 Hz, 1H), 3.33-3.25 (m,2H), 2.89-2.78 (m, 2H); ESI-MS: m/z [M + H]+ 178. |
| 61% |
With sulfuric acid; potassium nitrate at 0℃; for 2h; |
102
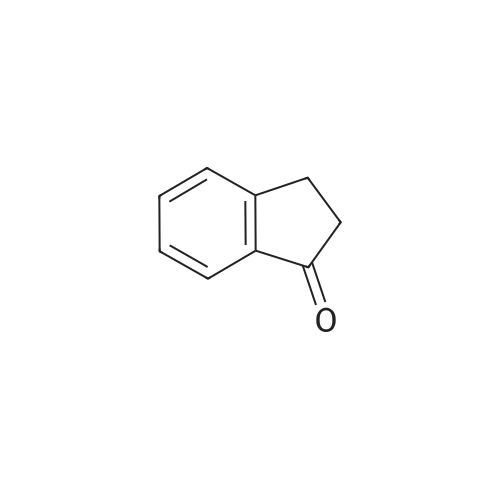
To a solution of 1-indanone (30 g, 0.23 mol) in cone. H2SO4 (200 mL) was added a solution of KNO3 (34 g, 0.34 mol) in cone. H2SO4 (100 mL) at 0 °C. The resulting mixture was stirred for 2h, and then poured into ice-H2O (3 L). The mixture was extracted with EtOAc (3x500 mL). The combined organic layers were washed with brine (3x500 mL), dried (Na2SO4), filtered, concentrated and purified via column chromatography to afford 6-nitro-indan-1-one (25 g, 61% yield). |
| 60% |
Stage #1: inden-1-one With sulfuric acid; potassium nitrate at 0℃; for 1h;
Stage #2: With water at 20℃; |
10
1-Indanone (5.00 g, 37.8 mmol) was dissolved in sulfuric acid (40 mL) , and thereto was added dropwise a solution of potassium nitrate (3.83 g, 37.8 mmol) in sulfuric acid (10 mL) under ice-cooling. The mixture was stirred for 1 hr under ice- cooling, ice was added to the reaction solution, and the mixture was stirred at room temperature overnight. The precipitated solid was collected by filtration, and purified by silica gel column chromatography (ethyl acetate/hexane=15/85->45/55) to give the title compound (4.01 g, yield 60%).1H-NMR (CDCl3) δ: 2.78 - 2.90 (2H, m) , 3.22 - 3.34 (2H, m) , 7.67 (IH, d, J = 8.5 Hz), 8.45 (IH, dd, J = 8.5, 2.3 Hz), 8.57 (IH, d, J = 2.3 Hz). |
| 54% |
With sulfuric acid; potassium nitrate In water at 0 - 15℃; for 1.5h; |
3
Preparation of compound 4 A solution of potassium nitrate (50.5g, 0. 5mol) in H2SO4 (200mL) was added, via a dropping funnel, to a solution of 1-INDANONE (60g, 0. 454mol) in concentrated sulfuric acid (500mL) cooled in an ice-water bath at a speed to maintain an internal temperature below 15°C. After stirring at 0°C for LH, the reaction mixture was poured into crushed ice and stirred for 30 min. The solid was filtered, washed with water, and air-dried. Purification by flash chromatography (TOLUENE/ETOAC, 95/5) gave compound 4 (43.5g, 54%) as a pale yellow solid. 1H NMR (CDC13, 400MHZ) : 2.81-2. 85 (2H, m, CH2), 3.28 (2H, t, J 6.1, CH2), 7.67 (1H, d, J 8.4, ArH), 8.45 (1H, d, J 8.4, ArH), 8.56 (1H, s, ArH). |
| 52% |
With sulfuric acid; potassium nitrate at 0 - 5℃; for 1.5h; |
|
|
(nitration); |
|
|
With nitric acid |
|
|
With sulfuric acid; potassium nitrate for 1h; |
|
|
With sulfuric acid; potassium nitrate |
|
|
With KNO3 In sulfuric acid |
129.A 6-nitro-1-indanone
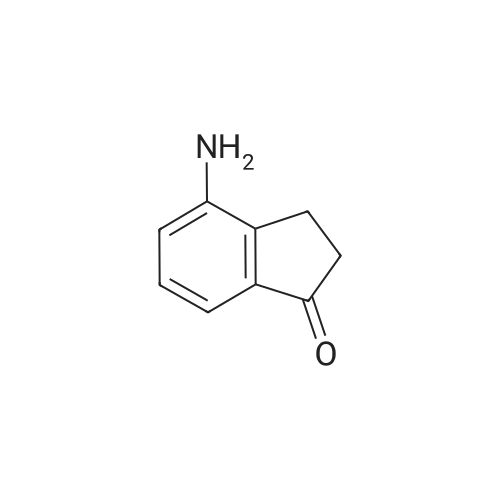
EXAMPLE 129A 6-nitro-1-indanone A solution of concentrated H2SO4 at 0° C. was treated with 1-indanone (6.00 g, 45.4 mmol) then treated dropwise with KNO3 (5.00 g, 49.94 mmol) in concentrated H2SO4 while maintaining the internal temperature at no more than 15° C. The reaction was stirred for 1 hour after the addition was complete, then poured onto ice. The resulting solids were collected by filtration, washed with water, and dried under vacuum to give a 4:1 mixture of 6-nitro- and 4-nitro-1-indanone (5.04 g, 63%). MS (ESI(+) m/e 178 (M+H)+, 195 (M+NH4)+; MS (ESI(-)) m/e 176 (M-H)-; 1H NMR (300 MHz, DMSO-d6, 6-nitro-1-indanone) δ 8.49 (dd, 1H), 8.29 (d, 1H), 7.87 (d, 1H), 3.25 (m, 2H), 2.78 (m, 2H); 1H NMR (300 MHz, DMSO-d6), 4-nitro-1-indanone) δ 8.51 (dd, 1H), 8.07 (dd, 1H), 7.74 (t, 1H), 3.53 (m, 2H), 2.76 (m, 2H). |
|
With sulfuric acid; potassium nitrate at 0 - 5℃; for 1.5h; |
14 Compound 14A:
2,3-Dihydro-1H-indene-1-one (10 g, 75.67 mmol, 9.09 mL) was dissolved in 100 mL of concentrated sulfuric acid, and KNO3 (8.03 g, 79.45 mmol) was added thereto at 0 °C. The reaction was carried out at 0-5 °C for 1.5 hours. After the reaction was completed, the reaction solution was poured into 300 mL of water. After filtration, the filter cake was dissolved in EtOAc, dried and then concentrated to give a crude product. The crude product was purified by column chromatography to give compound 14A. 1H NMR (400MHz, DMSO-d6) δ = 8.60 (d, J = 2.0 Hz, 1H), 8.48 (dd, J = 2.3, 8.5 Hz, 1H), 7.69 (d, J = 8.3 Hz, 1H), 3.34 -3.27 (m, 2H), 2.90-2.83 (m, 2H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping