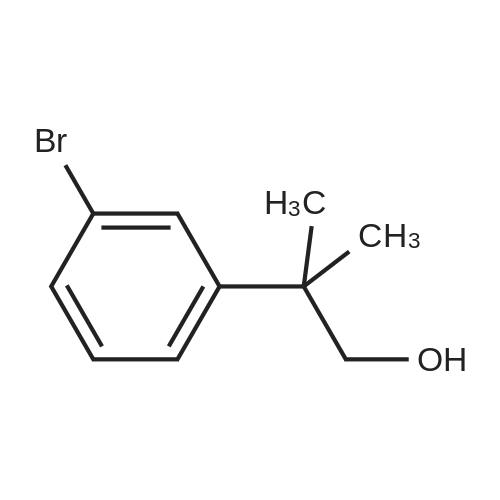
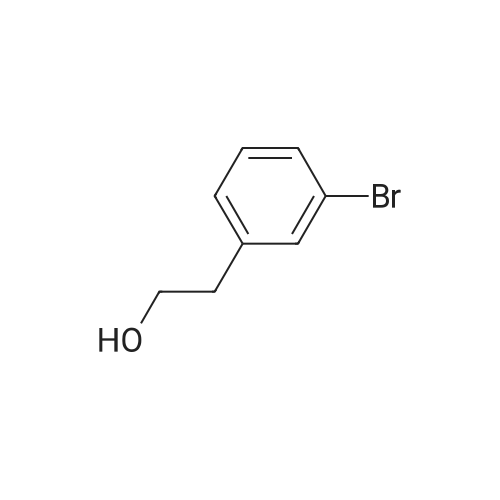
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of Labelled Compounds and Radiopharmaceuticals, 2007, vol. 50, # 3, p. 183 - 188
[2] Synthesis, 2006, # 18, p. 3085 - 3091
2
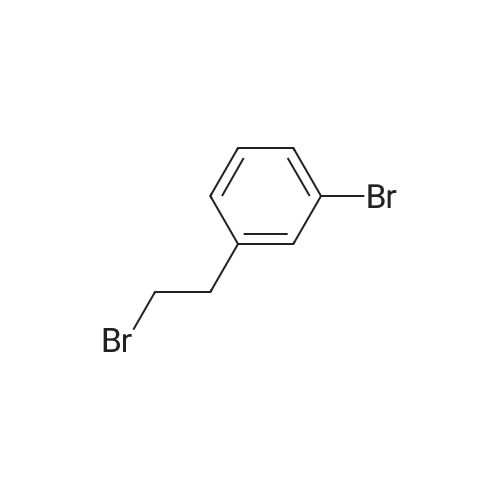
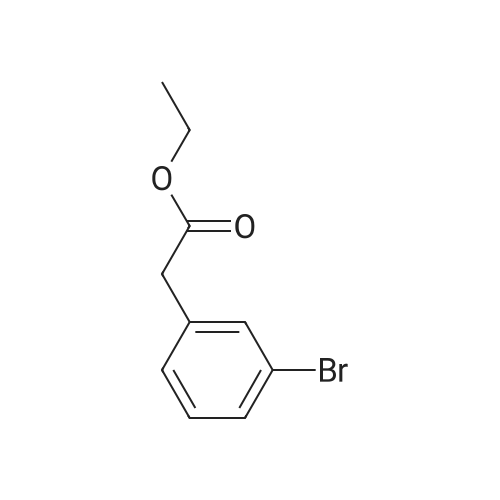
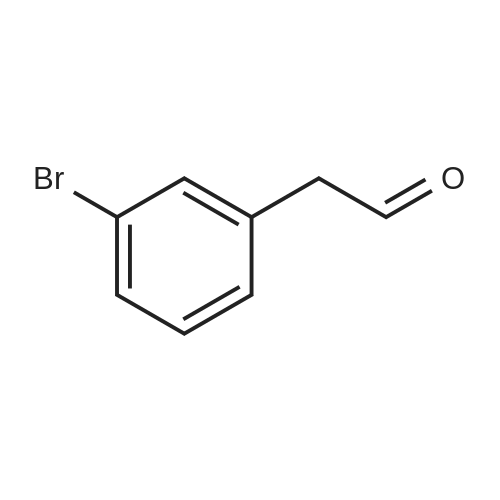
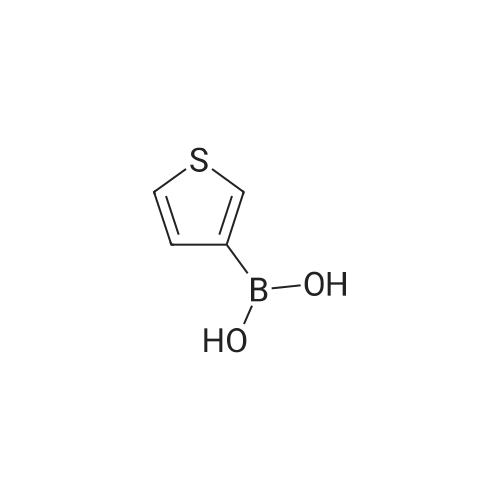
[ 1878-67-7 ]
[ 28229-69-8 ]
Yield
Reaction Conditions
Operation in experiment
79%
Stage #1: With dimethylsulfide borane complex In tetrahydrofuran at 0 - 20℃; for 20 h; Stage #2: With water In tetrahydrofuran
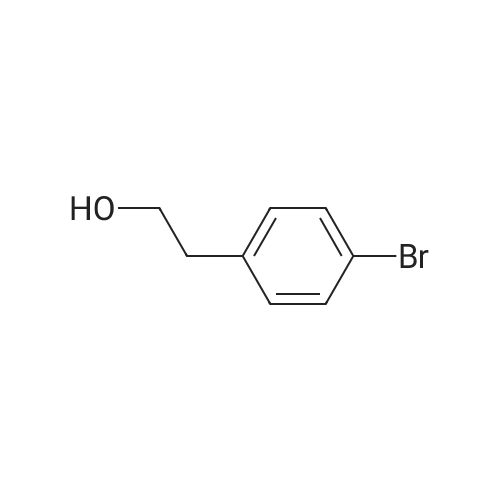
BH3-Me2S (7 ml_ of a 2M solution in THF, 14.0 mmol) was added to a cold (0 0C) solution of 3-bromophenylacetic acid (2.00 g, 9.3 mmol). The reaction mixture was allowed to warm to room temperature, stirred for 20 hours and re-cooled to 0 0C. Water (10 ml.) was added dropwise. The organic layer was separated, washed with brine (20 mL), dried (MgSO4), filtered and concentrated under reduced pressure to leave a colourless oil. Purification by column chromatography (50 percent EtOAc in heptane) afforded the title compound as a colourless oil (1.47 g, 79 percent yield). m/z 224/226 [M+H]+. 1H NMR (300 MHz, CDCI3) 7.41-7.35 (2H, m), 7.23-7.16 (2H, m), 3.87 (2H, t, J=6.5 Hz), 2.86 (2H, t, J=6.5 Hz).
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 11, p. 3018 - 3022
[2] Journal of Labelled Compounds and Radiopharmaceuticals, 2007, vol. 50, # 3, p. 183 - 188
[3] Patent: WO2008/53136, 2008, A1, . Location in patent: Page/Page column 38
[4] Journal of Medicinal Chemistry, 1991, vol. 34, # 9, p. 2882 - 2891
[5] Journal of Medicinal Chemistry, 1998, vol. 41, # 3, p. 358 - 378
[6] Journal of Medicinal Chemistry, 2006, vol. 49, # 5, p. 1562 - 1575
[7] Patent: WO2006/11631, 2006, A2, . Location in patent: Page/Page column 67-68
[8] Patent: US6316466, 2001, B1,
[9] Patent: US5827868, 1998, A,
[10] Patent: US5087635, 1992, A,
[11] Patent: WO2004/67521, 2004, A1, . Location in patent: Page 228-229
[12] Journal of the American Chemical Society, 2010, vol. 132, # 12, p. 4076 - 4077
[13] Patent: WO2007/69986, 2007, A1, . Location in patent: Page/Page column 89
[14] Journal of the American Chemical Society, 2011, vol. 133, # 43, p. 17142 - 17145
[15] Patent: US2014/57914, 2014, A1, . Location in patent: Paragraph 0356-0357
[16] Chemistry - A European Journal, 2014, vol. 20, # 47, p. 15325 - 15329
[17] Bioorganic and Medicinal Chemistry Letters, 2017, vol. 27, # 2, p. 131 - 134
3
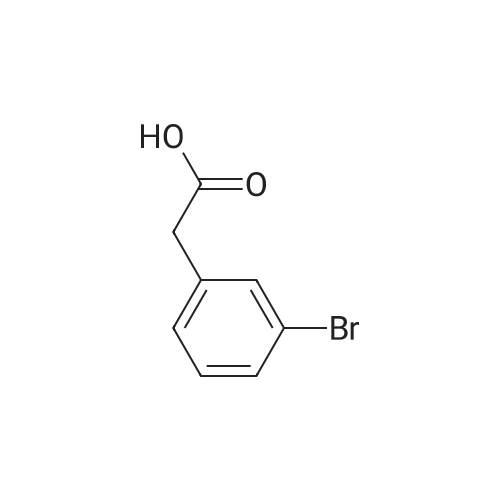
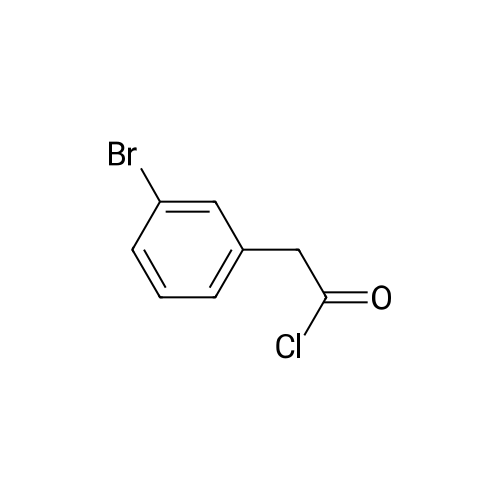
[ 2039-86-3 ]
[ 28229-69-8 ]
Reference:
[1] Patent: US6063789, 2000, A,
[2] ACS Catalysis, 2017, vol. 7, # 8, p. 5225 - 5233
4
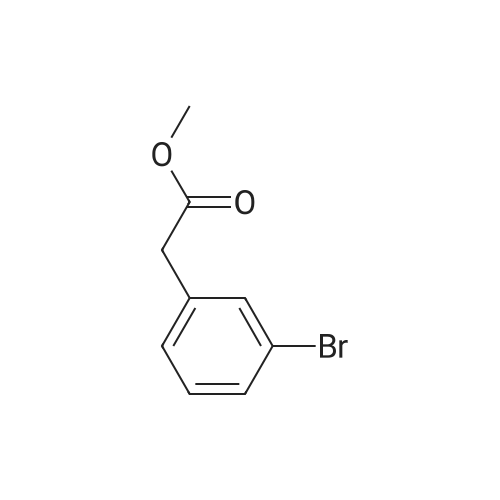
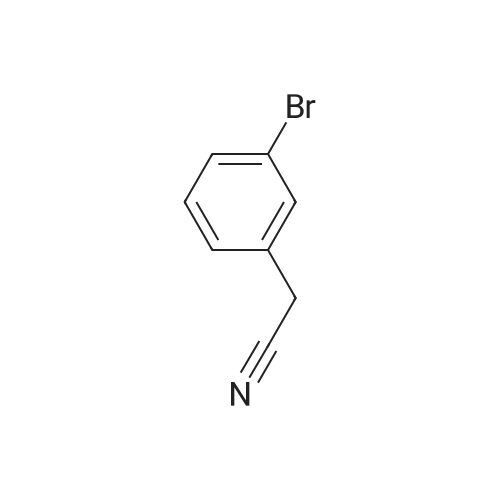
[ 150529-73-0 ]
[ 28229-69-8 ]
Reference:
[1] Chemistry - A European Journal, 2015, vol. 21, # 42, p. 14737 - 14741
5
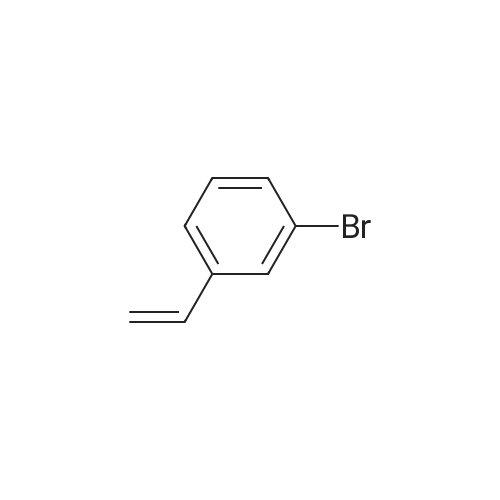
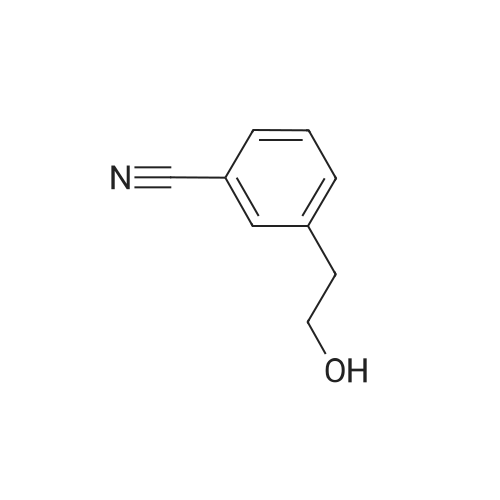
[ 14062-30-7 ]
[ 28229-69-8 ]
Reference:
[1] Journal of the American Chemical Society, 1957, vol. 79, p. 3710
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 11, p. 3018 - 3022
10
[ 28229-69-8 ]
[ 73183-34-3 ]
[ 651030-56-7 ]
Yield
Reaction Conditions
Operation in experiment
89%
With palladium bis[bis(diphenylphosphino)ferrocene] dichloride; potassium acetate In dimethyl sulfoxide at 120℃; for 18 h; Inert atmosphere
Stage 1: 2-[3-(4,4, 5, 5-Tetramethyl- 1, 3,2-dioxaborolan-2-yl)phenyl]ethanol To a solution of 2-(3-bromophenyl)-ethanol (3.0 g, 15 mmol) in anhydrous DMSO (30 mL) were added KOAc (4.39 g, 45 mmol), bis(pinacolato)diboron (5.68 g, 22 mmol) and PdCI2(dppf)2 (0.61 g, 0.74 mmol) and the reaction mixture was heated at 120°C under a nitrogen atmosphere for 18 hrs. The reaction was cooled to RT and EtOAc (60 mL) was added. The reaction mixture was filtered through Celite®, washing with EtOAc (500 mL). The filtrate was washed with sat NaHCO3 (150 mL), water (150 mL), brine (150 mL), dried over Mg504, filtered and concentrated in vacuo. The crude material was purified by automated column chromatography using EtOAc in heptane (gradient 0-100percent) and then column chromatography (33percent EtOAc/heptane) to give the title compound as a pale yellow oil (3.30 g, 89percent).1H NMR (300 MHz, ODd3) ö ppm: 7.70 (2H, m), 7.35 (2H, m), 3.89 (2H, t, J=6.6 Hz),2.90 (2H, t, J=6.6 Hz), 1.51 (1H, s), 1.37 (12H, s).
BH3-Me2S (7 ml_ of a 2M solution in THF, 14.0 mmol) was added to a cold (0 0C) solution of 3-bromophenylacetic acid (2.00 g, 9.3 mmol). The reaction mixture was allowed to warm to room temperature, stirred for 20 hours and re-cooled to 0 0C. Water (10 ml.) was added dropwise. The organic layer was separated, washed with brine (20 mL), dried (MgSO4), filtered and concentrated under reduced pressure to leave a colourless oil. Purification by column chromatography (50 % EtOAc in heptane) afforded the title compound as a colourless oil (1.47 g, 79 % yield). m/z 224/226 [M+H]+. 1H NMR (300 MHz, CDCI3) 7.41-7.35 (2H, m), 7.23-7.16 (2H, m), 3.87 (2H, t, J=6.5 Hz), 2.86 (2H, t, J=6.5 Hz).
Production Example 8; Synthesis of N-(4-{2-[3-(2-[amino(imino)methyl]amino}ethyl)phenyl]ethyl}-.,3-thiazol-2-yl)acetamide hydrochloride; Step 1; To a suspension of lithium aluminum hydride in drytetrahydrofuran (50 ml) was added (3-bromophenyl)acetic acid(10 g) in tetrahydrofuran (100 ml) under ice cooling. Themixture was refluxed for 2 hurs. After cooling, to thereaction mixture were added water and aqueous Rochelle salt.The mixture was stirred for another 30 min. Aqueous layer wasextracted with ethyl acetate. The organic layer was dried overanhydrous magnesium sulfate, and concentrated in vacua to give2-(3-bromophenyl)ethanol. This compound was used for the nextreaction without further purification.
With benzophenone; potassium; In tetrahydrofuran; methanol;
A. 2-(3-Bromophenyl)ethan-1-ol To a solution of 10.0 g (46.5 mmol, Aldrich) of 3-bromophenylacetic acid in 100 mL of dry tetrahydrofuran (distilled from potassium/benzophenone), cooled to 0 C., was added dropwise over 40 minutes 61 mL (61 mmol, 1M/tetrahydrofuran, Aldrich) of borane-tetrahydrofuran solution. The reaction was stirred at room temperature for 16 hours, then cooled to 0 C. and quenched by the dropwise addition of 10 mL methanol. The reaction mixture was concentrated in vacuo and azeotroped with two 10-mL portions of methanol to give 9.35 g (46.5 mmol, 100%) of alcohol A as a clear oil.
In tetrahydrofuran; (2S)-N-methyl-1-phenylpropan-2-amine hydrate;
Step 1--Preparation of 2-(3-bromophenyl)ethanol; reduction of 3-bromophenylacetic acid via diborane reduction A 2 liter 4 neck flask stirred under nitrogen, equipped with condenser and addition funnel, was charged with 75 gms of 3-bromophenylacetic acid (0.34 mole), in 100 ml of THF. Then 350 ml of 1M diborane-THF complex (0.35 mole) was added dropwise. Upon addition, gas evolved and the reaction was cooled to maintain the temperature below 10 C. After the addition was complete, the reaction was stirred at room temperature until, by thin layer chromatography, the reaction was complete. The reaction was quenched by adding ice water and the product was extracted into ether then washed with 5% NaOH, 5% HCl, water and dried over magnesium sulfate. After concentration, 77.4 gms of product resulted and was chlorinated directly. NMR (90 MHz): 2.7-2.9(m, 3H), 3.7-3.9(t, 2H) and 7.1-7.4(m, 4H).
3-Bromophenyl acetic acid (10 g, 0.0465 mol), dissolved in THF (50 mJL), was added dropwise added to a LAH (3.5 g, 0.93 mol) suspension in THF (100 mL), at 00C under a nitrogen atmosphere. The reaction mixture was stirred for 2 h at room temperature before being quenched with water (9.5 mL), 10% NaOH (9.5 mL) and then water (9.5 mL) again. Precipitated solid was filtered and the residue was washed with DCM. The filtrate was concentrated to afford 8.3 g of the sub-title compound as pale yellow liquid.
With borane-THF; In tetrahydrofuran; at 20℃;
Step 1 [0356] 2-(3-Bromophenyl)ethanol 2-(3-bromophenyl)acetic acid (10.0 g, 0.046 mol) in THF (150 mL), 1 M BH3-THF (100 mL, 100 mmol) was dropwise added at RT. The mixture was stirred overnight, concentrated, diluted with H2O (100 mL), and extracted with EtOAc (100 m×3). The combined organic layers were washed with H2O and brine, dried over Na2SO4, filtered, and concentrated to afford the crude product 2-(3-bromophenyl)ethanol (9.0 g, 97%) as a colorless oil, which was directly used for next step without further purification. MS (ES+) C8H9BrO requires: 200, 202. found: 201 [M+H]+, 203 [M+2+H]+(1:1).
With lithium aluminium tetrahydride; In tetrahydrofuran; at 0℃; for 4.5h;Reflux;
General procedure: To a mixture of LiAlH4 (15 mmol) in anhydrous THF (25 mL) in an ice-bath was added dropwise a solution of phenylacetic acids (15 mmol) in THF (8 mL). This mixture was stirred at room temperature for 30 min, and then heated to reflux for 4 h. After it was cooled to room temperature, water (0.5 mL) was added, and then NaOH (15%, 0.5 mL) and water (1.5 mL) were added in sequence. After stirring for another 30 min, the mixture was filtered, dried over anhydrous Na2SO4 and concentrated to give crude products. Pure phenylethyl alcohols were obtained in 50-85% yield by column chromatography. Alternative method: To a solution of phenylacetic acids (15 mmol) in MeOH (30 mL) was added SOCl2 (30 mmol). This mixture was heated to reflux for 3 h before evaporation. The residue was dissolved in DCM (30 mL), washed with aqueous NaHCO3, water and brine, dried over anhydrous Na2SO4, and concentrated to give 100% yield of crude methyl phenylacetates which were used to next step without further purification. To a solution of the methyl phenylacetates in THF (30 mL) was added NaBH4 (60 mmol). When the mixture was heated to gently reflux, MeOH (1.0 mL) was added dropwise from a syringe over 5 min. After refluxing for another 6 h, the mixture was cooled to room temperature and poured into 30 mL ice water, and extracted with EtOAc (30 mL × 2). The combined organic phase was washed with brine, dried over anhydrous Na2SO4, and concentrated to give crude products. Pure phenylethyl alcohols were obtained in 70-85% yield by column chromatography.
With diborane; In tetrahydrofuran;
Step 1 To a solution of 3-bromophenylacetic acid (10 g, 46.5 mmol) in tetrahydrofuran (100 ml) at 0 C. was added diborane (70 ml, 1.0 M solution in tetrahydrofuran). The reaction mixture was allowed to warm to room temperature. After 16 h, the reaction mixture was cooled to 0C. and water was added dropwise (50 ml). The organic layer was separated and washed with brine, dried over sodium sulfate and concentrated in vacuo. The residue was purified by flash chromatography (elution gradient: 40-60% ethyl acetate/hexane) to give 3-(2-hydroxyethyl)bromobenzene (9.0 g).
General procedure: To a solution of alcohol 4a (15.0 g, 74.4 mmol) in dichloromethane (225 mL) was added triphenylphosphine (24.9 g, 94.9 mmol) and imidazole (6.45 g, 94.7 mmol). After stirring at room temperature for 20 min, iodine (22.5 g, 88.7 mmol) was added and the resulting dark brown mixture was stirred for overnight at 0 C. A saturated solution of sodium thiosulfate (50 mL) was added and the mixture was allowed to warm to room temperature. The reaction mixture was extracted with dichloromethane, washed with brine, dried over Na2SO4, filtered, and concentrated under reduce pressure. Hexane was added to the residue and the white precipitate was filtered off. The filtrate was concentrated to give the crude 2-bromophenethyl iodide (23.0 g), which was used without further purification.
With 1H-imidazole; iodine; triphenylphosphine; In dichloromethane;
General procedure: To a solution of alcohol 4a (15.0 g, 74.4 mmol) in dichloromethane(225 mL) was added triphenylphosphine (24.9 g,94.9 mmol) and imidazole (6.45 g, 94.7 mmol). After stirring atroom temperature for 20 min, iodine (22.5 g, 88.7 mmol) wasadded and the resulting dark brown mixture was stirred for overnightat 0 C. A saturated solution of sodium thiosulfate (50 mL)was added and the mixture was allowed to warm to room temperature.The reaction mixture was extracted with dichloromethane,washed with brine, dried over Na2SO4, filtered, andconcentrated under reduce pressure. Hexane was added to theresidue and the white precipitate was filtered off. The filtrate wasconcentrated to give the crude 2-bromophenethyl iodide (23.0 g),which was used without further purification.To a solution of ethyl malonate (295 mL) was slowly added NaH(12.0 g, 30.0 mmol) at 0 C. After stirring for 30 min, a solution ofthe crude 2-bromophenethyl iodide (23.0 g) in THF (100 mL) wasadded. The reaction mixture was stirred at room temperature forovernight. After the completion of the reaction indicated by TLC,aqueous NH4Cl (50 mL) was added. The resulting mixture wasextracted with EtOAc, washed with brine, dried over Na2SO4,filtered, and concentrated. The crude product was purified by columnchromatography on silica gel (hexane/AcOEt 50:1) to givethe substituted ethyl malonate (17.0 g) as a colorless oil, which wasdissolved in 40 mL of 50% aqueous ethanol. To this solution, 9.30 g(165.8 mmol) of potassium hydroxide was added. After refluxingfor 6 h, the reaction mixture was diluted with water and acidifiedwith 15% hydrochloric acid solution. A colorless precipitate wascollected to afford 5a (13.5 g) in a total yield of 63% from 5a. Mp:157-158C. This data is in agreement with the reported value(156e158 C) [33].
With hydrogenchloride;titanium tetrachloride; In nitromethane; dichloromethane;
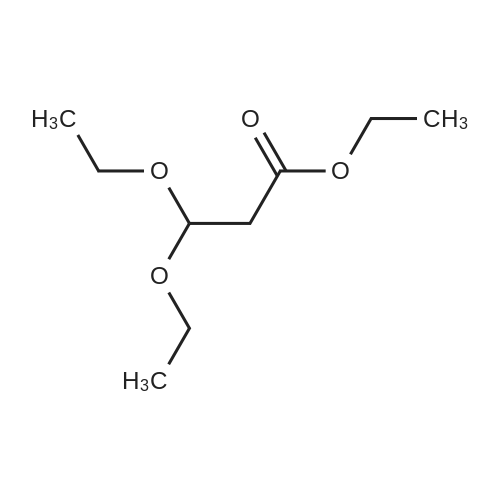
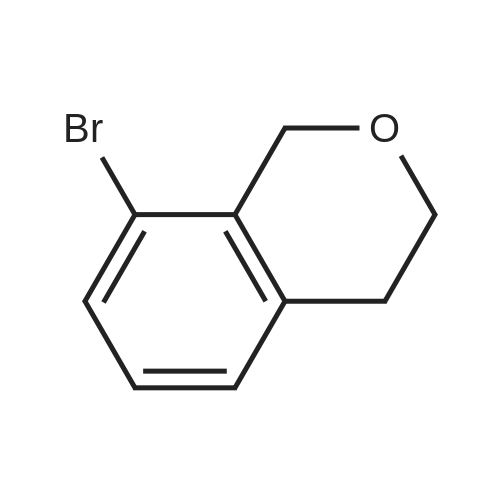
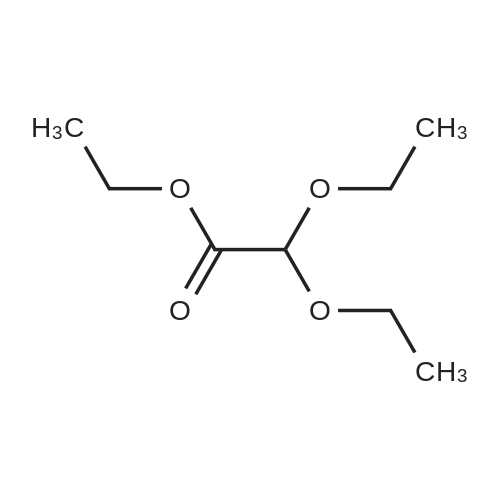
Titanium tetrachloride (1M in methylene chloride, 51 ml) is added over a period of 10 min to an ice-cooled mixture of <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong> (XLV, 4.34 g, 21.6 mmol) and ethyl 3,3-diethoxypropionate (LXXXV, 4.93 g, 25.9 mmol) in nitromethane (5 ml). After stirring for 10 min, the ice bath is removed and the mixture is allowed to stir at 20-250 for 5 hr, at which time it is poured onto ice/aqueous hydrochloric acid (~1N). The mixture is extracted with dichloromethane and backwashed with hydrochloric acid (0.5N)/saline and saline. The organic phase is separated dried over sodium sulfate, concentrated, and the residue chromatographed on silica gel eluding with ethyl acetate/hexane (5/95). The appropriate fractions are pooled and concentrated to give ethyl (6-bromoisochroman-1-yl)acetate (LXXVII), NMR (CDCl3) 1.28, 2.66-2.87, 2.96, 3.768, 3.807, 4.11, 4.21, 5.19, 6.93 and 7.28 δ.
With 1H-imidazole; In dichloromethane; at 20℃; for 2h;
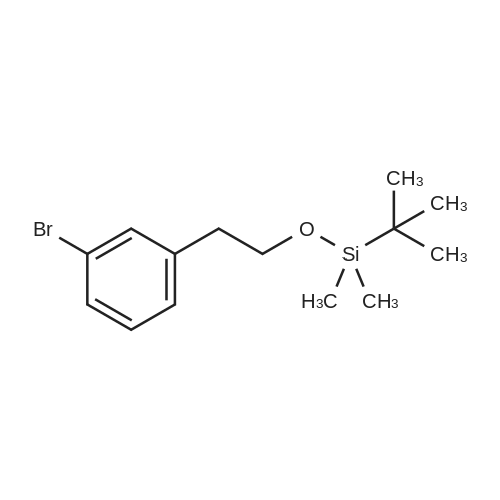
Intermediate 1A: (3 -Bromophenethoxy)(tert-butyl)dimethylsilane j00126j 2-(3-Bromophenyl)ethanol (1.89 g, 9.40 mmol) was dissolved in DCM (47.0 mL). Imidazole (2.56 g, 37.6 mmol) then TBS-Cl (2.83 g, 18.80 mmol) were added at ambient temperature. The reaction was stirred for 2 h. The reaction was diluted with DCM and washed with saturated NaHCO3, then water, then brine, dried (Na2SO4),filtered, and concentrated in vacuo to yield Intermediate 1A (2.93 g, 99%). 1H NMR (400 MHz, CHLOROFORM-cl) ö ppm 7.38 (1 H, s), 7.34 (1 H, ddd, J=6.40, 2.38, 2.26 Hz), 7.08 - 7.20 (2 H, m), 3.80 (2 H, t, J=6.78 Hz), 2.79 (2 H, t, J=6.78 Hz), 0.87 (9 H, s),-0.02 (6 H, s). MS (ESI) m/z: 315.0 (M+H).
95%
With 1H-imidazole; In N,N-dimethyl-formamide; at 20℃; for 16h;
To a stirred solution of 2- (3-bromophenyl)ethan-1-ol (30.0 g, 149.2 mmol) in DMF (150 mL) was added imidazole (15.24 g, 223.8 mmol) and TBSCl (26.99 g, 179.0 mmol) at rt. The reaction mixture was stirred at rt for 16h. The reaction mixture was neutralized by saturated LiCl solution (500 mL) and extracted with EtOAc (3x200 mL). The combined organics were dried over Na2SO4, filtered, concentrated under reduced pressure and purified by column chromatography (5% EtOAc in hexane) to afford the title compound (45.0 g, 95%) as a colorless oil. 1H NMR (400Mz, DMSO-d6): d 7.46-7.36 (m, 2H), 7.25-7.21 (m, 2H), 3.75 (t, 2H), 2.73 (t, 2H), 0.80 (s, 9H), -0.08 (s, 6H).
90%
With dmap; triethylamine; In dichloromethane; at 0 - 20℃; for 2h;
Triethylamine (1.3 mL, 9.5 mmol), DMAP (220 mg, 1.8 mmol) and terf-butyl(chloro)- dimethylsilane (1.10 g, 8.8 mmol) were added to a cold (0 C) solution of 2-(3- bromophenyl)ethanol (1.47 g, 7.3 mmol) in DCM (40 mL). The reaction mixture was stirred at 0 0C for 30 minutes, allowed to warm to room temperature, stirred for an additional 1.5 hour, washed with saturated NH4CI (40 mL), brine (40 mL), dried (MgSO4), filtered and concentrated under reduced pressure to leave a colourless oil. Purification by column chromatography (10 % EtOAc in heptane) afforded the title compound as a colourless oil (2.07 g, 90 % yield).1H NMR (300 MHz, CDCI3) 7.40-7.34 (2H, m), 7.18-7.15 (2H, m), 3.82 (2H, t, J=6.8 Hz) , 2.81 (2H, t, J=6.8 Hz), 0.90 (9H, s), 0.00 (6H, s).
89%
With 1H-imidazole; In dichloromethane; at 20℃; for 2.5h;
Add <strong>[28229-69-8]3-bromophenethyl alcohol</strong> (1.89g, 9.4mmol)And imidazole (2.56g, 36.16mmol) dissolved in 50mL of dichloromethane,Add tert-butyldimethylchlorosilane (2.83g, 18.78mmol) at room temperature,The system was then stirred at room temperature for 2.5 hours. TLC monitors that the reaction is complete.After the reaction is complete, water (30mLX2), saturated sodium bicarbonate solution (30mLX2),Wash with saturated brine (30mLX2), dry the organic phase over anhydrous sodium sulfate, filter and spin dry,Purified by column chromatography, the product 28.1 (2.64g, yield: 89%) was obtained.
With 1H-imidazole; In N,N-dimethyl-formamide; at 25℃; for 12h;
Step 2; To a solution of <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong> (7 g) in N,N-dimethylformamide (100 ml) were added tert-butyldimethylsilylchloride (5.77 g) and imidazole (2.84 g) at 25 C. The mixturewas stirred at 25 C for 12 hr. The reaction mixture waspoured into water (500 ml) and extracted with ethyl acetate(100 ml x2). The combined organic layer was dried overmagnesium sulfate and concentrated in vacuo. The residue waspurified by silica gel column chromatography with mixedsolvent of n-hexane and ethyl acetate to give [2-(3-- bromophenyl) ethoxy] (tert-butyl) dimethyls ilane as colorless oil.
With dmap; triethylamine; In dichloromethane;
Step 2 To a solution of <strong>[28229-69-8]3-(2-hydroxyethyl)bromobenzene</strong> (4.0 g, 20 mmol) in methylene chloride (100 ml) at 0 C. was added a solution of tert-butyldimethylsilyl chloride (3.6 g, 24 mmol), dimethylaminopyridine (0.61 g, 5 mmol) and triethylamine (3.6 ml, 25.9 mmol). After 1 h, the reaction mixture was washed with brine, saturated ammonium chloride, dried over sodium sulfate and concentrated in vacuo. The residue was purified by flash chromatography (elution gradient: 0-10% hexane/ethyl acetate) to give 3-(2-tert-butyl-dimethylsiloxyethyl)bromobenzene (6.0 g).
tetrakis(triphenylphosphine)palladium (0); In toluene;
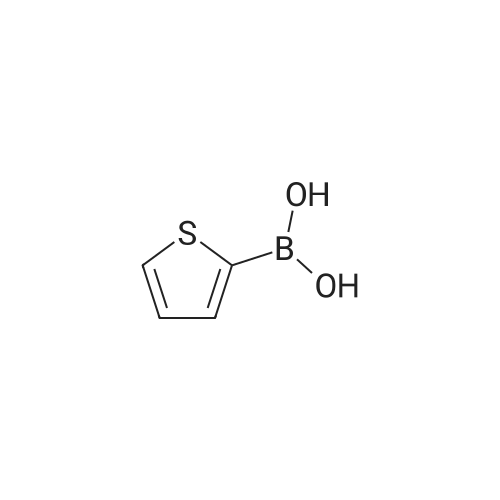
A reaction mixture of 2-(3-bromo-phenyl)-ethanol (1.0 eq.), furan-2-boranic acid (1.1 eq), 2N aqueous sodium carbonate, and tetrakis(triphenylphosphine)palladium(0) ("TTPP" or "(PPh3)4Pd") (0.1 eq) in toluene was stirred under nitrogen at 80 C. overnight. The mixture was diluted with ethyl acetate, washed with water and brine, dried over sodium sulfate, and concentrated. Purification by column chromatography (5-10% ethyl acetate/hexanes) gave 2-(3-furan-2-yl-phenyl)ethanol.
With Dess-Martin periodane; In dichloromethane; at 20℃; for 1h;
To a solution of <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong> (400 mg, 1.99 mmol) in dry CH2Cl2 (10 mL) wasadded Dess-Martin periodinane (848mg, 2.00 mmol) and the reaction mixture was stirred atroom temperature for 1 h. It was then washed with saturated sodium hydrogen carbonate solution, dried, and concentrated in vacuo to give the product (400 mg, 100%) which wasused in the next step without further purification.
98%
With Dess-Martin periodane; In dichloromethane; at 20℃; for 4h;
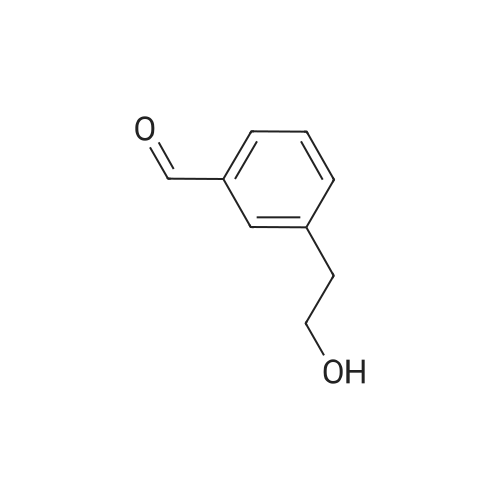
2-(3-Bromophenyl)ethanol (3.5 ml, 25 mmol) was dissolved in DCM (50 ml) and Dess-Martin periodinane (13 g, 0.03 mol) was added. The mixture was stirred at room temperature for 4 hours. The solvent was then partially evaporated and the residue filtered through a pad of Celite. Purification by chromatography on silica gel eluting with 10% Ethyl Acetate/ Hexane afforded 5 g (98%) of the aldehyde as a colourless oil: δH (360 MHz, CDCl3): 9.78 (1 H, s), 7.48 (1 H, d, J = 8.0 Hz), 7.42 (1 H, s), 7.23-7.26 (1 H, m), 7.18 (1 H, d, J = 7.6 Hz), 3.71 (2 H, d, J = 2.0 Hz).
96%
With Dess-Martin periodane; In dichloromethane; at 20℃; for 2h;
2-(3-Bromophenyl)ethanol (4.00g, 19.9mmol) was dissolved in DCM (30mL) and Dess-Martin periodinane (9.28g, 21.9mmol) was added in portions. The reaction mixture was stirred at ambient temperature for 2h before an aqueous solution of NaHCO3 (3.3equiv) and Na2S2O3 (6equiv) in water (30mL) was added and the resulting solution was stirred for five minutes before the two phases were separated. The aqueous phase was extracted with DCM. The organic extracts were combined and washed with water, dried over MgSO4 and evaporated to yield (3-bromophenyl)acetaldehyde (3.81g, 96%) as a colorless oil. 1H NMR (500MHz, CDCl3) δ 3.68 (d, J=2.1Hz, 2H), 7.13-7.17 (m, 1H), 7.22-7.25 (m, 1H), 7.37-7.40 (m, 1H), 7.43-7.47 (m, 1H), 9.75 (t, J=2.1Hz, 1H).
87%
With Dess-Martin periodane; In dichloromethane; at 0 - 20℃; for 2.5h;
Embodiment 30 2-(3-bromophenyl)acetaldehyde 2-(3-Bromophenyl)ethanol (676μL, 5mmol) was added to a flask, followed by addition of 10mL dry dichloromethane, then DMP (2.5g, 6mmol) was added under an ice bath. The reaction was continued for 2 h under the ice bath and another 0.5 h at room temperature, meanwhile TLC was used to monitor the reaction. After completion of the reaction, the reaction solution was cooled to 0C and the reaction was quenched with sodium thiosulfate solution. The mixture was diluted with ethyl acetate and washed with saturated sodium chloride solution. The organic phase was dried over anhydrous sodium sulfate, filtered, concentrated and purified by flash column chromatography (PE:EtOAc = 15:1) to give 2-(3-bromophenyl)acetaldehyde (0.86g, 87%). 1H NMR (300 MHz, CDCl3) δ 9.74 (1H, t, J = 2.0), 7.46 - 7.37 (2H, m), 7.30 - 7.14 (2H, m), 3.67 (2H, d, J = 2.0). 13C NMR (75 MHz, CDCl3) δ 198.9, 134.5, 133.0, 131.0, 130.9, 128.7, 123.3, 60.8. ESI(+)-MS: 199.1 [M+1]+.
5 g
With Dess-Martin periodane; In dichloromethane;
Step 2 [0358] 2-(3-Bromophenyl)acetaldehyde <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong> (9.0 g, 0.044 mol) in DCM (300 mL), Dess-Martin reagent (38.0 g, 0.090 mol) was added in portions. The reaction mixture was stirred overnight, and filtered. The filtrate was evaporated, and the residue was purified by silica gel column chromatography (Petroleum ether:EtOAc=5:1) to afford 2-(3-bromophenyl)acetaldehyde (5.0 g, 55%) as a yellow oil. MS (ES+) C8H7BrO requires: 198, 200. found: 199 [M+H]+, 201 [M+2+H]+(1:1).
With Dess-Martin periodane; In dichloromethane; at 0 - 20℃;
3-bromophenvDacetaldehvde To a solution of <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong> (4.9 g, 24.4 mmol) in DCM (20 ml) at 0 0C was added Dess Martin periodinane (10.8 g, 25 mmol) and the mixture was warmed <n="32"/>to RT and stirred overnight. The reaction was diluted with DCM (100 ml) and stirred with a saturated solution of sodium thiosulphate (100 ml) and saturated NaHCO3 (100 ml). The layers were separated and the organic layer was dried (MgSO4), filtered and evaporated under reduced pressure to leave the crude product as an orange oil (4.07 g, 84%). The product was used without purification. LCMS: m/z 200 [M+H]+.
2.5 g
With 2-Iodobenzoic acid; In dimethyl sulfoxide; at 20℃; for 3h;
2-iodobenzoic acid (4.2 g, 15.00 mmol) was added to an eggplant flask, and 15 mL of dimethyl sulfoxide was added. <strong>[28229-69-8]2-(3-bromophenyl)ethan-1-ol</strong> (1 g, 4.72 mmol) was added and stirred at room temperature for 3 hr. After the reaction was completed, the reaction solution was poured into a sodium thiosulfate solution, and a saturated sodium hydrogencarbonate solution was added. It was extracted 3 times with diethyl ether, and the organic phases were combined, washed twice with water and once with brine. The mixture was dried over anhydrous sodium sulfate, filtered over Celite funnel, and washed with diethyl ether. The organic phase was collected and concentrated on a rotary evaporator to afford crude product 2.5 g. Used directly in the next step.
With triethylamine; In dichloromethane; at 0 - 23℃;
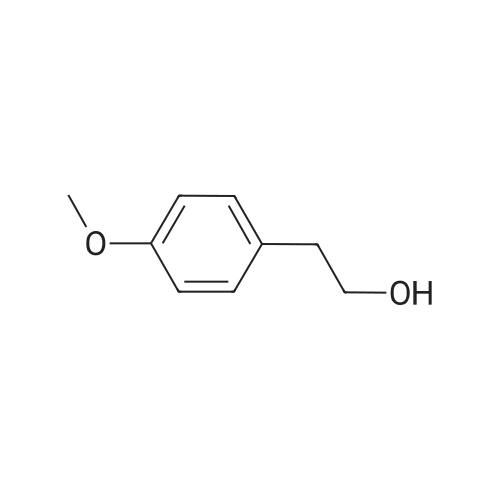
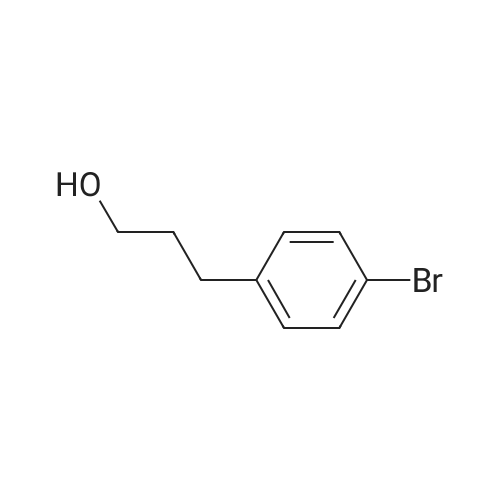
Triethylamine (5.2 ml, 37.3 mmol) was added to a solution of 4-bromophenethyl alcohol (5.0 g, 24.9 mmol) in CH2Cl2 (200 mL), and the resulting solution was cooled to 0 C. with an ice bath. Methanesulfonyl chloride (2.7 ml, 34.8 mmol) was added dropwise to the reaction mixture, and the resulting solution was allowed to warm slowly to room temperature overnight. The solution was washed with 0.1 N HCl and brine, and the combined organic extracts were dried over anhydrous MgSO4(s) and filtered, and the solvent was removed under reduced pressure. The crude yellow solid was purified by flash chromatography (silica gel, 70% v/v hexanes in CH2Cl2) to give mesylate 45 as a white solid in 98% yield. 1H NMR (CDCl3, 400 MHz) δ 7.42-7.40 (m, 2H), 7.21-7.16 (m, 2H), 4.41 (t, J=6.9 Hz, 2H), 3.03 (t, J=6.7 Hz, 2H), 2.90 (s, 3H) ppm; observed 13C NMR (CDCl3, 100.6 MHz) δ 138.87, 132.25, 130.51, 127.87, 123.92, 69.79, 37.67, 35.46 ppm; MS (ESI) m/z 300.9519 (MNa+[C9H11BrO3SNa+]=300.9595).
1-(3-{2-[Dimethyl-(1,1,2-trimethyl-propyl)-silanyloxy]-ethyl}-phenyl)-2-((1S,2R,3R,4R)-3-hydroxymethyl-7-oxa-bicyclo[2.2.1]hept-2-yl)-ethanol[ No CAS ]
With 1H-imidazole; In DMF (N,N-dimethyl-formamide); at 20℃;
3-Bromophenethyl alcohol was dissolved in 110 ml of N,N-dimethylformamide, and 16 ml of t-butyl chlorodiphenyl silane and 8.3g of imidazole were added. After stirring was continued at room temperature overnight, the solution was diluted with ethyl acetate, and washed successively with 1N hydrochloric acid and saturated brine. After drying the organic layer over anhydrous magnesium sulfate, the solvent was evaporated. The residue was purified by silica gel column chromatography, to give 22.1 g of [(3-bromophenethyl) oxy] (t-butyl)diphenyl silane in the 4:1 hexane-ethyl acetate fraction. Then, the obtained 22.1g of [(3-bromophenethyl)oxy](t-butyl)diphenyl silane was dissolved in 200 ml of tetrahydrofuran, and the mixture was cooled to -78 C under nitrogen atmosphere. 37 ml of Butyl lithium (1.52 M solution in hexane) was added thereto, and the mixture was stirred for 30 minutes, and then 10 ml of 4-formylmorpholine was added. After stirring was continued at -78C for 1 hour, IN-hydrochloric acid was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated. The residue was purified by silica gel column chromatography, to give 17 g of the title compound in the 4:1 hexane-ethyl acetate fraction.1H-NMR (CDCl3) δ: 0.12 (s, 6H) 0.93 (s, 9H) 4. 81(s, 2H) 7.49-7.53 (m, 1H) 7.60-7.62 (m, 1H) 7.77 (d, J=7. 6Hz, 1H) 7.87 (s, 1H) 10.02 (s, 1H)
3.27 g
With 1H-imidazole; In N,N-dimethyl-formamide; at 20℃;Cooling with ice;
Preparation Example 19 To a mixture of 1.5 g of <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong>, 1.0 g of imidazole and 15 mL of DMF was added 2.8 g of tert-butyl(chloro)diphenylsilane under ice cooling, and the followed by stirring for 1 hour at the same temperature then for a day at room temperature. Diethyl ether and water were added to the reaction mixture, followed by extraction with diethyl ether. The organic layer was washed with saturated brine and dried over anhydrous sodium sulfate, followed by concentration under reduced pressure. The residue was purified by silica gel column chromatography (hexane:ethyl acetate=7:3), thereby obtaining 3.27 g of [2-(3-bromophenyl)ethoxy](tert-butyl)diphenylsilane.
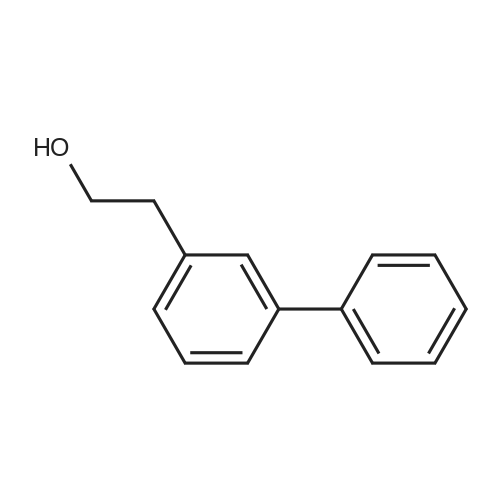
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water;Heating / reflux;
In a flask were combined the <strong>[28229-69-8]3-bromophenethyl alcohol</strong> (17.5 g, 87.1 mmol), phenyl boronic acid (12.7 g, 104.5 mmol), palladium tetrakistriphenylphosphine (2.0 g, 1.74 mmol), 2M cesium carbonate (105 mL, 210 mmol) and dimethoxyethylether (105 mL). Mixture stirred vigorously under nitrogen at reflux overnight. After cooling to ambient temperature, poured mixture into water (400 mL) and extracted with ethyl acetate (2×400 mL). Combined organics were washed with brine and dried over magnesium sulfate. Silica gel chromatography (ethyl acetate/hexane) provided the coupled product as a crystalline solid (15.04 g, 87.3%). NMR(CDCl3) δ 2.95 (t, 2H), 3.93 (q, 2H), 7.19-18 (m, 2H), 7.31-7.51 (m, 5H), 7.58 (d, 2H). GCMS EI+ 198 (M+).
87%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,2-dimethoxyethane; ethanol; water; for 4h;Reflux;
3-(2-Hydroxy-1-ethyl)-1,1'-biphenyl 2-(3-Bromophenyl)-1-ethanol (200 mg, 0.995 mmol) was dissolved in a mixed solvent of dimethoxyethane:ethanol (=5:1) (3.0 ml). To the solution, phenylboronic acid (242 mg, 1.985 mmol), a 2 M aqueous sodium carbonate solution (1.5 ml), and tetrakis(triphenylphosphine) palladium(0) (Pd(PPh)4, 56.0 mg, 0.048 mmol) were added, and the mixture was stirred for 4 hours under heating to reflux. After the reaction was confirmed by TLC to be complete, the reaction solution was filtered through celite, and the filtrate was neutralized by the addition of hydrochloric acid, followed by extraction with ethyl acetate (10 ml) three times. The organic layer was washed twice with brine and dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, and then, the residue was purified by silica gel column chromatography (hexane:ethyl acetate=8:2) to obtain 3-(2-hydroxy-1-ethyl)-1,1-biphenyl (172 mg, yield: 87%).
87%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,2-dimethoxyethane; ethanol; for 4h;Reflux; Inert atmosphere;
3-(2-Hydroxy-1-ethyl)-1,1'-biphenyl 2-(3-Bromophenyl)-1-ethanol (200 mg, 0.995 mmol) was dissolved in a mixed solvent of dimethoxyethane:ethanol (=5:1) (3.0 ml). To the solution, phenylboronic acid (242 mg, 1.985 mmol), a 2 M aqueous sodium carbonate solution (1.5 ml), and tetrakis(triphenylphosphine) palladium(0) (Pd(PPh3)4, 56.0 mg, 0.048 mmol) were added, and the mixture was stirred for 4 hours under heating to reflux. After the reaction was confirmed by TLC to be complete, the reaction solution was filtered through celite, and the filtrate was neutralized by the addition of hydrochloric acid, followed by extraction with ethyl acetate (10 ml) three times. The organic layer was washed twice with saturated saline and dehydrated over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, and then, the residue was purified using silica gel column chromatography (hexane:ethyl acetate=8:2) to obtain 3-(2-hydroxy-1-ethyl)-1,1'-biphenyl (172 mg, yield: 87%).
51%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium carbonate; In 1,4-dioxane; at 100℃; for 12h;Inert atmosphere;
A mixture of 2-(4-bromophenyl)ethan-l-ol (1 g, 4.97 mmol), phenylboronic acid (915 mg, 7.50 mmol), Pd(dppf)Cl2 (366 mg, 0.50 mmol), potassium carbonate (1.38 g, 9.98 mmol), and dioxane (20 mL) was stirred for 12 h at 100C under nitrogen. The solids were filtered out. The resulting solution was concentrated under vacuum. The residue was purified on a silica gel column eluting with ethyl acetate/petroleum ether (1/3) to afford the title compound (500 mg, 51%) as a white solid.
With copper(l) iodide;bis-triphenylphosphine-palladium(II) chloride; In tetrahydrofuran;Heating / reflux;
<strong>[28229-69-8]3-bromophenethyl alcohol</strong> (5.0 g, 24.9 mmol) and 2-(tributylstannyl)pyridine (13.6 g, 37.4 mmol) were combined in a round bottom flask with PdCl2(PPh3)2 (0.84 g, 1.2 mmol), CuI (0.23 g, 1.2 mmol) and anhydrous THF (100 mL) and heated to reflux. After refluxing overnight, additional PdCl2(PPh3)2 (0.84 g, 1.2 mmol) and CuI (0.23 g, 1.2 mmol) were added and the reaction refluxed overnight. The reaction was cooled to ambient temperature, Norit A charcoal added, the mixture stirred and then filtered through a bed of celite. Chromatography (on silica, ethyl acetate/hexanes) provided the alcohol as an orange oil (2.76 g, 55.8%). ESMS m/z=200 (M+H)+.
Example 1 4- ( (1 R)-2-ff2- (3-fr2- (Benzvloxv) ethoxvlmethvl} phenvl) ethvllamino-1-hydroxvethyl)-2- (hydroxymethyl) phenol acetate il 2-r2- (3-Bromophenvl) ethoxvltetrahydro-2H-pyran p-Toluenesulphonic acid monohydrate (0.40g) was added to a stirred solution of 2- (3- bromophenyl) ethanol (5.471 g) and dihydropyran (4.58g) in CH2CI2 (100ml) at 0C. The cooling bath was removed and the reaction mixture stirred at 20C for 4 h. Et3N (2ml) was added and the mixture evaporated under reduced pressure. The residue was purified by chromatography on a Biotage (90g) eluting with cyclohexane-Et20 (15: 1) to give the title compound (5.12g), ES+ve 302/304 (M+NH4) +
With toluene-4-sulfonic acid; In dichloromethane; at 20℃; for 2h;Inert atmosphere;
To a solution of <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong> (5.0 g, 25.0 mmol) in DCM (50 mL) were added 3,4-dihydro-2H-pyran (12.5 g, 62.0 mmol) and 4-methylbenzenesulfonic acid (0.3 g, 0.10 mmol) at RT. The reaction mixture was stirred for 2 hours, and then concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (PE/EtOAc: 2 to 4%) to afford the expected intermediate: 1H NMR (400 MHz, CDCl3) δ 7.47-7.31 (m, 2H), 7.26-7.13 (m, 2H), 4.61 (m, 1H), 4.07-3.43 (m, 4H), 2.91 (m, 2H), 1.97-1.46 (m, 6H).
With copper(l) iodide; potassium carbonate; N,N`-dimethylethylenediamine; In 1,4-dioxane; at 100℃; for 4.5h;Product distribution / selectivity;
Example 128 3- (3- { (1 R, 2S)-2- [4- (2-METHYL-5-QUINOLINYL)-1-PIPERAZINYL] CYCLOPROPYL} PHENYL)-1, 3- oxazolidin-2-one dihydrochloride and 3-(3-{(1S, 2R)-2-14-(2-METHYL-5-QUINOLINYL)-1- PIPERAZINYLICYCLOPROPYLPHENYL)-1, 3-OXAZOLIDIN-2-ONE DIHYDROCHLORIDE (E128) 3- [3- (2-HYDROXYETHYL) PHENYL]-1, 3-OXAZOLIDIN-2-ONE; A solution 2- (3-BROMOPHENYL) ETHANOL (1. 00G, 5. 00mmol), 2-oxazolidin-2-one (874mg, 10. 04MOL), copper (I) iodide (96mg, 0. 50MOL), N, N'-dimethyl-1, 2-ETHANEDIAMINE (60µL, 49mg, 0. 56MMOL) and potassium carbonate (1.04g, 7. 50MMOL) were suspended in dioxane (6 mL). The mixture was stirred under nitrogen for 2.5h at 100C. After addition of copper (I) iodide (96mg, 0. 50MMOL) and N, N'-dimethyl-1, 2-ethanediamine (60µL, 49mg, 0. 56MMOL) stirring was continued for another 2h at 100C. After cooling down to room temperature, the mixture was poured into saturated aqueous ammonium chloride solution and extracted with dichloromethane. The organic layer was dried over magnesium sulfate and concentrated under reduced pressure. The crude compound was purified by flash chromatography on silica gel, eluting with ethyl acetate-cyclohexane (1: 1 to 1: 0) affording the title compound in 64% yield (664mg). 1H-NMR (300 MHz, CDC13) 8 (ppm): 7.4 (s, 1H), 7.0-7. 3 (m, 2H), 7.85 (m, 1H), 4.25 (t, 2H), 3.85 (t, 2H), 3.65 (t, 2H), 2.65 (t, 2H), 1.50 (s, 1H).
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In toluene; for 1h;Heating / reflux;
2-(3-Thiophen-3-yl-phenyl)-ethanol; A mixture of 3-bromo-phenethyl-alcohol (1 g, 4.97 mmol), 3-thiophene-boronic-acid (1.9 g, 14.9 mmol, 3.0 equiv), Pd (PPh3) 4 (172 mg, 0.149 mmol, 0.03 equiv), and 2M Na2CO3 (10.8 mL, 22.4 mmol, 4.5 equiv) in toluene (40 mL) is heated to reflux 1 h, under an argon atmosphere. The resulting suspension is allowed to cool to RT and filtered through a pad of celite, washing the filter cake with CH2CI2 and water. The layers are separated. The organic phase is washed with brine, dried (Na2SO4), filtered and concentrated. Purification of the crude material by silica gel column chromatography (CH2CI2/Et2O, 95/5) affords the title compound: ES-MS: 205.0 [M+H] + ; single peak at tR= 4.27 min (System 2); Rf = 0.32 (CH2CI2/Et2O, 95/5)
5-methyl-7-phenylimidazo[5,1-f][1,2,4]triazin-2-amine hydrochloride[ No CAS ]
[ 28229-69-8 ]
2-{3-[(5-methyl-7-phenylimidazo[5,1-f][1,2,4]triazin-2-yl)amino]phenyl}ethanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium t-butanolate;tris-(dibenzylideneacetone)dipalladium(0); 2,2'-bis-(diphenylphosphino)-1,1'-binaphthyl; In 1,4-dioxane; at 160℃; for 0.25h;Microwave irradiation;
To a mixture of 5-methyl-7-phenylimidazo [5, 1-4 [1,2, 4] triazin-2-amine hydrochloride (0.026 g, 0.10 MMOL), 2- (3-BROMOPHENYL) ETHANOL (0.022 g, 0.11 MMOL), tris (DIBENZYLIDENEACETONE) DIPALLADIUM (2.7 mg, 0.0030 MMOL), (S)- (-)- 2,2'-bis (diphenylphosphino)-1, 1'-binaphthyl (5.6 mg, 0.0090 MMOL) and sodium t-butoxid (0.025 g, 0.23 MMOL) was added dioxane (1 mL). The solution was heated with microwave radiation to 160 C for 15 minutes. After cooling to room temperature, the solution was filtered, and silica gel (1 G) was added, followed by evaporation of the volatiles under reduced pressure. The pre-adsorbed solids were loaded into a solid loading cartridge and subjected to a gradient elution using ethyl acetate: hexanes (50: 50) to ethyl acetate: hexanes (100: 0) using a RediSep silica gel cartridge (4 g ; ISCO). The appropriate fractions were combined and concentrated under reduced pressure to give 2- {3- [ (5-METHYL-7-PHENYLIMIDAZO [5, 1-4 [1,2, 4] triazin-2- yl) amino] ethanol (0.0097 g) as a yellow SOLID. 1H NMR (CDCI3) : 8 8.75 (s, 1 H), 8.51-8. 45 (m, 2H), 7.74 (s, 1 H), 7.56-7. 26 (m, 5H), 7.04-6. 95 (m, 2H), 3.91 (t, J= 6.4 Hz, 2H), 2.92 (t, J= 6.4 Hz, 2H), 2.59 (s, 3H). MS m/z 346 (M+1).
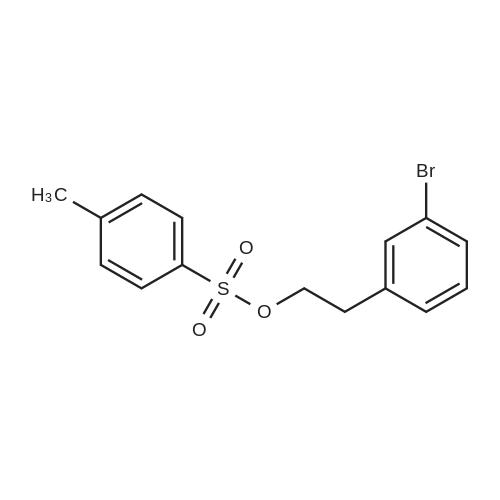
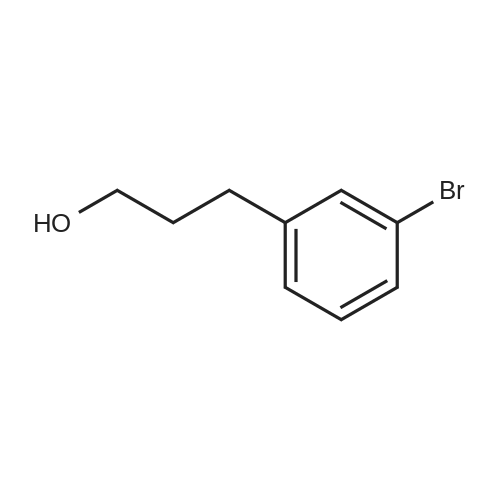
b 3-Bromobenzeneethanol 4-methylbenzenesulphonate 4-Methylbenzenesulphonyl chloride (4.38 g) was added portionwise to a solution of 3-bromobenzeneethanol [Example H(a)] (2.30 g, 11.4 mmol) in pyridine (30 ml) at 0 C., and the mixture stood at -20 C. for 16 h. The mixture was diluted with water, extracted with diethyl ether, the extracts combined, washed twice with aqueous 4N hydrochloric acid and once with saturated aqueous sodium chloride, dried over sodium sulphate and evaporated to afford a pale yellow oil (3.98 g). [M-OTs]+ 182; 360 MHz 1 H n.m.r (CDCl3) 7.66 (2H, d, J 8.3 Hz), 7.34 (1H, dm, J 7.9 Hz), 7.30-7.26 (2H, m), 7.20 (1H, d, J 1.5 Hz), 7.12 (1H, t, J 7.7 Hz), 7.05 (1H, d, J 7.7 Hz), 4.20 (2H, t, J 6.8 Hz), 2.91 (2H, t, J 6.8 Hz), 2.44 (3H, s).
With sodium hydroxide; dihydrogen peroxide; In tetrahydrofuran; water;
a 3-Bromobenzeneethanol Borane-dimethylsulphide complex (2.0M in THF, 49 ml) was added dropwise to a stirred solution of 3-bromostyrene (10 g, 55 mmol) in THF (130 ml) at 0 C. and the solution was stirred for 3 h at room temperature. 10% Aqueous sodium hydroxide was added cautiously, followed by hydrogen peroxide (30% wt. solution in water, 6 ml) dropwise. After a further 16 h, the mixture was diluted with water, extracted twice with ethyl acetate, the combined extracts washed with saturated sodium bisulphite solution and saturated sodium chloride solution, dried over sodium sulphate and evaporated to give a colourless oil. Purification by flash chromatography, eluding with 30% diethyl ether/hexane gave a colourless oil, [M+Si(CH3)3 derivative]+
tetrakis(triphenylphosphine) palladium(0); In N,N-dimethyl-formamide; at 100℃;
Zn(CN)2 (5.03 g, 0.028 mol) and Pd(PPh3)4 (2.3 g, 0.002 mol) were added to a solution of 2-(3-bromophenyl)ethanol (8.3 g, 0.412 mol; see step (i) above) in dry DMF (75 mL). The reaction mixture was stirred overnight at 100C before being cooled to room temperature and quenched with water. The resulting mixture was extracted with ethyl acetate and the organic layer was separated, dried over anhydrous Na2SO4 and then concentrated. The resulting crude product was purified by column chromatography, using 15% ethyl acetate / petroleum ether as eluent, to give 4.2 g of the sub-title compound as yellow liquid.
(1R,2R)/(1S,2S)-1-(3-bromophenethoxy)-2-(4-morpholinyl)cyclohexane monohydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
(iii) To sodium hydride, 80% oil dispersion, previously washed with hexanes (3 x 10 mL), (0.60 g, 25 mmol) in dry dimethylformamide (50 mL) was added via cannula a solution of <strong>[28229-69-8]3-bromophenethyl alcohol</strong> (4.0 g, 20 mmol) in dry dimethylformamide (50 mL). Addition was followed by evolution of a gas and the reaction mixture was stirred at room temperature for 3 hours. The mesylate as prepared in (ii) above was dissolved in dry dimethylformamide (50 mL) and the resulting solution was added quickly (2 min.) via cannula to the reaction mixture. The reaction mixture was heated to 85C for 2 hours, then the temperature was reduced to 45C and the reaction stirred overnight. The reaction mixture was poured into ice-water (800 mL) and extracted with ethyl acetate (3 x 200 mL). The combined organic extracts were backwashed with a saturated aqueous solution of sodium chloride (300 mL) and dried over sodium sulfate. Evaporation of the solvent in vacuo provided 8.0 g of an oil which was dissolved in ether (100 mL) and treated with a saturated solution of HCl in ether (100 mL). An oil precipitated and the solvent was evaporated in vacuo and the resulting crude hydrochloride salt was dissolved in water (200 mL). The acidic aqueous solution was extracted with ethyl ether (2 x 100 mL) and then basified to pH 10 with an aqueous solution of sodium hydroxide (50% w/v). The basic aqueous solution was extracted with ethyl ether (3 x 100 mL), the combined organic layers were dried over sodium sulfate and concentrated in vacuo to leave 2.9 g of the crude free aminoether. The crude product was purified by chromatography on silica gel 60 (70-230 mesh) with a mixture of ethyl acetate-dichloromethane (1:1, v/v) as eluent to provide the pure free base. The product was dissolved in ethyl ether (50 mL) and converted to the monohydrochloride salt by adding saturated solution of HCl in ethyl ether (50 mL). The solvent was evaporated in vacuo, the residue was dissolved in the minimum amount of cold ethanol and addition of ether triggered formation of crystals (0.53 g), m.p. 145-148C, having the elemental analysis indicated in Table 1.
With triethylamine;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; at 90℃; under 3750.38 Torr; for 10h;
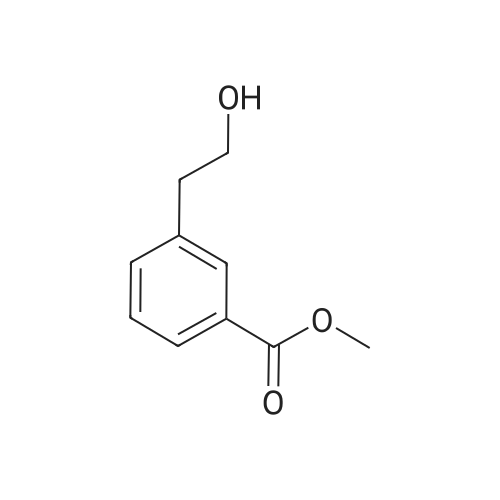
Intermediate 56; 3-(2-Hydroxy-ethyl)-benzoic acid methyl ester; 2-(3-Bromo-phenyl)-ethanol (5.00 g, 24.87 mmol), 1 ,1'-bis(diphenylphosphino) ferrocene-palladium dichloride (0.70 g, 0.85 mmol) and triethylamine (7.00 g, 69.18 mmol) in MeOH (30 ml.) was heated at 90 0C (internal temperature) over a period of 10 h under carbon monoxide atmosphere (5 bar). After cooling to RT the solution was concentrated in vacuo and then partitioned between ethyl acetate and water. The organic layer was washed with brine and evaporated in vacuo. The crude product was purified by flash silica chromatography eluting with ethyl acetate//-hexane (1 :1).Yield: 3.3 g (74%).1H NMR (400 MHz, CDCI3): delta 7.93 - 7.90 (m, 2H), 7.44 (d, J = 7.2 Hz, 1 H),7.39 (t, J = 8.1 Hz, 1 H), 3.92 (s, 3H), 3.89 (t, J = 6.2 Hz, 2H), 2.93 (t, J = 6.4
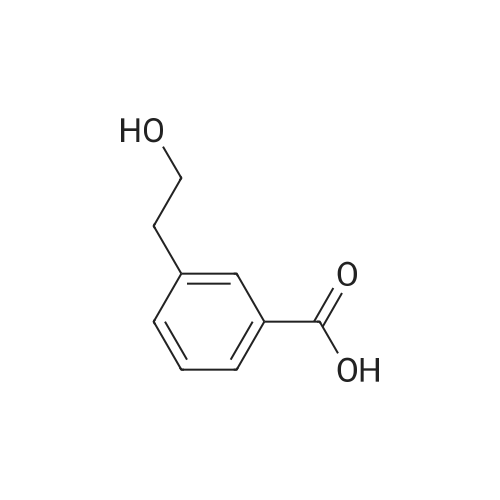
In a flame dried round-bottomed flask equipped with a magnetic stir bar and under inert atmosphere (N2), to a solution of 3-bromophenethyl alcohol (2.34 g, 11.29 mmol) and lambda/,lambda/,lambda/',lambda/'-tetramethylethylendiamine (3.24 ml_, 22.58 mmol) in Et2O (29.0 ml.) at -78 0C was added dropwise n-BuLi (14.0 ml. of a 1.6M solution in hexane, 22.59 mmol), maintaining the temperature at -78 0C. The reaction mixture was then stirred at -20 0C for2 h. Dry carbon dioxide gas was then bubbled for 10 min through the reaction mixture at -78 0C. The cooling bath was removed and the reaction mixture was stirred for 1 h. The reaction mixture was extracted with water (50 ml_). The aqueous layer was acidified to pH = 1 with 2N HCI and extracted with EA (2 x 75 ml_). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure to give the title compound as a beige solid: LC-MS-conditions 02: tR = 0.67 min.
With N-benzyl-trimethylammonium hydroxide; In methanol; acetonitrile; at 40℃; for 5h;Inert atmosphere;
m-Bromophenylethanol (2a) (80.4 g, 0.4 mol) was placed in acetonitrile (200 mL).tert-Butyl acrylate (76.9 g, 0.6 mol) was added, and benzyltrimethylammonium hydroxide (50.2 g, 0.12 mol, 40% in methanol) was added dropwise, and stirred at 40 C for 5 hours.Concentrated under reduced pressure to remove most of the reaction solvent.Water (200 mL) and diethylamine (50 mL) were added to the residue, and then stirred at room temperature for 30 min.Water (500 mL) was added and extracted with ethyl acetate (500 mL×2).The organic phase was washed with saturated brine (200 mL) and dried over anhydrous sodium sulfate. After filtration and concentration of the filtrate under reduced pressure,The residue was separated by silica gel column chromatography ( petroleum ether / ethyl acetate (v/v) = 12:1)2b (94.0 g, yield 71.4%) was obtained as a yellow liquid.
With N-benzyl-trimethylammonium hydroxide; In methanol; toluene; for 24h;Product distribution / selectivity;
Procedure B) A solution of <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong> (5 g) was stirred in toluene (30 mL) followed by the addition of Triton-B in methanol (0.57 mL). The volatiles were removed until ~10 mL remained. To this solution was added tert-butyl acrylate (3.94 mL) and the mixture was left to stir for 24 h. The solvent was evaporated and the residue was purified on silica eluting with isohexane-10% ethyl acetate/isohexane. The solvent was evaporated to afford the sub- titled compound as a colourless oil (6.7 g).1H NMR (300 MHz, CDCl3) δ 7.39 - 7.30 (m, 2H), 7.17 - 7.12 (m, 2H), 3.68 (t, J = 6.5 Hz, 2H), 3.64 (t, J = 6.9 Hz, 2H), 2.84 (t, J = 6.9 Hz, 2H), 2.48 (t, J = 6.5 Hz, 2H), 1.44 (t, 9H)
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 20℃;Cooling with ice;
Example 94; 2-{3-[2-(3-Cyano-phenyl)-ethoxy]-4-methoxy-benzoylamino}-indane-2-carboxylic acid; Step 1 : 3-[2-(3-Bromo-phenyl)-ethoxy]-4-methoxy-benzoic acid methyl ester; Methyl 3-hydroxy-4-methoxybenzoate (500 mg, 2.75 mmol) and triphenylphosphine (1.08 g, 4.12 mmol) were dissolved in THF (13 ml), the solution was cooled in an ice bath and 2-(3-bromophenyl)-ethanol (662 mg, 3.29 mmol) and DIAD (886 mg, 4.12 mmol) were added sequentially. Stirring was continued for 3 h at room temperature. The reaction mixture was evaporated to dryness and the residue purified by preparative RP HPLC (water/ACN gradient) to give 900 mg of the title compound. 1H-NMR: δ = 7.62-7.58 (m, 2H); 7.45 (d, 1 H); 7.41 (d, 1H); 7.35 (d, 1H); 7.28 (dd, 1 H); 7.08 (d, 1 H); 4.22 (t, 2H); 3.83 (s, 3H); 3.80 (s, 3H); 3.01 (t, 2H)
With tetra(n-butyl)ammonium hydrogensulfate; sodium hydroxide; In water; toluene; at 20℃; for 3h;
To a mixture of <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong> (3.40 g, 16.9 mmol), tert-butyl 2- bromoacetate (26.0 g, 133 mmol, 8.0 equiv.), tetra-w-butylammonium hydrogen sulfate (4.52 g, 13.3 mmol, 0.80 equiv.), and 84 mL of toluene was added 267 mL of 5N aqueous NaOH solution. The reaction mixture was stirred at room temperature for 3 h. The aqueous layer was then extracted with four portions of EtOAc, dried (MgSO4), and evaporated. The residue was purified by CC on silica gel with EtO Ac/petroleum ether 1:40 to afford the title compound (4.8 g, 90%) as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 7.42 (s, IH), 7.35- 7.39 (m, IH), 7.18-7.20 (m, 2H), 3.98 (s, 2H), 3.76 (t, 2H, J = 6.9 Hz), 2.94 (t, 2H, J = 7.2 Hz), 1.50 (s, 9H).
90%
With tetra(n-butyl)ammonium hydrogensulfate; sodium hydroxide; In water; toluene; at 20℃; for 3h;
tert-Butyl 2-(3-Bromophenethoxy)acetate To a mixture of <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong> (3.40 g, 16.9 mmol), tert-butyl 2-bromoacetate (26.0 g, 133 mmol, 8.0 equiv.), tetra-n-butylammonium hydrogen sulfate (4.52 g, 13.3 mmol, 0.80 equiv.), and 84 mL of toluene was added 267 mL of 5N aqueous NaOH solution. The reaction mixture was stirred at room temperature for 3 h. The aqueous layer was then extracted with four portions of EtOAc, dried (MgSO4), and evaporated. The residue was purified by CC on silica gel with EtOAc/petroleum ether 1:40 to afford the title compound (4.8 g, 90%) as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 7.42 (s, 1H), 7.35-7.39 (m, 1H), 7.18-7.20 (m, 2H), 3.98 (s, 2H), 3.76 (t, 2H, J=6.9 Hz), 2.94 (t, 2H, J=7.2 Hz), 1.50 (s, 9H).
Step 1: tert-butyl 2-(3-bromophenethoxy)acetate To a solution of <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong> (2 g, 9.9 mmol) in dry THF (15 mL) under argon at 0 C., was added dropwise NaHMDS 1M in THF (14.8 mL, 14.8 mmol). The reaction mixture was stirred at 0 C. for 30 min. Tert-butyl 2-bromoacetate (3.9 g, 19.9 mmol) in THF (2 mL) was slowly added. The reaction mixture was stirred at rt for 3 h. The completion of the reaction was monitored by anal. HPLC. The reaction was quenched with a solution of NH4Cl, washed with brine and dried over Na2SO4 and filtered. The filtrate was concentrated in vacuo to obtain the crude product which was purified by flash chromatography (DCM/MeOH) to obtain the title compound.
In dichloromethane; at 20℃; for 2.5h;Inert atmosphere;
General procedure: A mixture of the substituted phenylethyl alcohol (10 mmol), Chloromethyl methyl ether (15mmol) and N,N-diisopropylethylamine (20 mmol) in dry dichloromethane (25 mL)was stirred under nitrogen atmosphere for 2.5 h at rt. The reaction mixture wasthen washed with water (2×50 mL), dried (NaSO4) and the solvent wasremoved in vacuo. The crude MOM acetal was dissolved in dried CH3CN (25mL) and added to cooled (0 oC) solution of Trimethylsilyltrifluoromethanesulfonate (TMSOTf) (10 mmol). The reaction was carried outunder nitrogen atmosphere for 3 h. Then the mixture was quenched by theaddition of l M NaHCO3 (20 mL ). The orgnic phase was washed withbrine (2×50 mL), dried (NaSO4) and evaporated under reducedpressure. Purification by FC afforded relevant substituted isochromans.
To a solution of <strong>[28229-69-8]2-(3-bromophenyl)ethan-1-ol</strong> (1.00 g, 0.67 mL, 4.9 mmol) in anhydrous tetrahydrofuran (50 mL), sodium hydride (60% in mineral oil, 0.430 g, 10.8 mmol) was added and the resulting mixture was stirred for 15 min. at 0 C. A dropwise addition of benzyl bromide (1.67 g, 9.8 mmol) was then made at 0 C and the stirring continued for 12 h allowing the temperature to rise to room temperature. The reaction was quenched byaddition of water and the resulting mixture was extracted with dichloromethane (3 x 40 mL). The combined organic layers were dried over sodium sulfate, filtered and evaporated under reduced pressure. Purification via silica gel column chromatography using a mixture of 10% ethyl acetate in hexanes as eluent furnished 15 (yield: > 97%). 1HNMR (500 MHz, CDCl3) δ 7.43 - 7.38 (m, 1H), 7.38 - 7.32 (m, 3H), 7.31 - 7.29 (m, 3H),7.19 - 7.15 (m, 2H), 4.53 (s, 2H), 3.69 (t, J = 6.9 Hz, 2H), 2.91 (t, J = 6.9 Hz, 2H) ppm. 13C NMR (126 MHz, CDCl3) δ 141.69, 138.45, 132.18, 130.06, 129.53, 128.61, 127.84, 127.83, 122.58, 73.25, 70.84, 36.21 ppm.
89%
(126a) 1-[2-(benzyloxy)ethyl]-3-bromobenzene; N,N-dimethylformamide (20 mL) solution of <strong>[28229-69-8]3-bromophenethyl alcohol</strong> (2.04 g, 10.0 mmol) was cooled to 0C and was blended with sodium hydride (55% oily, 436 mg, 10.0 mmol) and the mixture was stirred at 0C for 15 minutes. Then the reaction mixture was blended with methyl iodide (1.40 mL, 12.0 mmol) and the mixture was stirred at room temperature for two hours. Water (50 mL) was added to the reaction liquid and extracted with ethyl acetate (50 mL) three times. The organic layers were combined and washed with water (50 mL) and a saturated saline solution (50 mL), the solvent was evaporated under reduced pressure after drying over sodium sulfate, the obtained residue was purified by silica gel column chromatography (hexane/toluene = 3:1) and the title compound was obtained (2.60 g, 89%). Colourless liquid IR (film) νmax 2858, 1568, 1475, 1361, 1203, 1103, 695 cm-1; 1H NMR(CDCl3, 400 MHz) δ 2.88 (2H, t, J = 7.0 Hz), 3.66 (2H, t, J = 7.0 Hz), 4.50 (2H, s), 7.11-7.15 (2H, m), 7.23-7.38 (7H, m); MS (EI) m/z: 292, 290 [M+], 262, 260, 184, 169, 91.
3-(2-Hydroxyethyl)benzaldehyde (13) A mixture <strong>[28229-69-8]2-(3-bromophenyl)ethanol</strong> (1.00 g, 0.67 mL, 4.9 mmol) and N,N,N',N'-tetramethylethylenediamine (1.5 mL) in anhydrous diethyl ether (10 mL) was cooled to -78 C. in a dry-ice acetone bath. n-Butyl lithium (2.4 M solution in hexanes, 4.0 mL, 9.8 mmol) was added dropwise and the resulting mixture was allowed to warm to -20 C. (over 1 h) and then cooled again to -78 C. Anhydrous N,N-dimethylformamide (5 mL) was added and the resulting mixture was allowed to reach room temperature and stirred for an additional 1 h. The reaction mixture was quenched with a saturated solution of ammonium chloride and the aqueous portion was extracted with ethyl acetate. The combined extracts were washed with brine, dried over magnesium sulfate, filtered and evaporated. The residue was purified by silica gel column chromatography using ethyl acetate-hexanes 2:3 as eluent to provide the desired compound as colorless oil. Yield: 88%. 1H NMR (CDCl3): δ 1.69 (broad s, 1H), 2.96 (t, J=6.5 Hz, 2H), 3.91 (t, J=6.5 Hz, 2H), 7.47-7.53 (m. 2H), 7.74-7.76 (m, 2H), 10.00 (s, 1H) ppm. 13C NMR (CDCl3): δ 39.0, 63.4, 128.4, 129.4, 130.1, 135.5, 136.9, 140.1, 192.6 ppm. IR: v 3381, 1696 cm-1.
78%
A mixture of n-butyl lithium (2.4 M solution in hexanes, 8.0 mL, 19.6 mmol) in anhydrous THF (20 mL) was cooled to -78C. To this reaction mixture, N123-B160033-070-1 (1.0 g, 4.97 mmol) in THF (15 mL) was added dropwise at -78 C and stirred at same temperature for 20 min. N,N-dimethylformamide (6 mL) was added dropwise at -78C and stirred at same temperature for 2 h. The reaction mixture was quenched with a saturated solution of ammonium chloride and the aqueous portion was extracted with ethyl acetate. The combined extracts were washed with brine, dried over magnesium sulfate, filtered and evaporated. The residue was purified by silica gel column chromatography using ethyl acetate:hexanes 2:3 as eluent to provide the desired compound. Colorless oil (580 mg , 78%).
53%
To a solution of N,N,N',N'-tetramethylethylenediamine (15.0 mL, 49.74 mmol, 1.0 eq) and dimethylformamide (40 mL, 497.36 mmol, 10.0 eq) in THF (150 mL) was added n- butyllithium (40 mL, 99.47 mmol, 2.0 eq) at -70 C under N2and stirred at -20 C for 1 h. Then to the reaction mixture was added <strong>[28229-69-8]3-bromophenethyl alcohol</strong> (10 g, 6.7 mL, 49.74 mmol, 1.0 eq) at -70 C and slowly warmed to 25 C and stirred for 2 h. The mixture was diluted with water and extracted with ethyl acetate. The combined organic phase was washed with brine, dried over sodium sulfate, filtered and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (PE/EtOAc 16-25) to afford the title compound (4 g, 26.64 mmol, 53% yield) as a yellow oil.
With palladium bis[bis(diphenylphosphino)ferrocene] dichloride; potassium acetate; In dimethyl sulfoxide; at 120.0℃; for 18.0h;Inert atmosphere;
Stage 1: 2-[3-(4,4, 5, 5-Tetramethyl- 1, 3,2-dioxaborolan-2-yl)phenyl]ethanol To a solution of 2-(3-bromophenyl)-ethanol (3.0 g, 15 mmol) in anhydrous DMSO (30 mL) were added KOAc (4.39 g, 45 mmol), bis(pinacolato)diboron (5.68 g, 22 mmol) and PdCI2(dppf)2 (0.61 g, 0.74 mmol) and the reaction mixture was heated at 120C under a nitrogen atmosphere for 18 hrs. The reaction was cooled to RT and EtOAc (60 mL) was added. The reaction mixture was filtered through Celite, washing with EtOAc (500 mL). The filtrate was washed with sat NaHCO3 (150 mL), water (150 mL), brine (150 mL), dried over Mg504, filtered and concentrated in vacuo. The crude material was purified by automated column chromatography using EtOAc in heptane (gradient 0-100%) and then column chromatography (33% EtOAc/heptane) to give the title compound as a pale yellow oil (3.30 g, 89%).1H NMR (300 MHz, ODd3) oe ppm: 7.70 (2H, m), 7.35 (2H, m), 3.89 (2H, t, J=6.6 Hz),2.90 (2H, t, J=6.6 Hz), 1.51 (1H, s), 1.37 (12H, s).
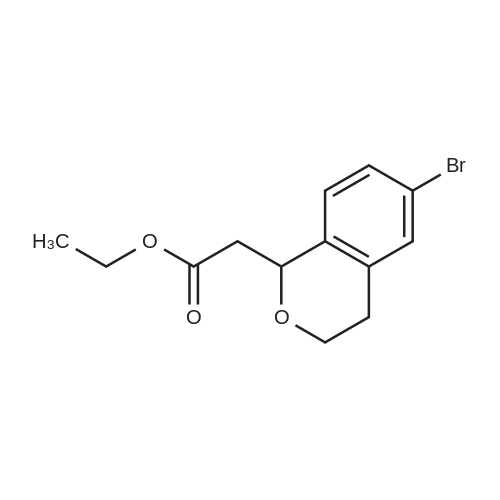
With titanium tetrachloride; In nitromethane; at 20℃; for 12h;Cooling with ice;
TiCl4 (7.48 g, 40 mmol) was added over a period of 10 min to an ice-cooled mixture of X-1 (2 g, 10 mmol) and X-1A (2.12 g, 12 mmol) in CH3NO2 (20 mL). The solution was allowed to stir at rt for 12 hrs. Then the mixture was poured into the HCl (aq. 1N) and extracted with DCM, dried over Na2SO4, concentrated in vacuo. The residue was purified by column chromatography to afford X-2 (0.5 g, yield 95%).
2-(3-bromophenyl)ethyl (E)-3-(3,4-dihydroxyphenyl)acrylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
46%
With ytterbium(III) triflate; In nitromethane; at 120℃;
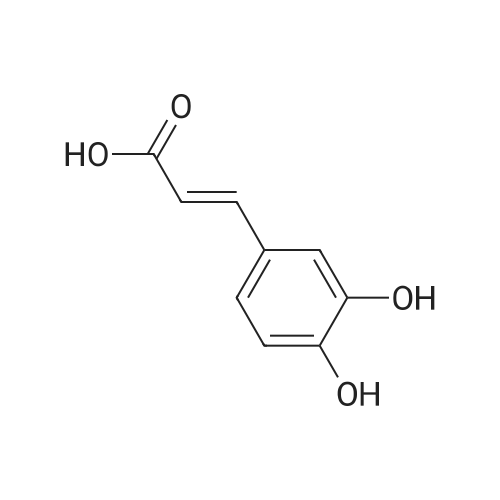
General procedure: To a mixture of caffeic acid fine powder (1.0 g, 5.56 mmol), various phenethyl alcohols (5.56 mmol) in CH3NO2 (125 mL) was added Yb(OTf)3 (34.4 mg, 0.056 mmol). After 5 min of ultrasonic shake, the mixture was stirred on a 120 C oil bath for 40-120 min. The reaction mixture was cooled to room temperature, washed with 2% NaHCO3 (30 mL) and brine, dried over anhydrous Na2SO4, and concentrated to give crude products, which were purified by column chromatography to give the compounds 1-26 in 18-61% yields.
45.5%
With ytterbium(III) triflate; In nitromethane; at 120℃; for 0.666667h;
General procedure: To a mixture of caffeic acid fine powder (1.0 g, 5.56 mmol, 1.0 equiv.),alcohol (5.56 mmol, 1.0 equiv.) in nitromethane (125 mL) was addedytterbium triflate (34.4 mg, 0.056 mmol, 0.01 equiv.). After 5 min inan ultrasonic bath the mixture without protective gas was stirred ona 120 C oil bath for a given time. The reaction mixture was cooled toroom temperature, washed with deionised water (30 mL), 2% NaHCO3(30 mL) and brine, dried over anhydrous Na2SO4 and evaporated underreduced pressure to give the crude product, which was purified on asilica gel column to give the compounds 1-5 and 8-30.
3-bromophenethyl 3-(3,4-dihydroxyphenyl)acrylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
63%
General procedure: Add 25 ml of ultra-dry THF, 1 mmol caffeic acid, 1.2 mmolR-OH, and 1.5 mmol triphenylphosphine (TPP) to thereaction flask, and then add 1.5 mmol diisopropyl azodicarboxylate(DIAD) in an ice bath. After reacting at roomtemperature for 36 h, the reaction was monitored by TLC.After the reaction, it was concentrated in vacuo to removeTHF, re-dissolved in absolute ethanol, added 3.5 mmolZnCl2, and reacted at room temperature for 18 h to removetriphenylphosphorus oxide and monitored by TLC. Afterthe reaction, the precipitate was removed by filtration. Thefiltrate was concentrated in vacuo, EA (3 × 30 ml) wasextracted three times, the organic layer was dried overanhydrous Na2SO4, and purified by column chromatography(PE:EA = 5:1).
With tetrakis(triphenylphosphine) palladium(0); sodium hydrogencarbonate; In 1,2-dimethoxyethane; for 48h;Reflux; Inert atmosphere;
3-Bromophenethyl alcohol (4 ml, 29.8 mmol) was dissolved in 1,2-dimethoxyethane (225 mL) and mixed with tetrakistriphenylphosphine palladium[0] (2.6 g, 2.25 mmol) at room temperature. The reaction mixture was then added to a solution of 2-thienyl-boronic acid (12.6 g, 99 mmol) and 1 N NaHCO3 (90 mL). The reaction mixture was heated to reflux under nitrogen atmosphere for 48 hours. The reaction mixture was partitioned with water and ethyl acetate. The organic layer was dried with MgSO4, filtered through a plug of silica and the solvent was evaporated in vacuo to yield a crude oil. The crude oil was purified via flash chromatography (30% EtOAc/hexane) to yield the title compounds as an oil.
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 0 - 20℃; for 5h;
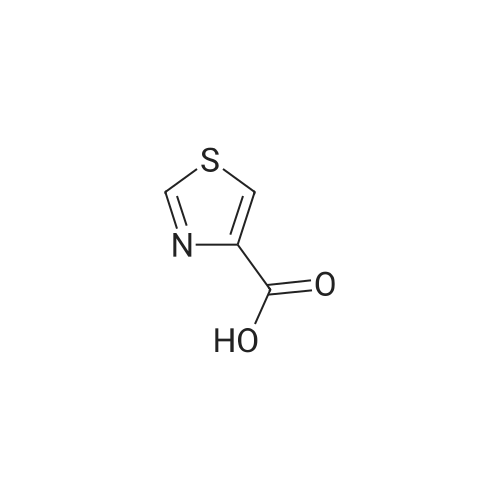
General procedure: 2.5mmol thiazole-4-carboxyli acid and 2.0mmol alcohol were dissolved in 25mL dichloromethane (DCM) in a dry flask with continuous stirring, followed by the addition of 2.5mmol 3-(3-dimethylaminopropyl) -1-ethylcarbodiimide hydrochloride. When the temperature of the reaction system cooled to 0°C, 0.2mmol 4-dimethylaminopyridine was added dropwise and reacted for 1hat 0°C. Then the temperature was elevated to room temperature for another 4h reaction. The reaction was stopped by adding 25mL saturated NaHCO3 solution and extracted twice with 20mL dichloromethane (2×20mL). The extracted organic layers were first dried by anhydrous Na2SO4, and then filtered and concentrated under vacuum distillation, obtaining the crude products. Finally, the crude products were further purified using column chromatography (ethy lacetate:petroleum ether, 1:5).
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 0 - 20℃; for 5h;
305.3mg benzoic acid (2.5mmol) and 313.6mg 3-chlorophenethylalcohol (2.0mmol) were dissolved in 25mL DCM in a dry flask with continuous stirring, followed by the addition of 418mg 3-(3-Dimethylaminopropyl)-1-Ethylcarbodiimide Hydrochloride (2.5mmol). When the temperature of the reaction system cooled to 0C, 50mg 4-Dimethylaminopyridine (0.2mmol) was added dropwise and reacted for 1hat 0C. Then the reaction temperature was elevated to room temperature for another 4h. The reaction was stopped by adding 25mL saturated NaHCO3 solution and extracted twice with 20mL dichloromethane (2×20mL). The extracted organic layers were first dried by anhydrous Na2SO4, and then filtered and concentrated under vacuum distillation, obtaining the crude products. Finally, the crude products were further purified using column chromatography (ethy lacetate:petroleum ether, 80:1) Yellow oil 76.9% yield; 95.22% purity. 1H NMR (300MHz, CDCl3) δ 8.00 (dt, J=8.5, 1.7Hz, 2H), 7.59-7.50 (m, 1H), 7.46-7.37 (m, 2H), 7.28 (s, 1H), 7.22 (dd, J=5.3, 2.8Hz, 2H), 7.16 (dd, J=2.5, 1.4Hz, 1H), 4.50 (t, J=6.9Hz, 2H), 3.04 (t, J=6.8Hz, 2H). 13C NMR (75MHz, CDCl3) δ 166.6, 143.0, 140.1, 134.4, 133.2, 130.3, 130.0, 129.72, 129.3, 128.6, 127.3, 127.0, 77.7, 77.2, 76.8, 65.1, 35.1. HRMS-ESI (m/z): calcd for C15H13ClO2 [M+H]+: 261.0677, found 261.0675.
tert-butyl 2-(3-bromophenethoxy)propanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With hydrogenchloride;
Step 1: tert-butyl 2-(3-bromophenethoxy)propanoate To a solution of <strong>[28229-69-8]2-(3-bromophenyl)ethan-1-ol</strong> (120 mg, 0.6 mmol) and tert-butyl 2-bromopropanoate (150 mg, 0.72 mmol) in dry DMF (5 mL) under argon, was added NaH (244 mg, 1.8 mmol). The reaction mixture was stirred at reflux. The completion of the reaction was monitored by anal. HPLC. The reaction was quenched with a solution of 0.5N HCl and diluted in AcOEt. The mixture was washed with saturated aqueous NaHCO3, brine and dried over Na2SO4 and filtered. The filtrate was evaporated in vacuo to obtain the title compound.
With glycerol; In dimethyl sulfoxide; at 0℃; for 0.5h;Inert atmosphere;
General procedure: (a) After mixing 5 mg / mL of titanium dioxide photocatalyst (P25, commercially available, Degussa), 10 mmol of benzaldehyde and 2 mL of methanol, and dispersing it by ultrasonic (electric power 80W) for 30 min to obtain a suspension; (b) The dispersed suspension is irradiated with LED light simulating sunlight (illumination wavelength is 365nm, optical power is 2.0W / cm2) and protected by nitrogen, and stirred at room temperature for 2h to obtain a reactant solution (containing benzyl alcohol. The gas chromatographic test of the reactant solution showed the spectrum shown in Figure 1, which shows that the benzaldehyde peak at 5.3min is basically not observed; that is, the conversion rate of benzaldehyde is 100%); (c) Extract the reactant solution (take the organic phase) and distill it to obtain benzyl alcohol (analyzed by GC (ie gas chromatography) and GC-MS (ie gas chromatography-mass spectrometry). The selectivity of benzyl alcohol is 91.4%).
1-(6-bromoisochroman-1-yl)-N-methylmethanamine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77%
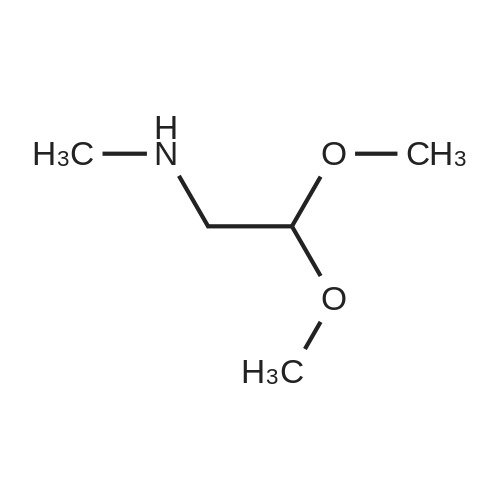
With trifluorormethanesulfonic acid; In neat (no solvent); at 0 - 60℃; for 16h;
To a solution of 2-(3-bromophenyl) ethanol (500 mg, 2.48 mmol) and 2,2-dimethoxy- N-methylethanamine (0.886 g, 7.44 mmol) was added trifluoromethanesulfonic acid (6 mL) at 0 C. The reaction mixture was heated to 60 C and stirred for 16 h. Upon completion, the reaction was cooled and neutralized with aqueous sodium bicarbonate to pH = 8 at 0 C. The aqueous mixture was extracted with ethyl acetate (3 chi 100 mL), washed with brine (1 chi 100 niL), and dried over anhydrous sodium sulfate. Purification by column chromatography afforded the desired product (500 mg, yield: 77%) as a yellow solid. MS (ESI): m/z 256 [M + H]+.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping