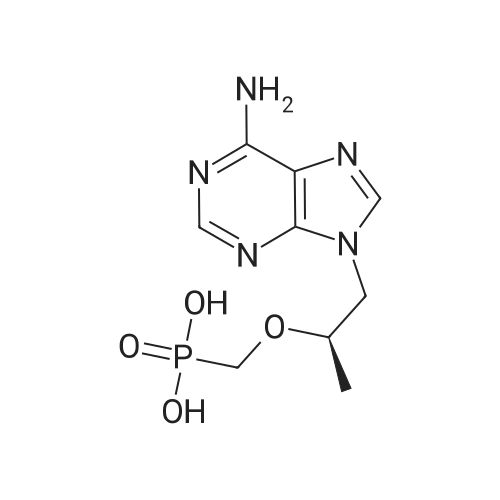
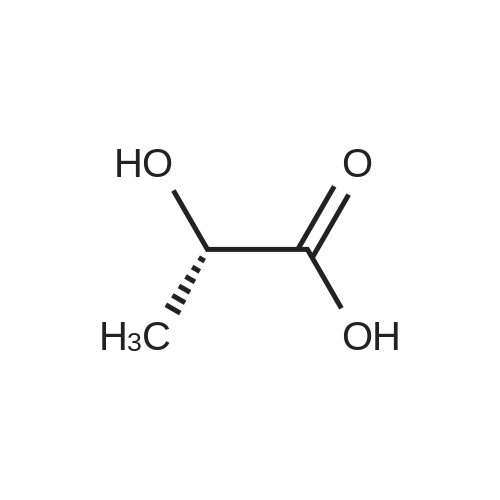
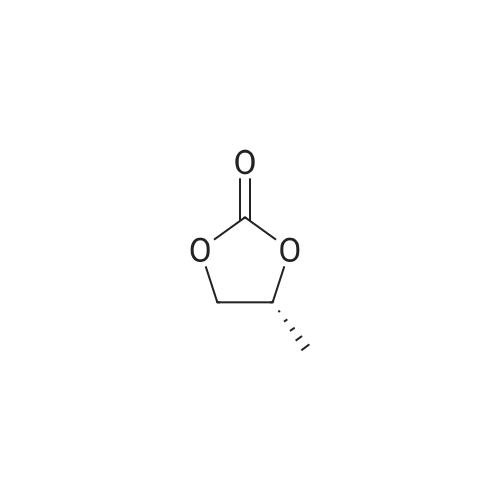
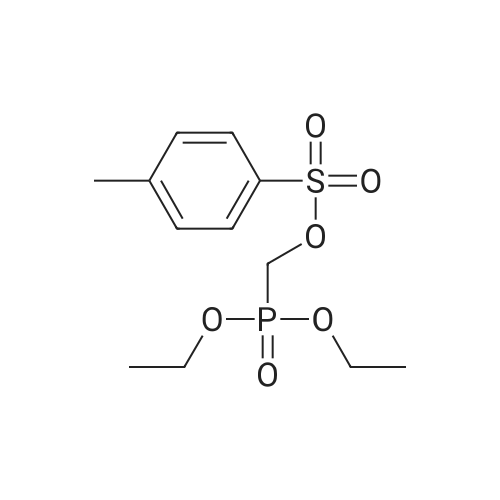
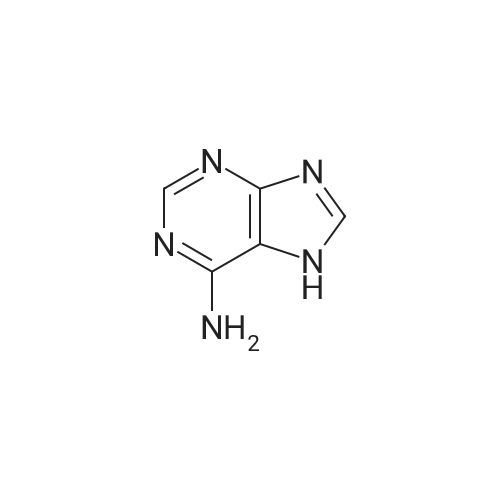
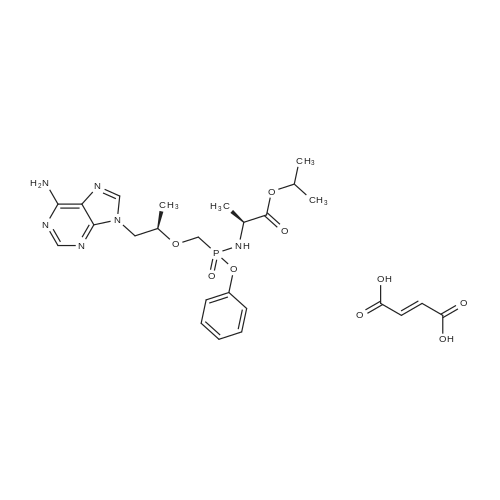
| 95% |
Stage #1: at -20℃; for 1 h;
Stage #2: at -20 - 20℃; for 2 h; |
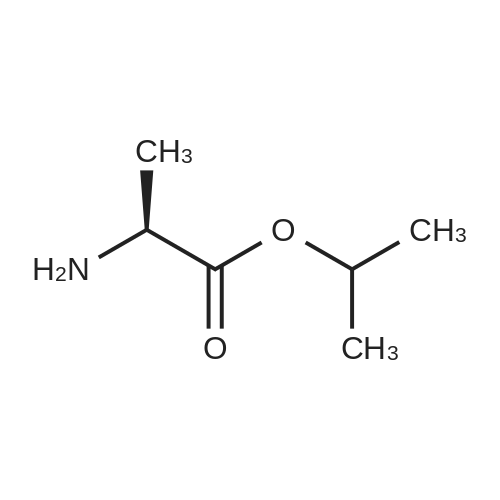
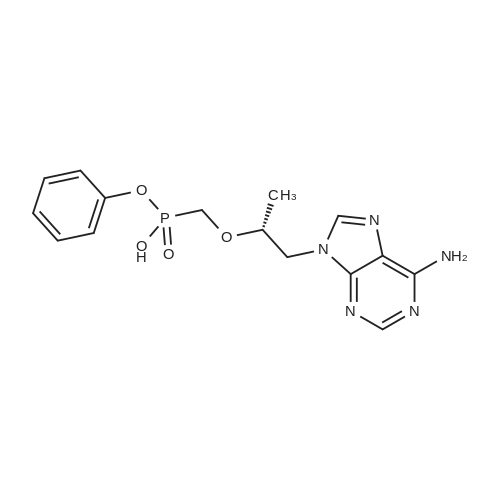
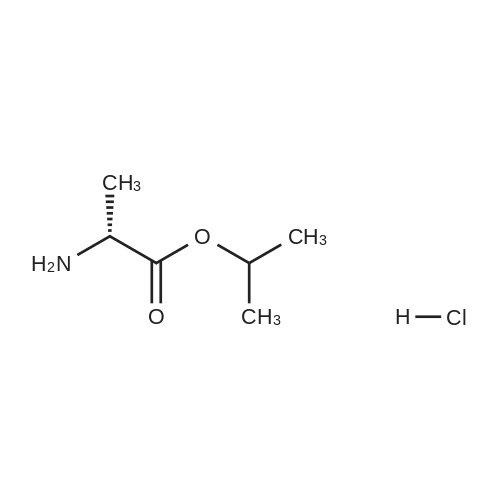
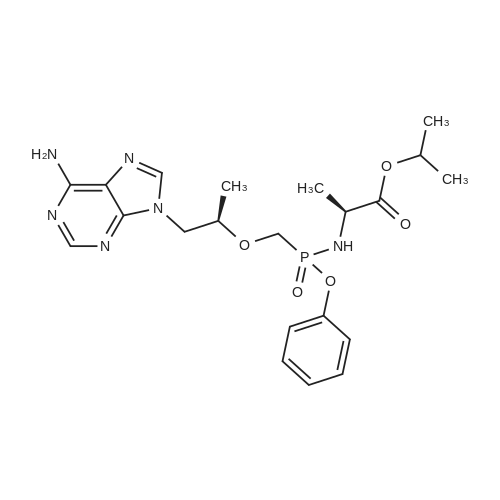
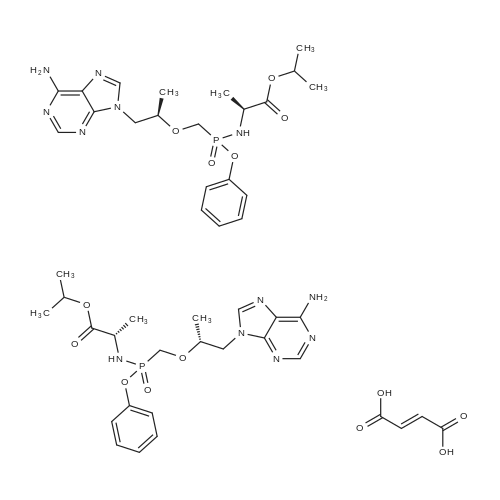
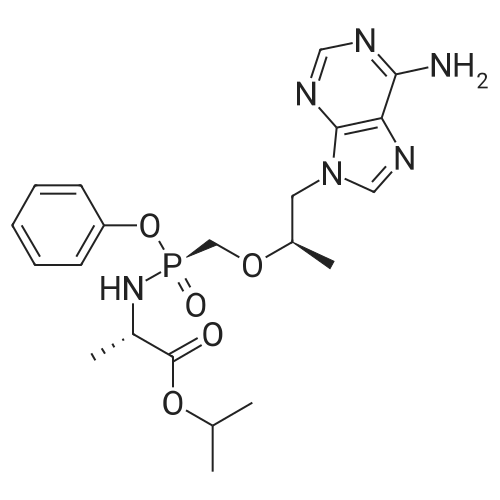
To a suspension of Ph-PMPA 8 (1 .47g, 4.05 mmol) in iPrOAc (25 mL) thionyl chloride (0.7mL, 10.11 mmol, 2.5 equiv) was added and the reaction mixture was stirred at 80°C for 2h.Then, sulfolane (2.2 mL) was added and the resulting mixture was stirred at 80°C for 24 — 72 h (until d.e. of 70percent was achieved). After cooling to the room temperature, the volatile constituents were removed by distillation at a reduced pressure (45°C, 10 mbar). The obtained mixture of the chlorides of formula 7a/7b was suspended in iPrOAc (25 mL) and addedduring 1 h to a cooled (—20°C) solution of iPr-L-alanine (2.65g, 20.23 mmol, 5 equiv) in iPrOAc (15 mL). The reaction was stirred for lh at —20°C, and then after heating for lh at 20°C. The reaction mixture was washed with an aqueous solution of NaI12P04, aqueous solution of KFICO3, water and a saturated solution of NaC1 and further dried over Na2SO4. The resulting solution was concentrated to the volume of 15 mL, inoculated with TA of formula laand left to crystallize at the room temperature. The product was aspirated on fit, washed with cold iPrOAc and dried in a vacuum drier (40°C, 200 Pa) for 8 h, providing 1.3 g (68percent) of Tenofovir Alafenamide of formula la in the form of white powder (chemical purity 95percent, d.e.70percent). Tenofovir Alafenamide (chemical purity 95percent, d.c. 70percent) was dissolved at 85°C in iPrOAc (5 mL/mmol), cooled during 2h to 22°C and stirred at 22°C for 2h. The product was aspirated on frit, washed with cold iPrOAc and dried in a vacuum drier (40°C, 200 Pa) for 8 h, providing Tenofovir Alafenamide in the yield of 92percent and d.c. of 99.0percent.Tenofovir alafenamide (d.e. 99.0percent) was dissolved at 85°C in iPrOAc (5 mL/mmol), cooled during 2h to 22°C and stirred at 22°C for 2h. The product was aspirated on fit, washed with cold iPrOAc and dried in a vacuum drier (40°C, 200 Pa) for 8 h, providing Tenofovir Alafenamide of formula la in the yield of 95percent and d.e. of 99.8percent.‘H NMR (500 MHz, DMSO) ö = 8.12 (d, J 16.6 Hz, 2H), 7.29 (t, J= 7.9 Hz, 211), 7.23 (s, 2H), 7.13 (t, J= 7.4 Hz, 1H), 7.04 (d, J= 8.4 Hz, 2H), 5.69 — 5.61 (m, 111), 4.84 (hept, J 6.3 Hz, 1H), 4.27 (dd, J= 14.4, 3.6 Hz, 111), 4.14 (dd, J= 14.4, 6.6 Hz, 111), 3.97 —3.90 (m, 1H), 3.90 —3.81 (m, 2H), 3.76 (dd, J 13.4, 9.8 Hz, 1H), 1.15 (d, J’= 6.3 Hz, 6H), 1.12 (d, J 7.1Hz, 3H), 1.06 (d, J= 6.2 Hz, 3H) ppm.‘3C NMR (126 MHz, DMSO) ö = 172.92, 172.89, 155.99, 152.44, 150.26, 150.19, 149.80, 141.40, 129.50, 124.36, 120.57, 120.54, 118.39, 75.55, 75.45, 67.91, 64.75, 63.52, 49.07,46.84, 21.45, 21.42, 20.34, 20.30, 16.66 ppm.31P NMR (202 MHz, DMSO) = 22.11 ppm. |
| 76.4% |
at -25 - -20℃; for 1.25 h; |
To a solution of isopropyl L-alanine ester 11 (4.50 equiv) in DCM (80 mL) at −25° C. was added a slurry containing compound 13a (1.00 equiv) that is at least 90percent diastereomerically pure in toluene (50 mL) over a minimum of 45 minutes, maintaining the internal temperature ≦−20° C. The mixture was then held at a temperature ≦−20° C. for at least 30 minutes, and the pH checked using water wet pH paper. If the pH was <4, it was adjusted with triethylamine to pH 4 to 7. The pot temperature was adjusted to room temperature (19° C. to 25° C.). The mixture was transferred to a separatory funnel and washed sequentially with 10percent w/v aqueous solution of sodium phosphate monobasic (2×50 mL), 15percent w/v aqueous solution of potassium bicarbonate (2×20 mL), and water (50 mL). The final organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to a viscous amber oil. The oil was dissolved in toluene/acetonitrile (4:1) (50 mL), and the solution was seeded with 9-{(R)-2-[((R,S)-{ [(S)-1-(isopropoxycarbonyl)ethyl]amino}phenoxyphosphinyl)methoxy]propyl}adenine (about 1 mg, 99:1 diastereomeric ratio) and stirred for 2 hours at ambient temperature. The resultant slurry was filtered and the filter cake was washed with toluene/acetonitrile (4:1) (15 mL) and dried in a vacuum oven at 40° C. for 16 hours to give the product, 9-{(R)-2-[((R,S)-[(S)-1-(isopropoxycarbonyl)ethyl]amino}phenoxyphosphinyl)methoxy]propyl}adenine (compound 15), as a white solid (10.0 g, 76.4percent, 97.5:2.5 diastereomeric ratio in favor of compound 16). 1H NMR (400 MHz, CDCl3): δ1.20-1.33 (m, 12H), 3.62-3.74 (m, 1H), 3.86-4.22 (m, 5H), 4.30-4.44 (m, 1H), 4.83-5.10 (m, 1H), 6.02 (br s, 3H), 7.18-7.34 (m, 5H), 7.98-8.02 (m, 1H), 8.32-8.36 (m, 1H); 31P NMR (162 MHz, CDCl3): δ21.5, 22.9. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping