|
|
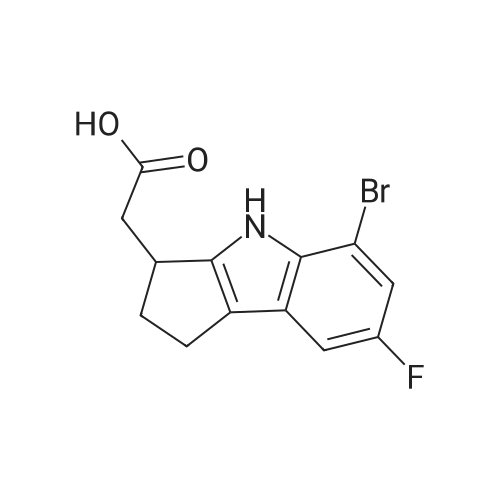
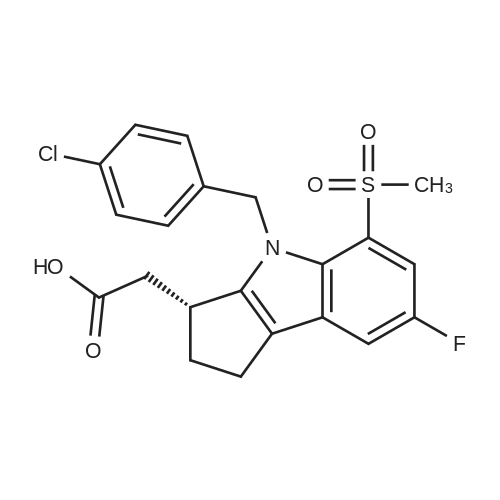
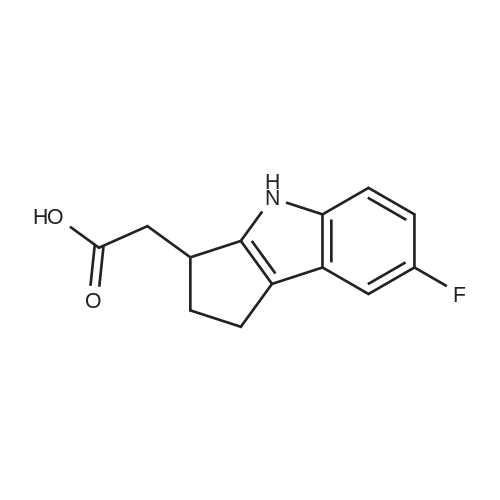
To a solution of 1.24 g of the ester from Step 1 in 14 mL of tetrahydrofuran (THF) at room temperature, 7 mL of MeOH followed by 7 mL of 2N NaOH were added. After 2.5 h, the reaction mixture was poured into a separatory funnel containing ethyl acetate (EtOAc)/ IN HCl. The phases were separated and the acidic phase was extracted twice with EtOAc. The organic layers were combined, washed with brine, dried over anhydrous Na2SO4 and evaporated to dryness to yield a crude oil that was used as such in the next step (>90% purity). 1H NMR (acetone-d6) delta 10.90 (br s, IH), 9.77 (br s, IH), 7.34 (dd, IH), 7.04 (dd, IH), 6.79 (td, IH), 3.56 (m, IH), 2.90-2.50 (m, 5H), 2.16 (m, IH). MS (-APCI) m/z 232.2 (M-H)". |
|
|
Step 2: ("+/-V(7-Fluoro-l,2.3,4-tetrahvdrocyclopenta[blindol-3-yl)acetic acid EPO <DP n="77"/>To a solution of 1.24 g of the ester from Step 1 in 14 mL of tetrahydrofuran (THF) at room temperature, 7 mL of MeOH followed by 7 mL of 2N NaOH were added. After 2.5 h, the reaction mixture was poured into a separatory funnel containing ethyl acetate (EtOAc)/lN HCl. The phases were separated and the acidic phase was extracted twice with EtOAc. The organic layers were combined, washed with brine, dried over anhydrous Na2SO4 and evaporated to dryness to yield a crude oil that was used as such in the next step (>90% purity).1H NMR (acetone-d6) delta 10.90 (br s, IH), 9.77 (br s, IH), 7.34 (dd, IH), 7.04 (dd, IH), 6.79 (td, IH), 3.56(m, IH), 2.90-2.50 (m, 5H), 2.16 (m, IH). MS (-APCI) m/z 232.2 (M-H)". |
|
With sodium hydroxide; water; In tetrahydrofuran; methanol; at 20℃; for 2.5h; |
To a solution of 1.24 g of the ester from Step 1 in 14 mL of tetrahydrofuran (THF) at room temperature, 7 mL of MeOH followed by 7 mL of 2N NaOH were added. After 2.5 h, the reaction mixture was poured into a separatory funnel containing ethyl acetate (EtOAc)/1N HCl. The phases were separated and the acidic phase was extracted twice with EtOAc. The organic layers were combined, washed with brine, dried over anhydrous Na2SO4 and evaporated to dryness to yield a crude oil that was used as such in the next step (>90% purity). 1H NMR (acetone-d6) delta 10.90 (br s, 1H), 9.77 (br s, 1H), 7.34 (dd, 1H), 7.04 (dd, 1H), 6.79 (td, 1H), 3.56 (m, 1H), 2.90-2.50 (m, 5H), 2.16 (m, 1H). MS (-APCI) m/z 232.2 (M-H)-. |
|
|
To a solution of 1.24 g of the ester from Step 1 in 14 mL of tetrahydrofuran (THF) at room temperature, 7 mL of MeOH followed by 7 mL of 2N NaOH were added. After 2.5 h, the reaction mixture was poured into a separatory funnel containing ethyl acetate (EtOAc)/1N HCl. The phases were separated and the acidic phase was extracted twice with EtOAc. The organic layers were combined, washed with brine, dried over anhydrous Na2SO4 and evaporated to dryness to yield a crude oil that was used as such in the next step (>90% purity).1H NMR (acetone-d6) delta 10.90 (br s, 1H), 9.77 (br s, 1H), 7.34 (dd, 1H), 7.04 (dd, 1H), 6.79 (td, 1H), 3.56(m, 1H), 2.90-2.50 (m, 5H), 2.16 (m, 1H). MS (-APCI) m/z 232.2 (M-H)-. |
|
|
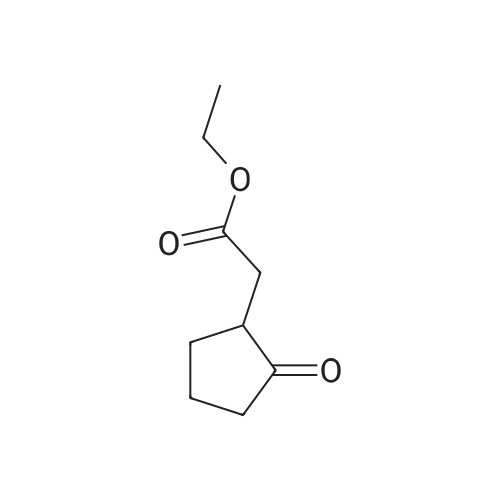
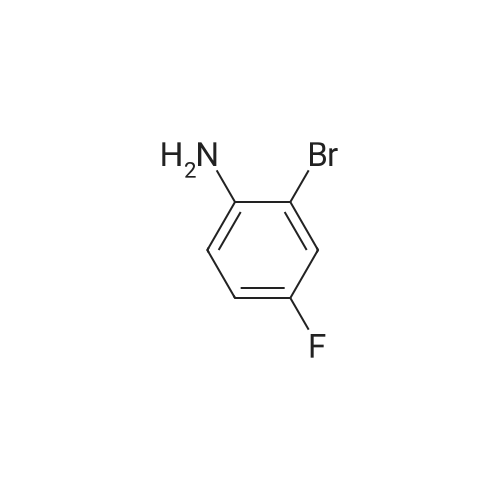
A 0.526 M solution of 2-bromo-4-fluoroaniline in xylene along with ethyl (2-oxocyclopentyl)acetate (1.5 eq) and sulfuric acid (0.02 eq) was heated to reflux for 20 hours. Water was azeotropically removed with a Dean-Stark apparatus. The reaction was followed by NMR and after 20 hours, an 80-85% conversion to the desired imine intermediate was generally observed. The reaction mixture was washed with IM sodium bicarbonate (0.2 volumes) for 15 minutes and the organic fraction was evaporated. The remaining syrup was distilled under vacuum (0.5 mm Hg). Residual xylenes distilled at 300C, then excess ketone and unreacted aniline were recovered in the 50-1100C range; the imine was recovered in the 110-1800C fraction as a light brown clear liquid with 83% purity.The imine intermediate was then added to a degased mixture of potassium acetate (3 eq), tetra-n-butylammonium chloride monohydrate (1 eq), palladium acetate (0.03 eq) and N,N-dimethylacetamide (final concentration of imine = 0.365 M). The reaction mixture was heated to 115C for 5 hours and allowed to cool to room temperature. 3N KOH (3 eq) was then added and the mixture was stirred at room temperature for 1 hour. The reaction mixture was diluted with water (1.0 volume), washed with toluene (3x0.75 volume). The aqueous phase was acidified to pH 1 with 3N HCl and extracted with tertbutyl methyl ether (2x0.75 volume). The combined organic fractions were washed with water (0.75 volume). To the clear light brown solution was added dicyclohexylamine (1 eq) and the solution was stirred at room temperature for 16 hours. The salt was filtered, washed with ethyl acetate, tertbutyl methyl ether and allowed to dry to give the title compound. Assay: 94 A%., 1H NMR (500 mHz, CDCl3) : delta 9.24 (s, 1H), 7.16-7.08 (m, 2H), 6.82 (t, 1H), 6.2 (br, 2H), 3.6-3.5 (m, 1H), 3.04-2.97 (m, 2H), 2.88-2.70 (m, 3H), 2.66 (dd, 1H), 2.45-2.37 (m, 1H), 2.13-2.05 (m, 2.05), 1.83 (d, 4H), 1.67 (d, 2H), 1.55-1.43 (m, 4H), 1.33-1.11 (m, 6H). |
|
|
Step 2: (+/-)-(7-Fluoro-l,2,3,4-tetrahvdrocvclopentaP3lindol-3-yl)acetic acidTo a solution of 1.24 g of the ester from Step 1 in 14 mL of tetrahydrofuran (THF) at room temperature, 7 mL of MeOH followed by 7 mL of 2N NaOH were added. After 2.5 h, the reaction mixture was poured into a separatory funnel containing ethyl acetate (EtOAc)/lN HCl. The phases were separated and the acidic phase was extracted twice with EtOAc. The organic layers were combined, EPO <DP n="61"/>washed with brine, dried over anhydrous Na2SO4 and evaporated to dryness to yield a crude oil that was used as such in the next step (>90% purity).1H NMR (acetone-d6) delta 10.90 (br s, IH), 9.77 (br s, IH), 7.34 (dd, IH), 7.04 (dd, IH), 6.79 (td, IH), 3.56 (m, IH), 2.90-2.50 (m, 5H), 2.16 (m, IH). MS (-APCI) m/z 232.2 (M-H)". |
|
|
DP EXAMPLE 17AAlternative procedure for (+/-)- [5-bromo-4-(4-chlorobenzyl)-7-fluoro-l, 23 A- te1tauahvdrocvclopenta|~b1indol-3-yl1acetic acid (Example 17, Step 4)Step 1 : (+/-)-7-fluoro-l,2,3,4-tetrahvdrocvclopentarb1indol-3-yl)acetic acid dicyclohexylamine(DCHA) salt A 0.526 M solution of 2~bromo-4-fluoroaniline in xylene along with ethyl (2- oxocyclopentyl) acetate (1.5 eq) and sulfuric acid (0.02 eq) was heated to reflux for 20 hours. Water was azeotropically removed with a Dean-Stark apparatus. The reaction was followed by NMR and after 20 hours, an 80-85% conversion to the desired imine intermediate was generally observed. The reaction mixture was washed with IM sodium bicarbonate (0.2 volumes) for 15 minutes and the organic fraction was evaporated. The remaining syrup was distilled under vacuum (0.5 mm Hg). Residual xylenes distilled at 300C, then excess ketone and unreacted aniline were recovered in the 50-1100C range; the imine was recovered in the 110-1800C fraction as a light brown clear liquid with 83% purity.The imine intermediate was then added to a degased mixture of potassium acetate (3 eq), tetra-n-butylammonium chloride monohydrate (1 eq), palladium acetate (0.03 eq) and N,N- dimethylacetamide (final concentration of imine = 0.365 M). The reaction mixture was heated to 1150C for 5 hours and allowed to cool to room temperature. 3N KOH (3 eq) was then added and the mixture was stirred at room temperature for 1 hour. The reaction mixture was diluted with water (1.0 volume), washed with toluene (3x0.75 volume). The aqueous phase was acidified to pH 1 with 3N HCl and extracted with tertbutyl methyl ether (2x0.75 volume). The combined organic fractions were washed with water (0.75 volume). To the clear light brown solution was added dicyclohexylamine (1 eq) and the solution was stirred at room temperature for 16 hours. The salt was filtered, washed with ethyl acetate, tertbutyl methyl ether and allowed to dry to give the title compound. Assay: 94 A%. EPO <DP n="64"/>IH NMR (500 mHz, CDC13) : delta 9.24 (s, IH), 7.16-7.08 (m, 2H), 6.82 (t, IH), 6.2 (br, 2H), 3.6-3.5 (m, IH), 3.04-2.97 (m, 2H), 2.88-2.70 (m, 3H), 2.66 (dd, IH), 2.45-2.37 (m, IH)5 2.13-2.05 (m, 2.05), 1.83 (d, 4H), 1.67 (d, 2H), 1.55-1.43 (m, 4H), 1.33-1.11 (m, 6H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping