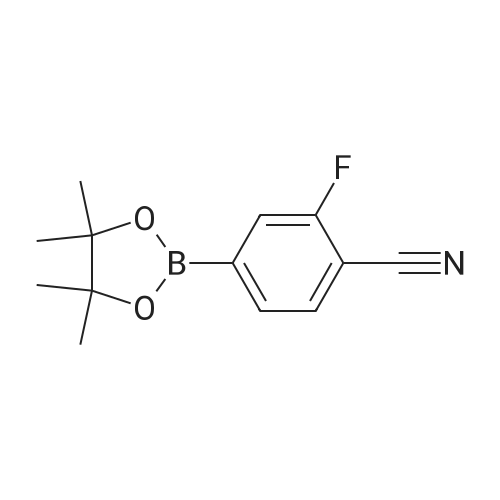
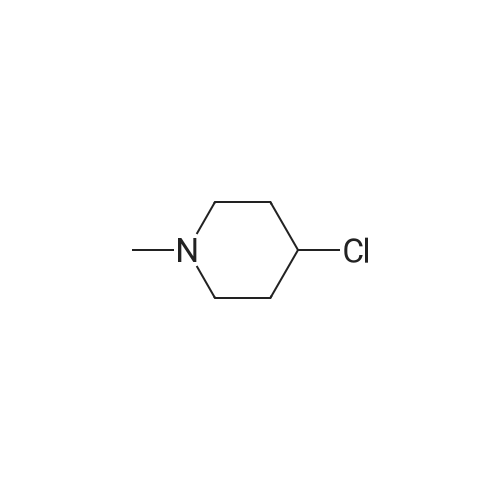
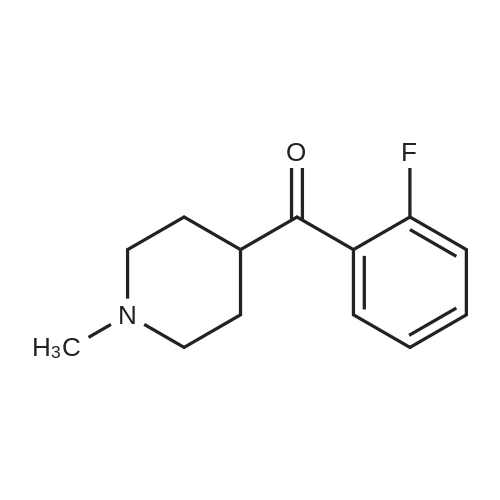
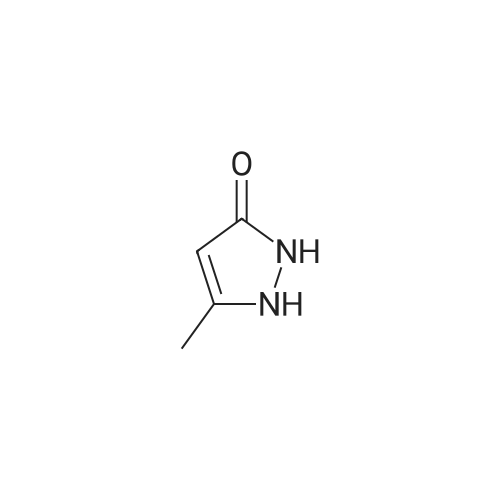
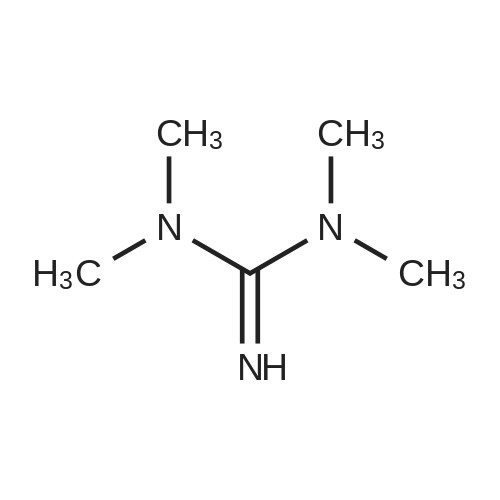
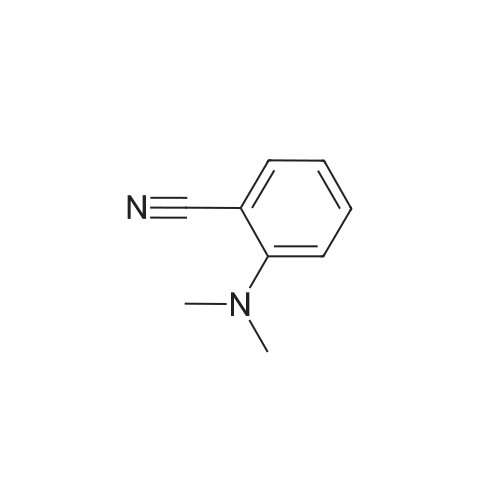
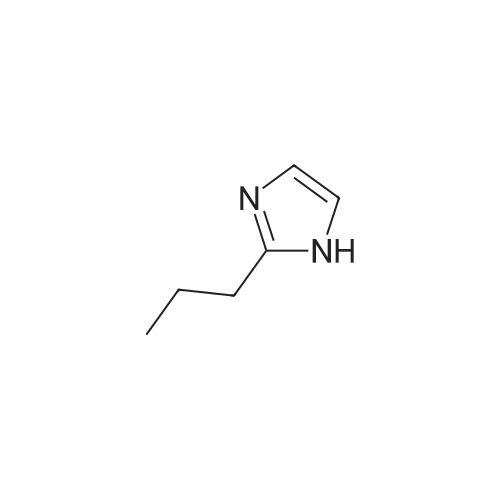
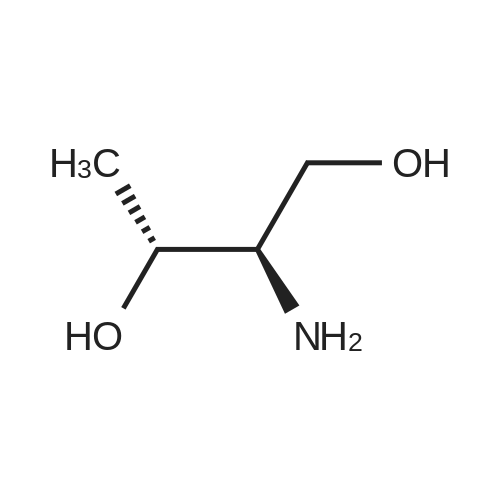
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With hydrazine In butan-1-ol for 5 h; Heating / reflux
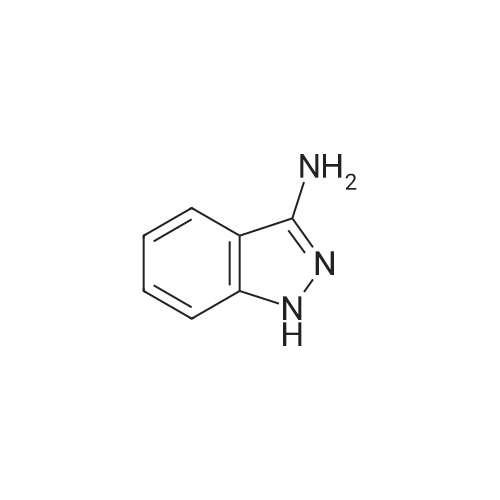
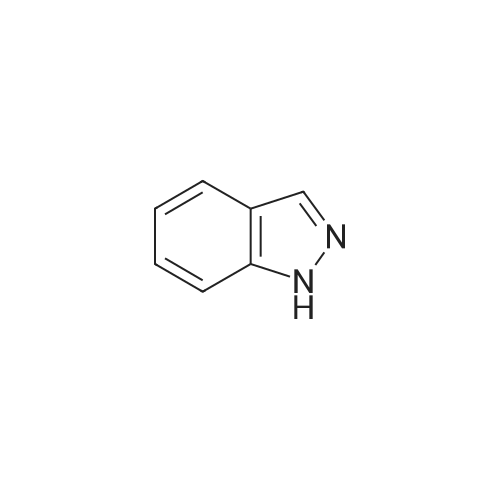
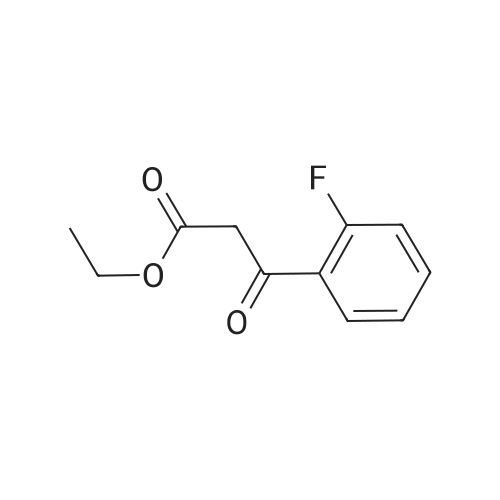
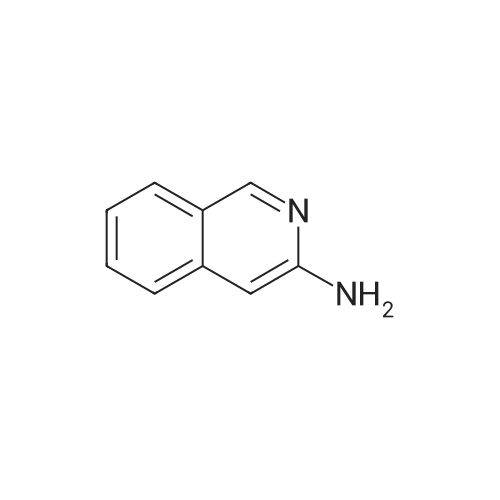
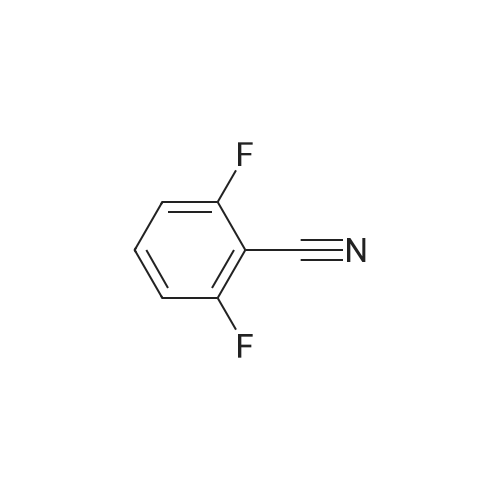
Example 21.1: Preparation of 3-Hydroxy-4-oxo-4,6-dihydro-pyrimido[l,2- 6]indazole-2-carboxylic acid methyl ester; <n="129"/>Step 1:2-Fluorobenzonitrile (605 mg, 5 mmol) and 85percent aqueous hydrazine hydrate (352mg, 6mmol) were mixed with 1-butanol (3 mL). The mixture was heated at reflux with stirring for 5 h then cooled to room temperature. The resulting precipitate was collected by filtration and washed with dichloromethane and the filter cake was dried in vacuo to give 3 -amino benzpyrazole (293 mg, 44 percent).1H NMR (300 MHz, D6-DMSO): δ 5.26-5.36 (brs, 2H), 6.84-6.93 (m, IH), 7.18-7.24 (m, 2H), 7.67 (dt, J=8.1, 0.9 Hz, IH), 11.33 (s, IH).
Reference:
[1] Chemical Communications, 2014, vol. 50, # 85, p. 12911 - 12914
[2] Advanced Synthesis and Catalysis, 2016, vol. 358, # 13, p. 2126 - 2133
[3] Patent: WO2008/77188, 2008, A1, . Location in patent: Page/Page column 127-128
[4] Organic Process Research and Development, 2011, vol. 15, # 3, p. 565 - 569
[5] Bioorganic and Medicinal Chemistry Letters, 2005, vol. 15, # 23, p. 5293 - 5297
[6] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 1, p. 321 - 329
[7] Journal of Medicinal Chemistry, 2012, vol. 55, # 7, p. 3088 - 3100
[8] Journal of Medicinal Chemistry, 2013, vol. 56, # 5, p. 1946 - 1960
[9] Patent: CN105523955, 2016, A, . Location in patent: Paragraph 0167; 0168; 0286
[10] Patent: US2008/103182, 2008, A1, . Location in patent: Page/Page column 13
2
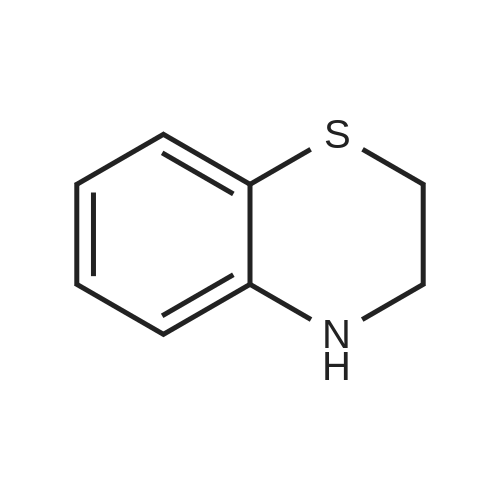
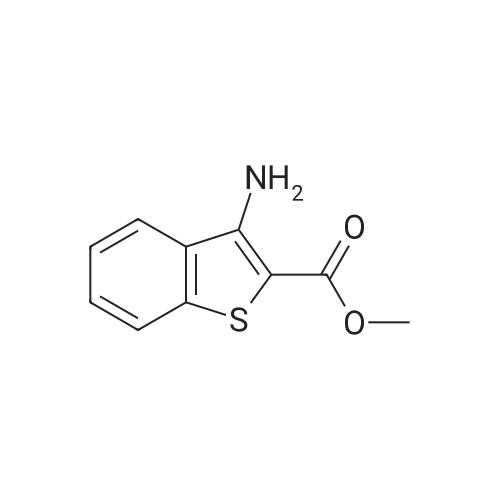
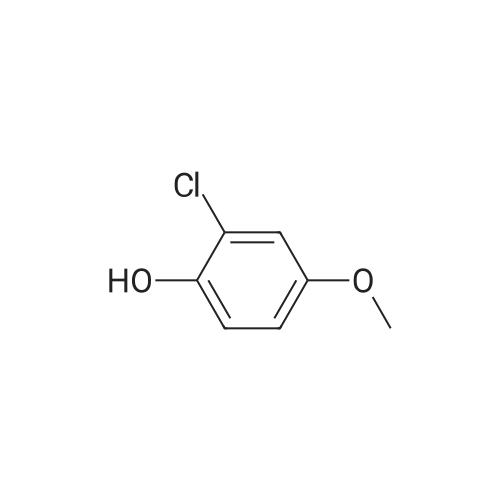
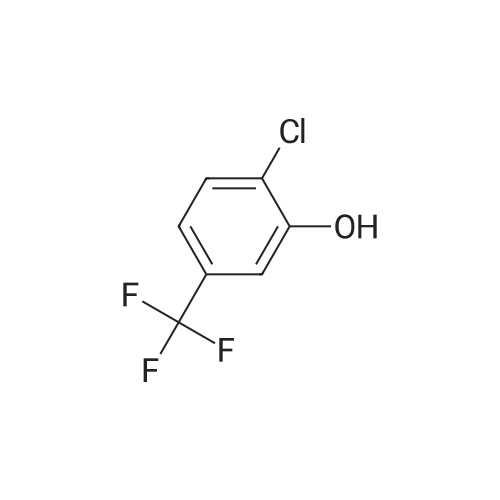
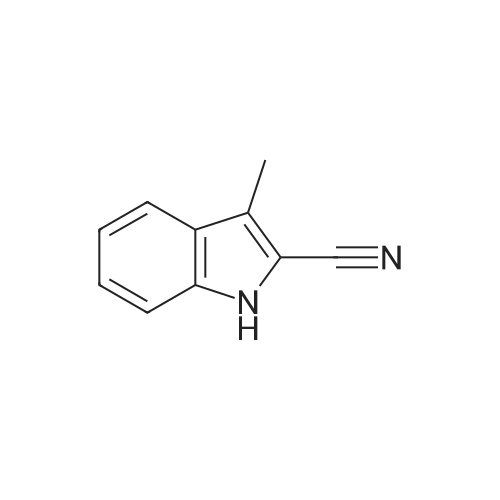
[ 394-47-8 ]
[ 302-01-2 ]
[ 874-05-5 ]
Yield
Reaction Conditions
Operation in experiment
8.2%
at 160℃; for 0.333333 h; Irradiation
To 2-fluorobenzonitrile (1 mL, 9.08 mmol) in EtOH (10 mL) was added hydrazine (0.855 mL, 27.2 mmol) and the reaction was heated to 1600C in a microwave for 20 min. LCMS m/z 134.1(M + H)+. The solvents were removed in vacuo and the residue was quenched with water (10 mL) and ethyl acetate (20 mL). The organic layer was washed with water (10 mL), brine (10 mL), dried (MgSO4) to afford 140A (O. lg, 8.2percent) as an oil which was used directly in the next step.
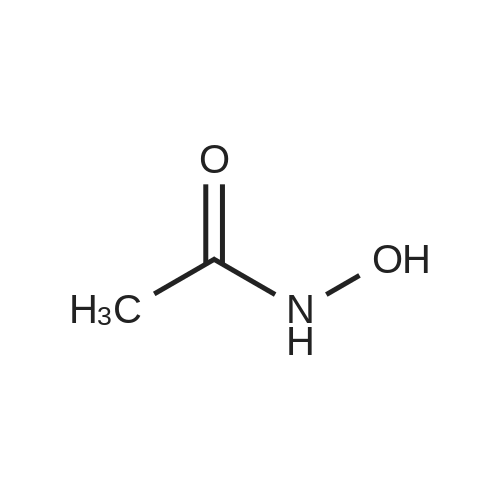
With potassium <i>tert</i>-butylate; acide acetohydroxamique In N,N-dimethyl-formamide at 20℃; for 5.5 h;
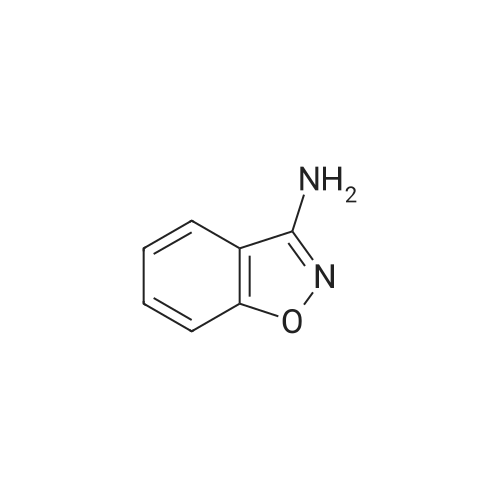
Example 1; N-1,2-Benzisoxazol-3-yl-4-(3-phenyl-1,2,4-thiadiazol-5-yl)piperazine-1-carboxamide; (1) 1,2-Benzisoxazol-3-amine; To a solution of acetohydroxamic acid (10.0 g, 133 mmol) in N,N-dimethylformamide (150 ml) was added potassium tert-butoxide (14.9 g, 133 mmol), and the mixture was stirred at room temperature for 30 minutes. 2-Fluorobenzonitrile (18.0 g, 133 mmol) was added thereto, followed by stirring at room temperature for 5 hours. The reaction mixture was poured to water and extracted with ethyl acetate. The extract wase washed with water and dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was recrystallized from hexane to give 4.80 g (27.0percent) of the desired product as a solid. 1H-NMR (CDCl3) δ; 4.43 (2H, br s), 7.23 - 7.28 (1H, m), 7.43 (1H, d, J = 9.3 Hz), 7.50 - 7.56 (2H, m).
Reference:
[1] Patent: EP1813606, 2007, A1, . Location in patent: Page/Page column 24
[2] Journal of Organic Chemistry, 2000, vol. 65, # 10, p. 2924 - 2932
[3] Journal of Heterocyclic Chemistry, 1989, vol. 26, p. 1293 - 1298
[4] Patent: EP2460799, 2012, A1,
[5] Bioorganic and Medicinal Chemistry, 2015, vol. 23, # 18, p. 6157 - 6165
[6] Patent: WO2008/77188, 2008, A1,
4
[ 546-88-3 ]
[ 394-47-8 ]
[ 36216-80-5 ]
Yield
Reaction Conditions
Operation in experiment
53.7%
With potassium carbonate In N,N-dimethyl-formamide at 80℃;
2- fluorobenzonitrile (6.1g, 50.0mmol), acetohydroxamic acid (5.6g, 75.0mmol) and potassium carbonate (10.4g, 75mmol) was dissolved in DMF (40mL), the mixture was stirred overnight and heated to 80 .With ethyl acetate (100mL × 2) and extracted.The combined organic phases were washed with water (100 mL × 2), saturated brine (100 mL), dried over anhydrous sodium sulfate.Filtered, and the solvent was evaporated under reduced pressure and the crude product purified by column chromatography (petroleum ether / ethyl acetate (v / v) = 4/1), as a white solid (3.6g, 53.7percent).
Reference:
[1] Journal of Medicinal Chemistry, 2012, vol. 55, # 7, p. 3135 - 3143
[2] Journal of Heterocyclic Chemistry, 1988, vol. 25, p. 1173 - 1177
10
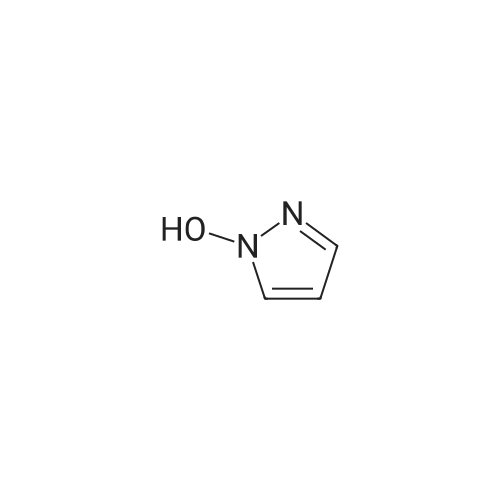
[ 394-47-8 ]
[ 89-99-6 ]
Yield
Reaction Conditions
Operation in experiment
60%
With potassium borohydride; copper dichloride In water; isopropyl alcohol at 60℃; for 12 h;
General procedure: General procedure for the reduction of nitrile by KBH4and CuCl2:To a 10 mL round-bottomed flask was added 2-(4-chlorophenyl)acetonitrile (0.1 5 g, 1 mmol), KBH4 (0.17 g, 3mmol), CuCl2 (0.03 g, 0.25 mmol) and 80 percent isopropanol (1.6mL isopropanol and 0.4 mL water). The reaction completed in8 h at 60 °C as evidenced by TLC (DCM:MeOH 10:1). Thereaction mixture was cooled to 25 °C and removed the solvent.Ethyl acetate (5 mL) was added to the residue, washed withwater (1 mL) and brine (1 mL). The organic layer were dried withanhydrous Na2SO4, filtered and evaporated in vacuo to affordthe crude product.
Reference:
[1] Journal of the American Chemical Society, 2015, vol. 137, # 28, p. 8888 - 8891
[2] Synlett, 2001, # 10, p. 1623 - 1625
[3] Small, 2018, vol. 14, # 37,
[4] ChemCatChem, 2017, vol. 9, # 4, p. 559 - 563
[5] Asian Journal of Chemistry, 2015, vol. 27, # 10, p. 3564 - 3566
[6] Journal of Medicinal Chemistry, 2017, vol. 60, # 19, p. 7965 - 7983
11
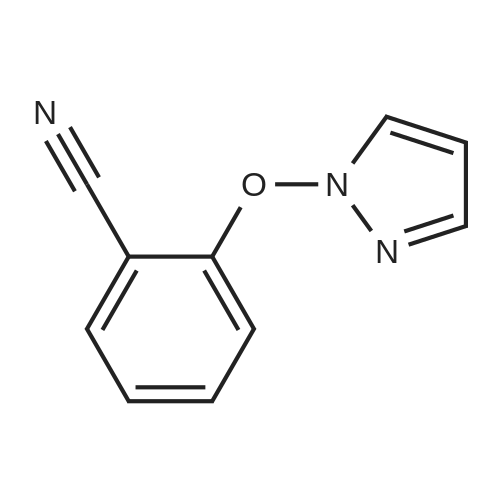
[ 96-35-5 ]
[ 394-47-8 ]
[ 57805-85-3 ]
Yield
Reaction Conditions
Operation in experiment
48%
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 12 h; Heating / reflux
Preparation Example 1: Preparation of 3-amino-benzofuran-2-carboxylic acid methyl ester0.22 g (1.18 mmol) of σ-fluorobenzonitrile was dissolved in 5 ml of N5N- dimethylformamide, 0.16 ml (2.18 mmol) of methyl glyconate and 0.62 g (4.54 mmol) of potassium carbonate were added thereto at room temperature, and the mixture was refluxed with heating for 12 hrs. After the reaction was completed, the reaction mixture was diluted with 10 ml of ethyl acetate, washed with water, dried over anhydrous magnesium sulfate, and concentrated under a reduced pressure. The residue was subjected to silica gel column chromatography <n="19"/>(hexane:ethyl acetate = 4:1) to obtain the title compound (0.10 g, 48percent yield).1H NMR (300MHz, DMSO-d6): δ 3.97(s, 3H), 4.98(s, 2H), 7.23-7.28(m, IH), 7.44-7.47(m, 2H), 7.56(d, IH)Mass(m/e, M+): 192
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 22, p. 6362 - 6365
[2] Patent: WO2009/48274, 2009, A2, . Location in patent: Page/Page column 16-17
12
[ 394-47-8 ]
[ 623-51-8 ]
[ 34761-09-6 ]
Yield
Reaction Conditions
Operation in experiment
92%
Stage #1: With N-ethyl-N,N-diisopropylamine In N,N-dimethyl-formamide at 20℃; for 0.5 h; Stage #2: With potassium carbonate In N,N-dimethyl-formamide at 80℃; for 20 h;
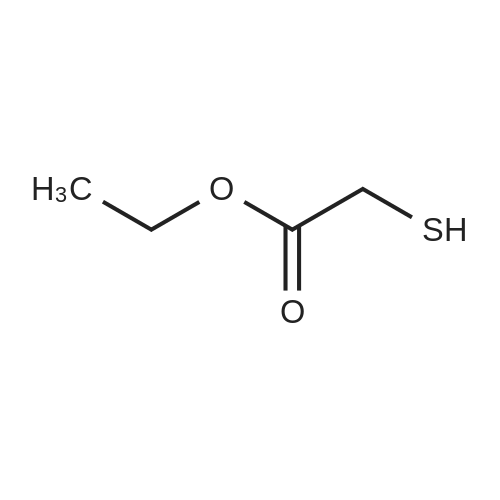
Method 1 : 4-Aminobenzothienof3,2-dlpyrimdineSynthesis 1AEthyl 3-amin -carboxylate2-Fluorobenzonitrile (8.25 mmol), diisopropylethylamine (8.25 mmol) and ethyl2-mercaptoacetate (8.25 mmol) in anhydrous Ν,Ν-dimethylformamide (5 mL) was allowed to stir for 30 min at room temperature. Potassium carbonate (8.25 mmol) was added and the solution was heated to 80°C for 20 h. Ice-water was added and the resulting precipitate was filtered off, washing with water to obtain the carboxylate as a white solid (92percent).LCMS rt 7.43, M+H 222; 1H-NMR (CDCI3) δ 7.43 (1H, d, J 8.1 Hz), 7.65 (1H, d, J 8.1 Hz), 7.49-7.44 (1H, m), 7.39-7.34 (1H, m), 4.36 (2H, q, J 7.1 Hz), 1.40 (3H, t, J 7.1 Hz).
86%
Stage #1: With sodium hydride In dimethyl sulfoxide at 20℃; for 0.25 h;
Ethyl thioglycolate 6 (11 mmol) was added to a suspension of sodium hydride (15 mmol) in dry DMSO (10 ml). The mixture was left under stirring for 15 min at rt followed by fluoro-benzonitrile 5 (10 mmol) addition. After stirring over night, the reaction mixture was quenched with ice, forming a yellow precipitate which was filtered to yield aminobenzo[b]thiophene-2-carboxylate.
Reference:
[1] Patent: WO2012/131297, 2012, A1, . Location in patent: Page/Page column 85
[2] Bioorganic and Medicinal Chemistry Letters, 2014, vol. 24, # 15, p. 3291 - 3297
[3] Journal of Heterocyclic Chemistry, 1997, vol. 34, # 4, p. 1163 - 1172
13
[ 394-47-8 ]
[ 15332-09-9 ]
[ 95-52-3 ]
Reference:
[1] Research on Chemical Intermediates, 2012, vol. 38, # 7, p. 1619 - 1628
[2] Research on Chemical Intermediates, 2012, vol. 38, # 7, p. 1619 - 1628
14
[ 394-47-8 ]
[ 271-44-3 ]
[ 15332-09-9 ]
[ 95-52-3 ]
Reference:
[1] Research on Chemical Intermediates, 2012, vol. 38, # 7, p. 1619 - 1628
15
[ 394-47-8 ]
[ 3759-28-2 ]
Reference:
[1] Journal of Medicinal Chemistry, 2000, vol. 43, # 14, p. 2703 - 2718
16
[ 394-47-8 ]
[ 17417-09-3 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2015, vol. 90, p. 537 - 546
[2] Journal of Heterocyclic Chemistry, 1997, vol. 34, # 4, p. 1163 - 1172
[3] Chemical Biology and Drug Design, 2018, vol. 91, # 1, p. 39 - 49
17
[ 394-47-8 ]
[ 445-28-3 ]
Yield
Reaction Conditions
Operation in experiment
83%
With Acetaldehyde oxime; [(eta.(5)-pentamethylcyclopentadienyl)Rh(H2O)3](OTf)2 In water at 50℃; for 6 h; Schlenk technique
A solution of 2-fluorobenzonitrile (121 mg, 1 mmol), [Cp * Rh (H2O)3] [OTf]2(3.0 mg, 0.005 mmol, 0.5 molpercent), acetaldehyde oxime (65 mg, 1.1 mmol) and water (1 ml) were successively added to a 25 ml Schlenk reaction flask.The reaction mixture was reacted at 50 ° C for 6 hours, then cooled to room temperature, and the water was removed by rotary evaporation to remove the title product. The yield was 83percent
Reference:
[1] Tetrahedron Letters, 2010, vol. 51, # 12, p. 1589 - 1591
[2] RSC Advances, 2014, vol. 4, # 3, p. 1102 - 1106
[3] Journal of Organic Chemistry, 2015, vol. 80, # 8, p. 4148 - 4151
[4] New Journal of Chemistry, 2018, vol. 42, # 19, p. 15572 - 15577
[5] Catalysis Science and Technology, 2016, vol. 6, # 12, p. 4398 - 4409
[6] ChemSusChem, 2011, vol. 4, # 1, p. 104 - 111
[7] Chemistry - A European Journal, 2017, vol. 23, # 60, p. 15210 - 15221
[8] Chemistry - A European Journal, 2008, vol. 14, # 22, p. 6601 - 6605
[9] Chemistry - A European Journal, 2010, vol. 16, # 32, p. 9808 - 9817
[10] RSC Advances, 2014, vol. 4, # 108, p. 63466 - 63474
[11] Patent: CN104744288, 2017, B, . Location in patent: Paragraph 0040; 0041; 0042; 0043
[12] Organometallics, 2011, vol. 30, # 20, p. 5442 - 5451
[13] ChemCatChem, 2017, vol. 9, # 7, p. 1349 - 1353
[14] Tetrahedron Letters, 2000, vol. 41, # 19, p. 3747 - 3749
[15] Organometallics, 2010, vol. 29, # 17, p. 3955 - 3965
[16] ACS Catalysis, 2014, vol. 4, # 6, p. 1901 - 1910
[17] European Journal of Medicinal Chemistry, 1990, vol. 25, # 8, p. 673 - 680
[18] Journal of Medicinal Chemistry, 2009, vol. 52, # 17, p. 5295 - 5298
[19] Tetrahedron Letters, 2010, vol. 51, # 13, p. 1639 - 1641
[20] Bioorganic and Medicinal Chemistry Letters, 2015, vol. 25, # 24, p. 5682 - 5686
Stage #1: at 0 - 25℃; for 46 h; Stage #2: With hydrogenchloride In tetrahydrofuran; water at 20 - 25℃; for 1 h;
Under nitrogen atmosphere, 30 ML of THF was added to 6.09 g (10 mmol, 1.0 equivalent) of (BrZnCH2COOEt*THF)2.. Under argon atmosphere, a solution of 1.21 g (10 mmol) of 2-fluorobenzonitrile in 5 ML of THF was added dropwise while stirring at 0.similar.5°C. The mixture was stirred at 20°C.similar.25°C for 46 hours. 15 ML of 10percent hydrochloric acid added dropwise at 20°C or lower, and the mixture was stirred at 20.similar.25°C for 1 hour, followed by dilution with 50 ML of ethyl acetate.. Then, the layers were separated. The organic layer was washed successively with 15 ML of 1N hydrochloric acid, 20 ML of an aqueous saturated sodium chloride solution, 20 ML (*2) of an aqueous saturated sodium bicarbonate solution, and 20 ML of an aqueous saturated sodium chloride solution.. After washing, the organic layer was dried with anhydrous magnesium sulfate.. Concentration under reduced pressure afforded 1.94 g of the desired product (yield 92percent).1H NMR (CDCl3), (ppm): δ [1.26 (t, J=7.1 Hz), 1.34 (t,J=7.1 Hz)] (3H), [3. 98 (s), 5.84 (s), 12.6 (s)] (2H), 4.17-4.28 (2H, m), 7.08-7.97 (4H, m).
Reference:
[1] European Journal of Organic Chemistry, 2006, # 12, p. 2753 - 2765
[2] Patent: WO2006/97220, 2006, A1, . Location in patent: Page/Page column 34
[3] Journal of Medicinal Chemistry, 1990, vol. 33, # 4, p. 1230 - 1241
23
[ 394-47-8 ]
[ 4039-32-1 ]
[ 57075-81-7 ]
Reference:
[1] Patent: US6265426, 2001, B1,
24
[ 394-47-8 ]
[ 79544-27-7 ]
Reference:
[1] Chemical Communications, 2014, vol. 50, # 64, p. 8974 - 8977
25
[ 394-47-8 ]
[ 447464-03-1 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 21, p. 6253 - 6257
26
[ 394-47-8 ]
[ 159589-71-6 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 2005, vol. 53, # 7, p. 764 - 769
27
[ 394-47-8 ]
[ 775304-60-4 ]
Reference:
[1] Advanced Synthesis and Catalysis, 2017, vol. 359, # 10, p. 1708 - 1716
28
[ 394-47-8 ]
[ 33240-34-5 ]
[ 111982-45-7 ]
Yield
Reaction Conditions
Operation in experiment
40%
Stage #1: With copper(I) bromide In tetrahydrofuran at 60℃; for 15 h; Stage #2: With sulfuric acid; water In tetrahydrofuran at 0℃; for 15 h;
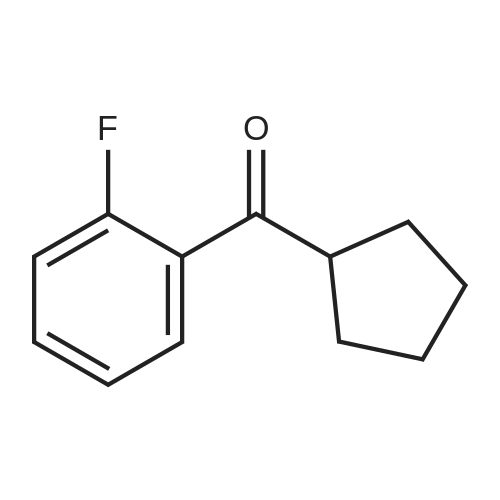
Preparation 100; Cvclopentvl- (2-fluoro-phenyl)-methanone Stir 2-Fluorobenzonitrile (5. 0g, 41.01 mmole) in 80 ml of THF with a 2 molar cylcopentyl magnesium bromide THF solution (20.51 ml, 41.01 mmole) and CuBr (0. 100g, 0.697 mmole) for 15 hrs at 60 °C under argon gas. Add al5percent solution of sulfuric acid to the reaction at 0 °C and stir for 15 hrs. Extract the reaction mixture three times with diethyl ether. Combine organic layers and dry over MgS04 and concentrate Purify residue via column chromatography using mixture of Ethyl Acetate and hexanes ; to give 3.085 grams. Yield 40percent MS (ES) = 192.15 (M+1) +.
To a suspension of Mg (5.57 [G,] 0.23 mol) in THF (25 mL) was added a few drops of bromoethane to initiate reaction. <strong>[5570-77-4]4-Chloro-1-methylpiperidine</strong> (24.5 g, 0.18 mol) in THF (80 mL) was then added dropwise. After addition the reaction was heated under reflux for 1 hour and then allowed to attain rt. The [AVAILABLE 2-FLUOROBENZONITRILE] (22.2 g, 0.18 mol) in THF (44 mL) was added and the mixture was heated under reflux for 3 hours and stirred at rt overnight. The reaction was treated with a ammonium chloride solution and water. After extraction with [ET20,] the organic layer was dried over [NA2SO4,] filtered and evaporated off. The crude oil was purified by distillation under vaccum [(T=110XB0;C)] to give the title compound as a yellow oil (10.0 g, 18 [MMOL)] in a 25% yield ; LC-MS: M+H [C13H, 7NFO] 222.
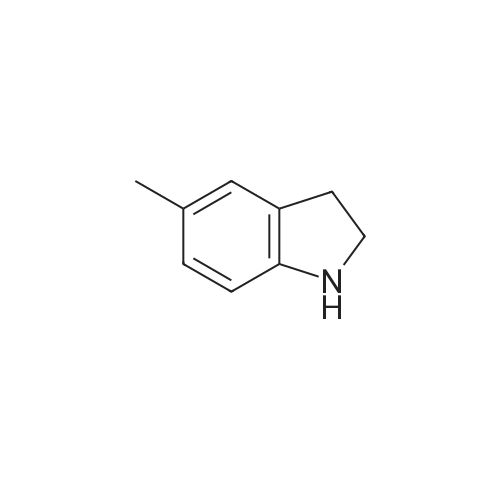
EXAMPLE 54 2-(5-Methylindolin-1-yl)benzonitrile A slurry of <strong>[65826-95-1]5-methylindoline</strong> (31 g, 0.23 mole), sodium hydride (11.3 g, 60% in oil) and dimethylsulfoxide (120 ml) was stirred at room temperature for 1 hour. A solution of o-fluorobenzonitrile (31 gm, 0.25 mole) in dimethylsulfoxide (25 ml) was added dropwise at a temperature below 20 C. Upon completion of the addition, the mixture was stirred for 2 hours at room temperture. The reaction mixture was partitioned between methylene chloride (700 ml) and ice-water (700 ml). The dichloromethane solutions were separated. The aqueous phase was extracted with dichloromethane (2 times, 600 ml). The combined dichloromethane solutions were washed with 2N hydrochloric acid (2 times, 500 ml), water (500 ml), brine (2 times, 400 ml), dried over anhydrous sodium sulfate, filtered, and concentrated. The residue was dissolved in ethanol (300 ml) and heptane (100 ml), and chilled in a freezer. The precipitate was collected. The mother liquor was concentrated and purified by flash chromatography on a silica gel column (400 g, 230-400 mesh) eluted with hexane:dichloromethane (3:1, 3 l); hexane:dichloromethane (1:1, 2 l) and dichloromethane (2 l). The fractions containing product were pooled and concentrated to yield 33 g (73% overall yield). The analytical sample was prepared by high-pressure liquid chromatography (Water Associates Prep 500, 10 g, 2 chamber, elution with hexane:dichloromethane, 9:1, 12 l) followed by crystallization from ethanol, mp 59-60 C. ANALYSIS: Calculated for C16 H14 N2: 82.02%C 6.02%H 11.96%N. Found: 82.47%C 6.08%H 12.10%N.
With Acetaldehyde oxime; [(eta.(5)-pentamethylcyclopentadienyl)Rh(H2O)3](OTf)2; In water; at 50℃; for 6h;Schlenk technique;
A solution of 2-fluorobenzonitrile (121 mg, 1 mmol), [Cp * Rh (H2O)3] [OTf]2(3.0 mg, 0.005 mmol, 0.5 mol%), acetaldehyde oxime (65 mg, 1.1 mmol) and water (1 ml) were successively added to a 25 ml Schlenk reaction flask.The reaction mixture was reacted at 50 C for 6 hours, then cooled to room temperature, and the water was removed by rotary evaporation to remove the title product. The yield was 83%
With dihydrogen peroxide; potassium carbonate; In water; dimethyl sulfoxide; at 0 - 25℃; for 0.5h;
A stirred solution of 2-fluoro-benzonitrile (0.88 g) and K2CO3 (0.15 g) in DMSO (3 mL), cooled in an ice bath, was added 35% H2O2 (1 mL). The mixture was allowed to warm up to 25 C for 30 min, poured into H2O (50 mL), and the resulting solid was collected and washed with Et2O to give 23b as white solid: mp 109-111 C (lit.7 116-119C); 1H NMR (400 MHz, Acetone-d6) delta: 7.91 (td, J = 7.7, 1.8 Hz, 1H), 7.57 (tdd, J = 7.2, 5.2, 1.9 Hz, 1H), 7.30 (tt, J = 6.2, 3.1 Hz, 1H), 7.24 (dd, J = 11.5, 8.3 Hz, 1H).
With potassium carbonate; In N,N-dimethyl-formamide; at 130℃; for 10h;
To a suspension of2-fluorobenzonitrile (10.0 g, 82.6 mmol) and phenol (7.7g,5.1 mmol) in DMF (80 mL) was added K2C03 (22.8 g, 165 mmol) at rt. The reaction was stirred at 130 oc for 10 h. The reaction mixture was diluted with water, extracted with EtAOAc (100 mLx3), washed with brine (100 mL), dried (Na2S04), and concentrated togive 15.0 g, (93%) of the title compound as a yellow oil. 1NMR (400 MHz, CDCh): 8 6.85 (1H, d, J = 8.4 Hz), 7.07-7.14 (3H, m), 7.22 (1H, t, J = 7.2 Hz), 7.40 (2H, t, J = 8.0Hz), 7.47 (1H, td, J = 2.0, 8.4 Hz), 7.65 (1H, dd, J = 1.2, 8.0 Hz).
9.8 g
With 18-crown-6 ether; potassium carbonate; In N,N-dimethyl acetamide; at 110℃; for 16h;
To a solution of compound B1 (5.00 g, 41.3 mmol) and phenol (5.80 g, 61.9 mmol) in DMA (50 mL) was added l8-crown-6 (1.10 g, 4.13 mmol) and K2C03 (11.4 g, 82.6 mmol), the reaction mixture was stirred at H0C for 16 hours to give a brown mixture. LCMS showed the reaction was complete. To the reaction mixture was added H20 (50 mL), the reaction mixture was extracted with EtOAc (50 mL x 3), the combined organic phase was washed with H20 (40 mL x 2) and brine (100 mL), dried over anhydrous Na2S04, filtered and concentrated under reduced pressure to give a brown oil, which was purified by Combi Flash to give compound B2 (9.80 g) as a yellow oil.
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃;
2- fluorobenzonitrile (6.1g, 50.0mmol), acetohydroxamic acid (5.6g, 75.0mmol) and potassium carbonate (10.4g, 75mmol) was dissolved in DMF (40mL), the mixture was stirred overnight and heated to 80 .With ethyl acetate (100mL × 2) and extracted.The combined organic phases were washed with water (100 mL × 2), saturated brine (100 mL), dried over anhydrous sodium sulfate.Filtered, and the solvent was evaporated under reduced pressure and the crude product purified by column chromatography (petroleum ether / ethyl acetate (v / v) = 4/1), as a white solid (3.6g, 53.7%).
Method 1 : 4-Aminobenzothienof3,2-dlpyrimdineSynthesis 1AEthyl 3-amin -carboxylate2-Fluorobenzonitrile (8.25 mmol), diisopropylethylamine (8.25 mmol) and ethyl2-mercaptoacetate (8.25 mmol) in anhydrous Nu,Nu-dimethylformamide (5 mL) was allowed to stir for 30 min at room temperature. Potassium carbonate (8.25 mmol) was added and the solution was heated to 80C for 20 h. Ice-water was added and the resulting precipitate was filtered off, washing with water to obtain the carboxylate as a white solid (92%).LCMS rt 7.43, M+H 222; 1H-NMR (CDCI3) delta 7.43 (1H, d, J 8.1 Hz), 7.65 (1H, d, J 8.1 Hz), 7.49-7.44 (1H, m), 7.39-7.34 (1H, m), 4.36 (2H, q, J 7.1 Hz), 1.40 (3H, t, J 7.1 Hz).
86%
Ethyl thioglycolate 6 (11 mmol) was added to a suspension of sodium hydride (15 mmol) in dry DMSO (10 ml). The mixture was left under stirring for 15 min at rt followed by fluoro-benzonitrile 5 (10 mmol) addition. After stirring over night, the reaction mixture was quenched with ice, forming a yellow precipitate which was filtered to yield aminobenzo[b]thiophene-2-carboxylate.
With potassium tert-butylate; acide acetohydroxamique; In N,N-dimethyl-formamide; at 20℃; for 5.5h;
Example 1; N-1,2-Benzisoxazol-3-yl-4-(3-phenyl-1,2,4-thiadiazol-5-yl)piperazine-1-carboxamide; (1) 1,2-Benzisoxazol-3-amine; To a solution of acetohydroxamic acid (10.0 g, 133 mmol) in N,N-dimethylformamide (150 ml) was added potassium tert-butoxide (14.9 g, 133 mmol), and the mixture was stirred at room temperature for 30 minutes. 2-Fluorobenzonitrile (18.0 g, 133 mmol) was added thereto, followed by stirring at room temperature for 5 hours. The reaction mixture was poured to water and extracted with ethyl acetate. The extract wase washed with water and dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was recrystallized from hexane to give 4.80 g (27.0%) of the desired product as a solid. 1H-NMR (CDCl3) delta; 4.43 (2H, br s), 7.23 - 7.28 (1H, m), 7.43 (1H, d, J = 9.3 Hz), 7.50 - 7.56 (2H, m).
With potassium borohydride; copper dichloride; In water; isopropyl alcohol; at 60℃; for 12h;
General procedure: General procedure for the reduction of nitrile by KBH4and CuCl2:To a 10 mL round-bottomed flask was added 2-(4-chlorophenyl)acetonitrile (0.1 5 g, 1 mmol), KBH4 (0.17 g, 3mmol), CuCl2 (0.03 g, 0.25 mmol) and 80 % isopropanol (1.6mL isopropanol and 0.4 mL water). The reaction completed in8 h at 60 C as evidenced by TLC (DCM:MeOH 10:1). Thereaction mixture was cooled to 25 C and removed the solvent.Ethyl acetate (5 mL) was added to the residue, washed withwater (1 mL) and brine (1 mL). The organic layer were dried withanhydrous Na2SO4, filtered and evaporated in vacuo to affordthe crude product.
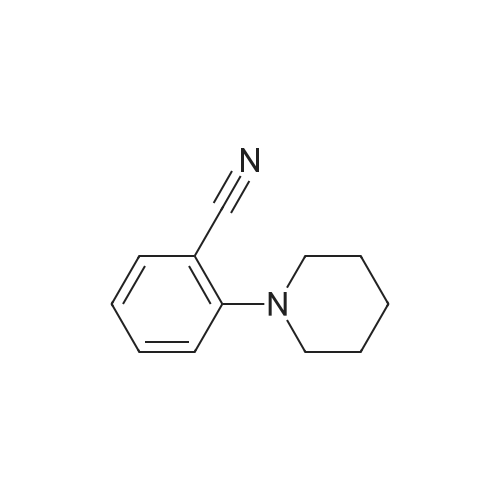
REFERENCE EXAMPLE 14 2-Piperidinobenzonitrile By using the same method as described in Reference Example 12, the subject compound is synthesised from 2-fluorobenzonitrile and piperidine. 1H-NMR (CDCl3) delta (ppm): 1.54-1.62 (2H, m), 1.70-1.79 (4H, m), 3.11-3.16 (4H, m), 6.90-6.99 (2H, m), 7.40-7.51 (2H, m).
REFERENCE EXAMPLE 14 2-Piperidinobenzonitrile According to a similar manner to that in Reference Example 12, the title compound was synthesised from 2-fluorobenzonitrile and piperidine. 1 H-NMR (CDCl3) delta (ppm): 1.54-1.62(2H, m), 1.70-1.79(4H, m), 3.11-3.16(4H, m), 6.90-6.99(2H, m), 7.40-7.51(2H, m).
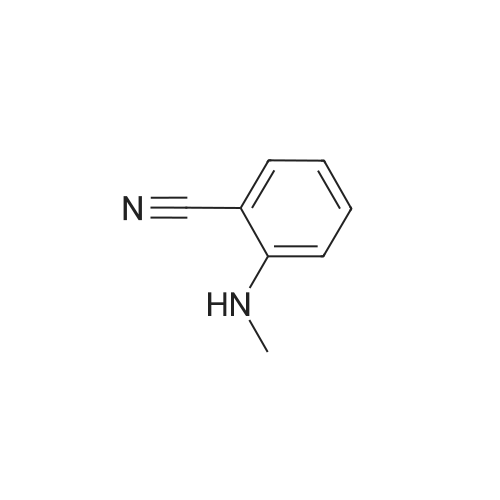
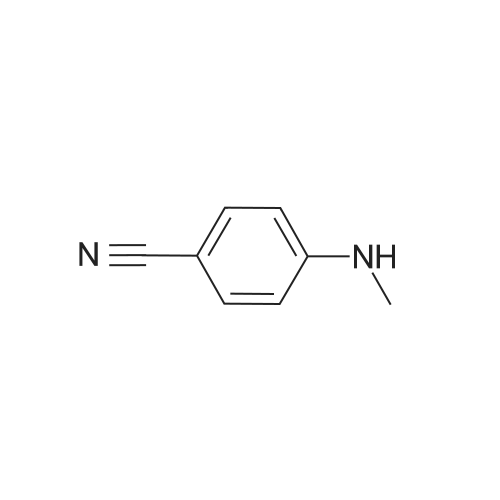
With methylamine; In chloroform; water; acetonitrile;
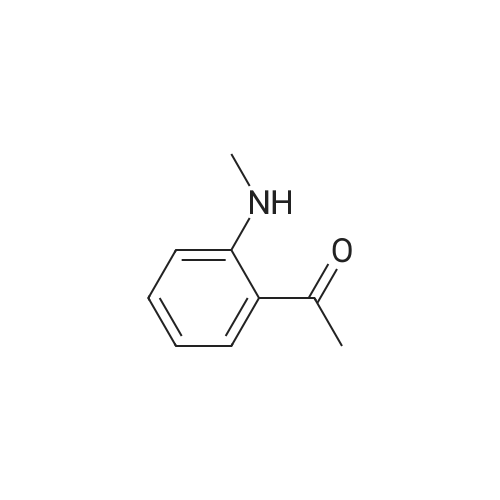
REFERENCE EXAMPLE 1 2-Methylaminobenzonitrile 2-Fluorobenzonitrile (10.0 g, 82.8 millimole) is dissolved in acetonitrile (100 ml), and then 40%-methylamine aqueous solution (200 ml) is added. The solution is stirred for one night at 60 degrees Celsius. In addition, the above methylamine aqueous solution (100 ml) is added, and the solution is stirred for 9 hours at 60 degrees Celsius. The reaction solution is concentrated under reduced pressure, water is added to the obtained residuum, and extraction is performed with chloroform. After the organic layer is dried (magnesium sulfate anhydride), the desiccant is separated through filtration, and the organic layer is concentrated under reduced pressure. The obtained oily material is purified using silica gel chromatography. (At the beginning, it is eluted with chloroform/hexane=1/2. Gradually increasing the contents of the chloroform, at the end, it is eluted with chloroform/hexane={fraction (1/1)}.) Thus, the subject compound (5.38 g, 49%) is obtained as oily material. 1H-NMR (CDCl3) delta (ppm): 2.87 (3H, d, J=5.0 Hz), 4.70 (1H, q, J=5.0 Hz), 6.61-6.69 (2H, m), 7.35-7.43 (2H, m).
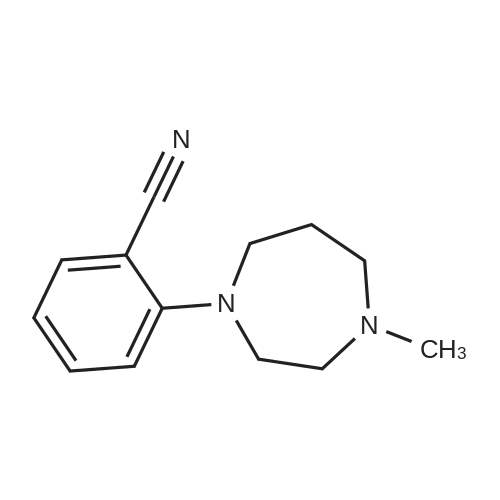
REFERENCE EXAMPLE 94 2-(4-Methyl-1-homopiperazinyl)benzonitrile By using the same method as described in Reference Example 12, the subject compound is synthesised from 2-fluorobenzonitrile and 4-methylhomopiperazine. 1H-NMR (CDCl3) delta (ppm): 2.01-2.10 (2H, m), 2.41 (3H, s), 2.63-2.67 (2H, m), 2.80-2.84 (2H, m), 3.59-3.68 (2H, m), 3.69-3.71 (2H, m), 6.71-6.78 (1H, m), 6.85 (1H, d, J=8.6 Hz), 7.33-7.45 (1H, m), 7.47 (1H, d, J=7.9 Hz).
REFERENCE EXAMPLE 94 2-(4-Methyl-1-homopiperazinyl)benzonitrile According to a similar manner to that in Reference Example 12, the title compound was synthesised from 2-fluorobenzonitrile and 4-methylhomopiperazine. 1 H-NMR (CDCl3) delta (ppm): 2.01-2.10(2H, m), 2.41(3H, s), 2.63-2.67(2H, m), 2.80-2.84(2H, m), 3.59-3.68(2H, m), 3.69-3.71(2H, m), 6.71-6.78(1H, m), 6.85(1H, d, J=8.6 Hz), 7.33-7.45(1H, m), 7.47(1H, d, J=7.9 Hz).
REFERENCE EXAMPLE 1 2-Methylaminobenzonitrile 2-Fluorobenzonitrile (10.0 g, 82.8 mmol) was dissolved in acetonitrile (100 ml), and 40% aqueous methylamine solution (200 ml) was added thereto, followed by stirring at 60 C. overnight. 40% aqueous methylamine solution (100 ml) was further added thereto, followed by stirring at 60 C. for 9 hours. The reaction mixture was concentrated under reduced pressure, then water was added to the resulting residue, the mixture was extracted with chloroform. After the organic layer was dried (over anhydrous magnesium sulfate), the drying agent was filtered off, and the organic layer was concentrated under reduced pressure. The resulting oily substances were purified by silica gel column chromatography (the substances were eluted with an increasing concentration of chloroform from chloroform/hexane=1/2 to chloroform/hexane=1/1) to give the title compound (5.38 g, 49%). 1 H-NMR (CDCl3) delta (ppm): 2.87(3H, d, J=5.0 Hz), 4.70(1H, q, J=5.0 Hz), 6.61-6.69(2H, m), 7.35-7.43(2H, m).
With potassium tert-butylate; In dimethyl sulfoxide;
Reference Example 41 At room temperature, 11.4 g of <strong>[4714-62-9]4-methylaminobenzonitrile</strong> dissolved in dimethyl sulfoxide was added dropwise to a mixture of 11.6 g of potassium tert-butoxide and 100 ml of dimethyl sulfoxide. After 20 minutes of stirring, 10.5 g of fluorobenzonitrile was added to the reaction mixture, and the stirring was continued for 30 minutes at room temperature. The whole mixture was poured into water, and the thus formed precipitate was collected by filtration, washed with water and ethanol in that order and then dried to obtain 17.5 g of N,N-bis(4-cyanophenyl)methylamine. Nuclear magnetic resonance spectrum (CDCl3, TMS internal standard)
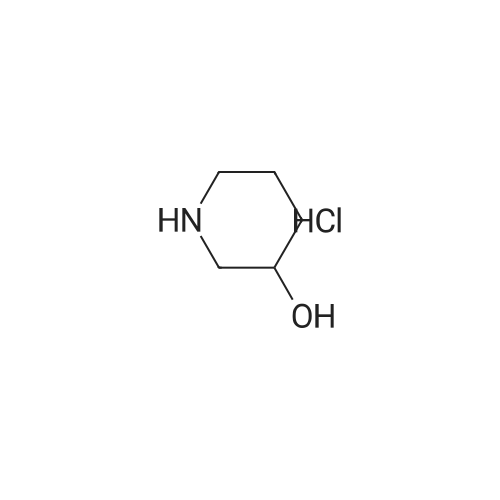
(RS)-3-[1-(Benzocyclobuten-1-ylmethyl)-4-piperidyl]-1,2-benzisoxazole hydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50%
With ammonium chloride; In tetrahydrofuran; water;
EXAMPLE 27 (RS)-3-[1-(Benzocyclobuten-1-ylmethyl)-4-piperidyl]-1,2-benzisoxazole hydrochloride STR25 STAGE A: 2-Fluorobenzoyl-1-methylpiperidine The magnesium compounds is prepared from 0.067 mole of <strong>[5570-77-4]4-chloro-1-methylpiperidine</strong> and 0.055 gram-atom of magnesium in tetrahydrofuran. The reaction is initiated with a few drops of bromoethane. 0.07 mole of 2-fluorobenzonitrile is then introduced into the magnesium compounds. The mixture is brought to reflux for 2 hours and then left overnight at room temperature. It is hydrolyzed with a solution of 15.3 g of ammonium chloride, 45 g of ice and 50 ml of water. The mixture is brought to reflux for 3 hours. It is allowed to cool. It is extracted several times with ethyl ether. The oil obtained is dried. Yield: 50%. Proton nuclear magnetic resonance spectrum (solvent CDCl3): 1.7-2.15 ppm, m, 6H; 2.25 ppm, s, 3H; 2.85 ppm, m, 2H; 3.1 ppm, m, 1H; 7.0-7.3 ppm, m, 4H.
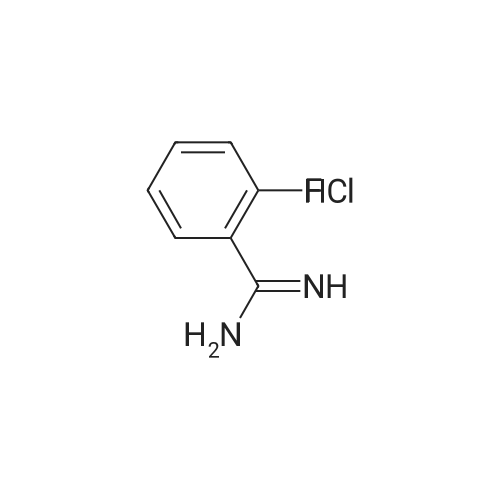
EXAMPLE 200 2-Fluorobenzamidine hydrochloride salt (1:1) To a solution of lithium-bis(trimethylsilyl)amide (10 g, 59.8 mmol) in ether (200 ml) in a flame-dried flask, was added 2-fluorobenzonitrile (7.26 g, 59.8 mmol) dropwise over 45 min. at r.t. The mixture was stirred for a further 4 h. at r.t. then cooled to -10 C. and 6N HCl in ethanol (40 ml) added dropwise over 30 min. (exothermic). After allowing the reaction mixture to warm to r.t. overnight the precipitate was filtered and washed with ether then dried under high vacuum at 40 C. overnight. To remove LiCl the dried solid was re-suspended in ethanol (200 ml) and filtered, then the volume reduce volume to ca. 30 ml. The solution was filtered then triturated with ether, and stirred 30 min. at 4 C. Filtration of the precipitate, washing with ether and high vacuum drying overnight at 40 C., afforded the title compound (7.8 g, 44.7 mmol, 75% yield) as a white solid. MS: m/e=138 (M+).
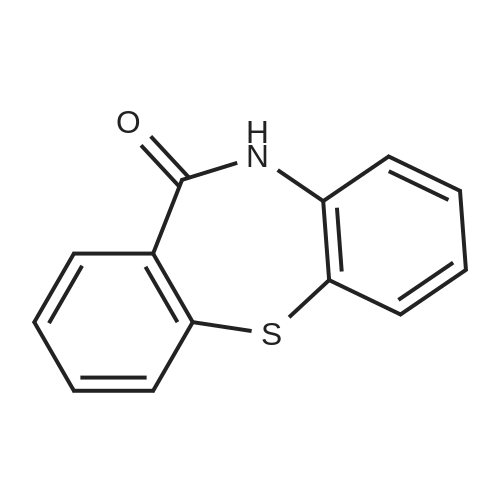
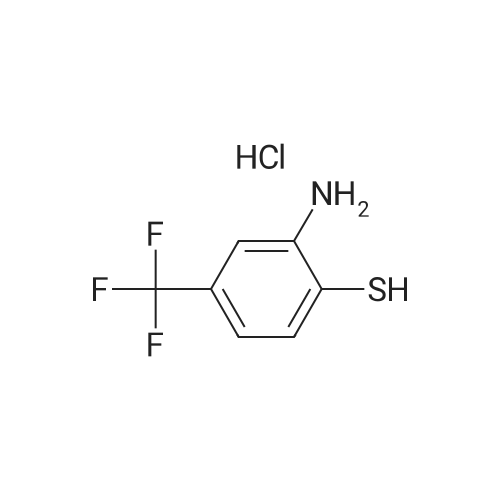
EXAMPLE 6 8-Trifluoromethyl-10,11-dihydro<strong>[3159-07-7]dibenzo[b,f][1,4]thiazepin-11-one</strong> Using a procedure analagous to that of Example 3, 4-trifluoromethyl-2-aminothiophenol hydrochloride (11.48 g, 50 mmol) was combined with 2-fluorobenzonitrile in DMF as solvent (100 ml) to give crude 2-(4-trifluoromethyl-2-aminophenylthio) benzonitrile (6.84 g). Hydrolysis and subsequent condensation gave the title compound as off-white crystals from ethanol (1.1 g, m.p. 233-235 C., Rf 0.21, CH2 Cl2 /MeOH 50:1).
With potassium tert-butylate; dimethyl sulfoxide; In acetone;
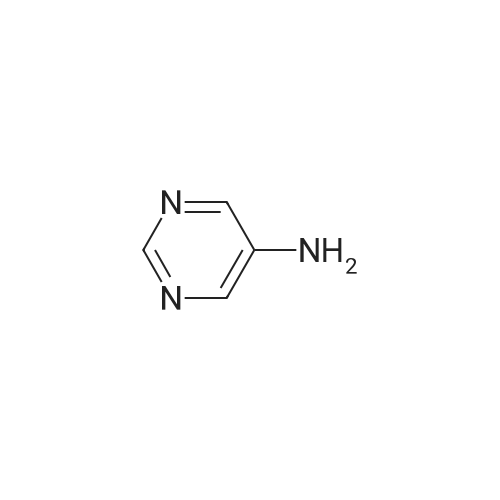
REFERENCE EXAMPLE 9 To 10 ml of dimethyl sulfoxide was added 1.40 g (12.5 mmol) of potassium tert-butoxide, and to the solution was added 951 mg (10.0 mmol) of <strong>[591-55-9]5-aminopyrimidine</strong> while cooling with water. After stirring at room temperature for 1 hour, 0.54 ml (5.0 mmol) of 2-fluorobenzonitrile was added thereto while cooling with water, and the mixture was stirred at room temperature for 30 minutes. The reaction mixture was poured into ice-water, and the mixture was adjusted to pH 7.0 with 1 N hydrochloric acid, and filtered. The filter residue was dissolved in 200 ml of acetone, and the acetone solution was filtered. The filtrate was dried over magnesium sulfate, and the solvent was removed by distillation under reduced pressure to obtain 0.53 g (2.70 mmol) of 5-[N-(2cyanophenyl)amino]pyrimidine. Mass Spectrum (m/z): 196 (M+) NMR Spectrum (DMSO-d6, TMS internal standard) delta: 7.12 (1 H, t, J=7 Hz), 7.35 (1 H, d, J=8 Hz), 7.57-7.60 (1 H, m), 7.76 (1 H, dd, J=8 Hz, 1 Hz), 8.61 (2 H, s), 8.78 (1 H, s), 8.84 (1 H, s)
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 12h;Heating / reflux;
Preparation Example 14: Preparation of 3-amino-benzo[Z>]thiophene-2- carboxylic acid methyl ester0.90 ml (8.26 mmol) of °-fluorobenzonitrile was dissolved in 6 ml of N3N- dimethylformamide, 0.90 ml (9.91 mmol) of methyl thioglyconate and 3.42 g(24.78 mmol) of potassium carbonate was added thereto at room temperature, and the mixture was refluxed with heating for 12 hrs. After the reaction was completed, the reaction mixture was diluted with 10 ml of ethyl acetate, washed with water, dried over anhydrous magnesium sulfate, and concentrated under a reduced pressure. The resulting residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 4:1) to obtain the title compound (1.53 g,89% yield).1H NMR (300MHz, CDCl3); delta 3.90(s, 3H), 5.87(br5 2H), 7.35(t, IH), 7.45(t, IH), 7.63(d, IH), 7.72(d, IH) <n="25"/>MS(m/e, M+): 207
88%
With potassium tert-butylate; In N,N-dimethyl-formamide; at 20℃; for 1.25h;
To a solution of 2-fluorobenzonitrile (9.0 g, 74.3 mmol) and methyl thioglycolate (13.3 mL, 148.6 mmol) in DMF (50 mL) at 0 C. was added potassium tert-butoxide portionwise (16.7 g. 149 mmol) over 15 min with vigorous stirring. The orange solution was warmed to rt and stirred for 1 h before being poured into 700 mL of vigorously stirring ice water. The precipitate was filtered and air-dried to give a pale yellow solid (13.6 g, 88%) that was used without purification. 1H NMR (CD3OD): 8.16-8.12 (m, 1H), 7.85-7.82 (m, 1H), 7.54-7.49 (m, 1H), 7.43-7.38 (m, 1H), 7.19 (s, 2H), 3.79 (s, 3H).
70%
With triethylamine; In dimethyl sulfoxide; at 100℃; for 3h;
Example 46: 3-Hydroxy-l-methyl-lH-benzo[4,51thienof3,2-dlpyrimidine-2,4-dione Step a.) 3-Amino-benzo[b]thiophene-2-carboxylic acid methyl ester 2-Fluorobenzonitrile (10 g, 83 mmol) was placed in a round bottom flask equipped with a stir bar followed by anhydrous DMSO (83 ml). Next, Et3N (35 ml, 249 mmol) and methylthioglycolate (83 mmol, 7.42 ml) were added and the reaction mixture was heated to 100 "C. After 3 h, the mixture was poured into ice water and the precipitate was filtered and dried providing 12 g (70%) of the title compound as a tan solid:
13.5 g
With potassium tert-butylate; In N,N-dimethyl-formamide; at 0 - 20℃;
Into a 250-mL round-bottom flask, was placed a solution of 2-fluorobenzonitrile (10 g, 82.57 mmol, 1 equiv), methyl 2-sulfanylacetate (17.5 g, 164.87 mmol, 2 equiv) in DMF (30 mL). This was followed by the addition of a solution of t-BuOK (18.51 g, 164.96 mmol, 2 equiv) in DMF (50 mL) dropwise with stirring at 0 C. The resulting mixture was stirred for 1 h at room temperature and poured into water/ice. The solid was collected by filtration and dried to afford the title compound as a yellow solid (13.5 g) which was used without any purification. MS: (ES, m/z): 208 [M+H]+.
A solution of acetoxime (0.055 mol) in DMF (50 ml) was stirred at RT, then KtBuO (0.055 mol) was added and the solution stirred for another 30 min. at RT. 2- cyanofluorobenzene (0.050 mol) was added drop wise and the reaction mixture stirred for another hour at RT. The reaction mixture was poured out in a solution of 200 ml isopropylether and 200 ml saturated NH4Cl solution and stirred vigorously for 10 minutes. After completion, the organic layer was separated, washed with water, dried (MgS04), filtered off and the solvent was evaporated dry. The residue (Yield: 9g) was dissolved in ethanol (100 ml). A solution of 2N HCl (100 ml) was added and the reaction mixture refluxed for 1 hour. After completion the solvent was evaporated, the aqueous residue alkanized with a solution of KzCO3 and extracted with EtOAc. The organic layer was separated, washed with water, dried (MgS04), filtered off and the solvent was evaporated dry. Quantitative Yielding 1-2-Benzisoxazole-3-amine 4.7 g (70%), Melting Point 111C (intermediate 3).
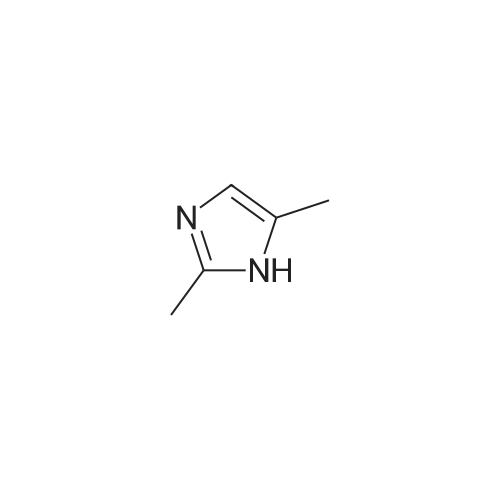
EXAMPLE 29 Preparation of 2-(2,4-dimethyl-1H-imidazol-1-yl)benzonitrile 6 Sodium hydride (8.65 grams 60%, 0.216 mol) was stirred in 75 mL of anhydrous dimethylformamide. To this was added dropwise a solution of 2,4-dimethylimidazole (20.75 grams, 0.216 mol) in 100 mL ofDMF. After stirring at ambient temperature for 1 hour a solution of 2-fluorobenzonitrile (23.0 mL, 0.216 mol) in 75 mL of DMF was added dropwise. This was stirred at 50 C for 2 hours and then at ambient temperature for 16 hours. The mixture was then poured into water and the product extracted with ethyl acetate. The organic layer was washed with water and dried over sodium sulfate. The crude product was chromatographed (silica gel) and eluted with 19:1 dichloromethane-methanol then 9:1 dichloromethane-methanol to afford the product as a solid. LCMS data confirmed structure.
Example 29 Preparation of 2-(2,4-dimethyl-1H-imidazol-1-yl)benzonitrile 6 Sodium hydride (8.65 grams 60%, 0.216 mol) was stirred in 75 mL of anhydrous dimethylformamide. To this was added dropwise a solution of 2,4-dimethylimidazole (20.75 grams, 0.216 mol) in 100 mL of DMF. After stirring at ambient temperature for 1 hour a solution of 2-fluorobenzonitrile (23.0 mL, 0.216 mol) in 75 mL of DMF was added dropwise. This was stirred at 50 C. for 2 hours and then at ambient temperature for 16 hours. The mixture was then poured into water and the product extracted with ethyl acetate. The organic layer was washed with water and dried over sodium sulfate. The crude product was chromatographed (silica gel) and eluted with 19:1 dichloromethane-methanol then 9:1 dichloromethane-methanol to afford the product as a solid. LCMS data confirmed structure.
Preparation of 2-(2,4-dimethyl-1H-1-yl)benzonitrile Sodium hydride (8.65 g 60%, 0.216 mol) were stirred in anhydrous dimethylformamide in 75 . Thus in the 100 2,4- dimethyl DMF was added dropwise to a solution of imidazole (20.75 g, 0.216 mol). After stirring for 1 hour at room temperature, it was added dropwise a solution of benzonitrile (23.0 , 0.216 mol) of 2-fluoro-75 of DMF. This was stirred for 2 hours at 50 , was stirred at room temperature for 16 hours. The mixture was poured into water and the product was extracted with ethyl acetate. The organic layer was washed with water, dried over sodium sulfate. The crude product was 19: 1 dichloromethane-methanol followed by 9: 1 dichloromethane-treated by chromatography (silica gel) eluting with methanol to give the product as a solid. LCMS data were confirmed the structure.
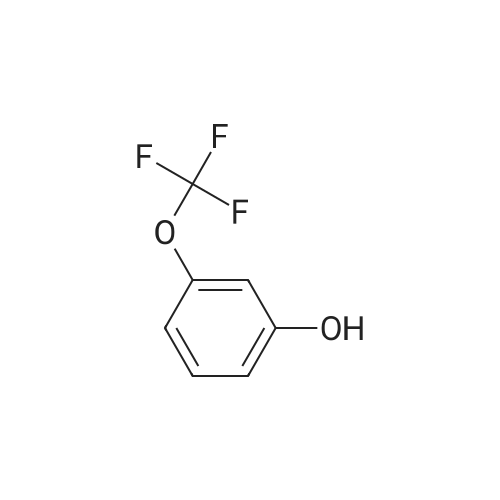
2-(3-(Trifluoromethoxy)phenoxy)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In n-heptane; water; dimethyl sulfoxide; ethyl acetate;
Step 1 2-(3-(Trifluoromethoxy)phenoxy)benzonitrile A mixture of potassium fluoride on alumina (40percent w/w, Aldrich, 11 g), 2-fluorobenzonitrile (6.0 ml, 56 mmol), <strong>[827-99-6]3-trifluoromethoxyphenol</strong> (10 g, 56 mmol), and 18-crown-6 (1.48 g, 5.6 mmol) in dimethylsulphoxide (40 ml) was heated to 140° C. for 20 hours. The reaction mixture was cooled to room temperature and diluted with ethyl acetate (300 ml). The solid was filtered off through a plug of celite. The solution was washed with a mixture of water (100 ml) and brine (100 ml). The aqueous phase was extracted with ethyl acetate (2*50 ml). The combined organic layers were washed with water (100 ml) and dried over magnesium sulphate. The solvent was removed in vacuo. The crude product was purified by flash chromatography on silica (200 g), using ethyl acetate/heptane (1:3) as eluent, to give 9.80 g of 2-(3-(difluoromethoxy)phenoxy)benzonitrile. 1H NMR (CDCl3): delta 6.95 (m, 2 H); 7.00 (d, 1 H); 7.10 (d, 1 H); 7.25 (t, 1 H); 7.45 (t, 1 H); 7.55 (t, 1 H); 7.70 (d, 1 H). MS: Calc for [M+H]+: 280; Found: 280.
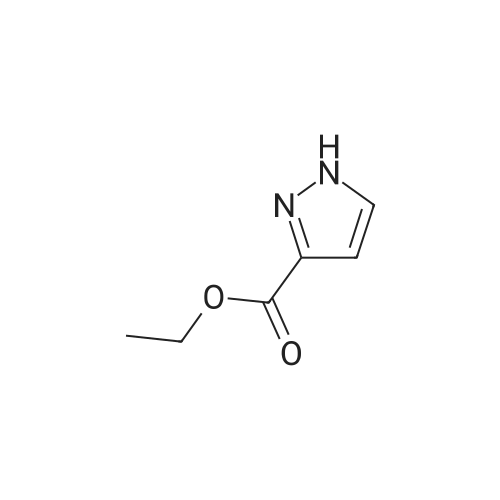
General procedure: A mixture of benzonitrile 1 (10.0 mmol) and methylhydrazine (2.8 mL, 50.0 mmol) in EtOH (10.0 mL) was heated to reflux overnight. The mixture was cooled to rt and then concentrated. H2O (10.0 mL) and EtOAc (20.0 mL) were added to the residue. The organic layer was washed with H2O (10.0 mL), brine (10.0 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was subjected to silica-gel chromatography by using EtOAc/hexanes (1:1) as eluent to give the product 2.
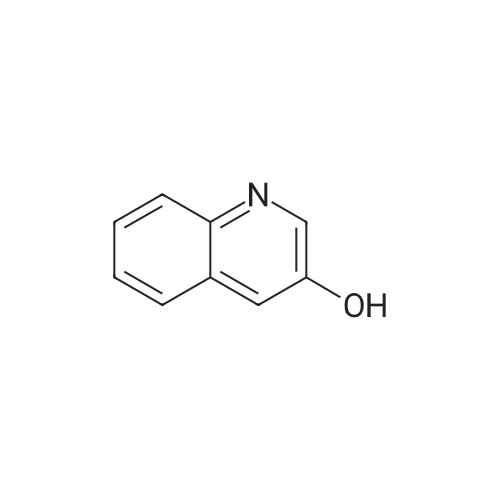
Example 16 Synthesis of 3-(2-cyanophenoxy)quinoline After 0.27 g of <strong>[580-18-7]3-hydroxyquinoline</strong> was dissolved in N-methyl-2-pyrrolidone, 0.087 g of sodium hydride (60% oil dispersion) was added under ice-cooling, and the mixture was stirred for 30 minutes. At the same temperature, 0.27 g of 2-fluorobenzonitrile was added and then the mixture was stirred for 6 hours at room temperature. After the reaction mixture was poured into ice water, extracted with ethyl acetate, and washed with saturated saline solution, the resultant was dried with magnesium sulfate. After the solvent was evaporated under reduced pressure, the crude product was purified by silica gel column chromatography to obtain 0.37 g of 3-(2-cyanophenoxy)quinoline (Compound Number 62).
2-(3-methyl-1H-indazol-1-yl)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89%
With caesium carbonate; In N,N-dimethyl-formamide; for 12h;
Step 1: Cs2CO3 (1.56 g, 4.8 mmol) was added to a solution of <strong>[3176-62-3]3-methyl indazole</strong> (0.634 g, 4.8 mmol) and 2-fluorobenzonitrile (1 mL, 9.6 mmol) in DMF (20 mL). After 12 h the mixture was diluted with sat. NH4C1 and a precipitate formed. The solid was filtered, washed with H20,hexane and dried under vacuum to give 2-(3-methyl-1H-indazol-1-yl)benzonitrile (1 g, 89 %) which was used without purification.
With caesium carbonate; In N,N-dimethyl-formamide; at 20℃; for 12h;
Step 1 : CS2CO3 (1.56 g, 4.8 mmol) was added to a solution of <strong>[3176-62-3]3-methyl indazole</strong> (0.634 g, 4.8 mmol) and 2-fiuorobenzonitrile (1 mL, 9.6 mmol) in DMF (20 mL) and the mixture stirred at RT for 12 h. The mixture was diluted with sat. NH4C1 and a precipitate formed. The solid was filtered, washed with H20, hexane and dried under vacuum to give 2-(3-methyl-lH-indazol-l- yl)benzonitrile (1 g, 89 %) which was used without purification.
1-(2-cyanophenyl)-3-methyl-1H-indole-2-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With caesium carbonate; In N,N-dimethyl-formamide; at 60.0℃;
Step 1 : A mixture of 3-methyl-lH-indole-2-carbonitrile (500 mg, 3.2 mmol), 2- fluorobenzonitrile (388 mg, 3.2 mmol) and CS2CO3 (1.05g, 3.2 mmol) in DMF (10 mL) was heated at 60 C overnight. The mixture was poured into H20 and a precipitate formed. The solid was collected by filtration, washed with H20 and hexane, and dried under vacuum to give l-(2- cyanophenyl)-3-methyl-lH-indole-2-carbonitrile (780 mg, 94.6 %) as an off white solid that was used without purification.
With caesium carbonate; In dimethyl sulfoxide; at 120℃; for 16h;
General procedure: 4.4.1. Ethyl 5-amino-2-phenylpyrazolo[1,5-a]quinoline-3-carboxylate (13aa). A mixture of 2-fluorobenzonitrile 6a (121 mg,1.00 mmol), 12a (280 mg, 1.20 mmol) and Cs2CO3 (980 mg,3.00 mmol) in DMSO (5.0 mL) was stirred at 120 C for 16 h. Aftermonitoring the end of the reaction on TLC, the mixture was cooledto room temperature and diluted with water. The resulting mixturewas extracted with ethyl acetate twice. The combined organiclayers were washed with water twice, dried over MgSO4 and thesolvent was removed in vacuo to afford a residue. The residue waspurified by flash column chromatography (hexane:EtOAc1:1) onsilica gel to afford pyrazolo[1,5-a]quinoline 13aa (137 mg, 41%yield).
With caesium carbonate; In dimethyl sulfoxide; at 120℃; for 16h;
General procedure: 4.3.1. Ethyl 5-amino-2-methylpyrazolo[1,5-a]quinoline-3-carboxylate (7a). Condition C: A mixture of 2-fluorobenzonitrile6a (121 mg, 1.00 mmol), 1H-pyrazole 4 (202 mg, 1.20 mmol) andCs2CO3 (980 mg, 3.00 mmol) in DMF (5.0 mL) was stirred at 120 Cfor 16 h. After monitoring the end of the reaction on TLC, themixture was cooled to room temperature and diluted with water.The resulting mixture was extracted with ethyl acetate twice. Thecombined organic layers werewashed with water twice, dried overMgSO4 and the solvent was removed in vacuo to afford a residue.The residue was purified by flash column chromatography(hexane:EtOAc1:1) on silica gel to afford 7a (124 mg, 46% yield).Condition D: The reaction was carried out in DMSO instead of DMFunder the same conditions as that of condition C to afford 7a (175 mg, 65% yield).
General procedure: A mixture of DMF (2.00 mL, 25.8 mmol) and 10 M KOH (0.500 mL, 5.00 mmol) were heated at reflux for 5 min. The corresponding aryl halide (1.00 mmol) was added and the resulting mixture was heated for 0.5 h at 95 C. The mixture was treated with additional 10 M KOH (0.500 mL,5.00 mmol) and heated for an additional 0.5 h. The reaction was monitored by TLC and additional portions 10M KOH (0.500 mL, 5.00 mmol) were added in 0.5 h intervals until completion was evident. The reaction mixture was then cooled, diluted with water and vacuum filtered (for solid products) or extracted (for product oils) with Et2O (3x). For reaction products that were extracted, the organic fractions were combined, dried over anhydrous Na2SO4, and concentrated under reduced pressure to give the final product.
With 5% active carbon-supported ruthenium; oxygen; In toluene; at 150℃; under 3750.38 Torr; for 4h;Autoclave;
General procedure: In a typical process, into a 25 ml autoclave equipped with a magnetic stirrer were added 5percentRu/AC (0.03 mmol, 3 molpercent), benzylamine (1 mmol), 5 mL toluene at room temperature successively. After which the resulting reaction mixture was heated at 150 °C for 4 h under 0.5 MPa of oxygen atmosphere. The final reaction conversion and selectivity towards the corresponding nitriles were determined by Gas Chromatograph. After reaction, the product was purified by column chromatography of the reaction mixture on neutral alumina using hexanes/dichloromethane (80:20) or hexanes/EtOAc (30:1) as eluent.
ethyl 5-ainobenzo[4,5]imidazo[1,2-a]quinoline-6-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
52%
With potassium carbonate; In dimethyl sulfoxide; at 120℃; for 16h;
General procedure: To extend the scope of the 2-methyl-1H-benzimidazole substrates for our cascade reaction, we nextexamined the reaction of 2-fluorobenzonitrile (2a) with a variety of 2-methyl-1H-benzimidazolederivatives 1 under the conditions (Cs2CO3, DMSO, 120 C). The results are shown in Table 2. As shown in Table 2, almost all of the tested combinations successfully produced the desiredbenzimidazo[1,2-a]quinolines 8ba-8la with moderate to good isolated yields, though an undeniableamount of overreaction product 9 was isolated in most entries. Unsymmetrical 1H-benzo[d]imidazolessuch as 1b, 1e, 1f and 1g exist as an equilibrium mixture of their tautomers. Therefore, the SNAr sequenceof our cascade reaction with these substrates theoretically provides a regioisomeric mixture of thecorresponding adducts. Our survey revealed that the regiochemical outcome is highly controlled uponutilizing 1e, 1f and 1g having a substituent at the 4-position to give 8ea, 8fa and 8ga without detectingtheir regioisomers (entries 5-7). However, no reigioselectivity was observed with the cascade reactionwith 1b bearing methyl substituent at the 5-position (entry 2). These results suggest to us that the lesssterically congested nitrogen atom of 1H-benzo[d]imidazoles preferably reacted with 2-fluorobenzonitrile(2a) in the SNAr reaction. 2-Methyl-1H-benzimidazole derivatives 1h, 1i, and 1j, having an electrondonating group (-CH3, -OCH3,-SCH3), reacted with 2a to give 8ha, 8ia, and 8ja in modest yields underthe conditions, respectively (entries 8-10). When the benzimidazoles 1k and 1l possessing an electronwithdrawing group (-CN, -CO2Et) at the 2-methyl group were treated with 2a under the conditions, alarge amount of insoluble unidentified product was produced. In these reactions, the desired cascadeproduct 8la was not detected while the cascade product 8ka was isolated in a modest yield (entries 11 and12). The cascade products 8ka and 8la were obtained in good yields upon replacing Cs2CO3 with K2CO3.(entries 11 and 12). In our SNAr/Knoevenagel cascade reaction with 1k and 1l, we found the Knoevenagelcondensation between aldehydes and the active methylene of 1k and 1l occured preferably to the SNArreaction in the first step, and the desired cascade adducts could not be obtained.14 These comparativeresults indicate that the SNAr reaction occured preferably to the Dieckmann-Thorpe type reaction in thepresent cascade reaction upon fine-tuning the base used.
5-amino-9,10-dichrolobenzimidazo[1,2-a]quinoline[ No CAS ]
2-{(9,10-dichlorobenzo[4,5]imidazo[1,2-a]quinolin-5-yl)amino}benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
43%; 19%
With caesium carbonate; In dimethyl sulfoxide; at 120℃; for 16h;
General procedure: To extend the scope of the 2-methyl-1H-benzimidazole substrates for our cascade reaction, we nextexamined the reaction of 2-fluorobenzonitrile (2a) with a variety of 2-methyl-1H-benzimidazolederivatives 1 under the conditions (Cs2CO3, DMSO, 120 C). The results are shown in Table 2. As shown in Table 2, almost all of the tested combinations successfully produced the desiredbenzimidazo[1,2-a]quinolines 8ba-8la with moderate to good isolated yields, though an undeniableamount of overreaction product 9 was isolated in most entries. Unsymmetrical 1H-benzo[d]imidazolessuch as 1b, 1e, 1f and 1g exist as an equilibrium mixture of their tautomers. Therefore, the SNAr sequenceof our cascade reaction with these substrates theoretically provides a regioisomeric mixture of thecorresponding adducts. Our survey revealed that the regiochemical outcome is highly controlled uponutilizing 1e, 1f and 1g having a substituent at the 4-position to give 8ea, 8fa and 8ga without detectingtheir regioisomers (entries 5-7). However, no reigioselectivity was observed with the cascade reactionwith 1b bearing methyl substituent at the 5-position (entry 2). These results suggest to us that the lesssterically congested nitrogen atom of 1H-benzo[d]imidazoles preferably reacted with 2-fluorobenzonitrile(2a) in the SNAr reaction. 2-Methyl-1H-benzimidazole derivatives 1h, 1i, and 1j, having an electrondonating group (-CH3, -OCH3,-SCH3), reacted with 2a to give 8ha, 8ia, and 8ja in modest yields underthe conditions, respectively (entries 8-10). When the benzimidazoles 1k and 1l possessing an electronwithdrawing group (-CN, -CO2Et) at the 2-methyl group were treated with 2a under the conditions, alarge amount of insoluble unidentified product was produced. In these reactions, the desired cascadeproduct 8la was not detected while the cascade product 8ka was isolated in a modest yield (entries 11 and12). The cascade products 8ka and 8la were obtained in good yields upon replacing Cs2CO3 with K2CO3.(entries 11 and 12). In our SNAr/Knoevenagel cascade reaction with 1k and 1l, we found the Knoevenagelcondensation between aldehydes and the active methylene of 1k and 1l occured preferably to the SNArreaction in the first step, and the desired cascade adducts could not be obtained.14 These comparativeresults indicate that the SNAr reaction occured preferably to the Dieckmann-Thorpe type reaction in thepresent cascade reaction upon fine-tuning the base used.
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; N,N-dimethylethylenediamine; copper(l) chloride; In toluene; at 80℃; for 24h;Sealed tube; Schlenk technique;
General procedure: A mixture of benzylamine 1a (107.0 mg, 1.0 mmol), CuCl (5.0 mg, 0.05 mmol, 5 molpercent), 2,2,6,6-tetramethyl-1-piperidyloxy (TEMPO, 7.8 mg, 0.05 mmol, 5 molpercent), and N,N-dimethylethane-1,2-diamine (DMEDA, 4.4 mg, 0.05 mmol, 5 molpercent) in toluene (0.5 mL) sealed in a Schlenk tube (100 mL) with an air balloon was stirred at 80 °C for 24 h. The reaction was then monitored by TLC and/or GC-MS. After completion of the reaction, solvent was evaporated under vacuum. The residue was purified by scosh column chromatography on silica gel using petroleum ether and ethyl acetate (0?100/1) as the eluent, giving product 2a in 80percent isolated yield
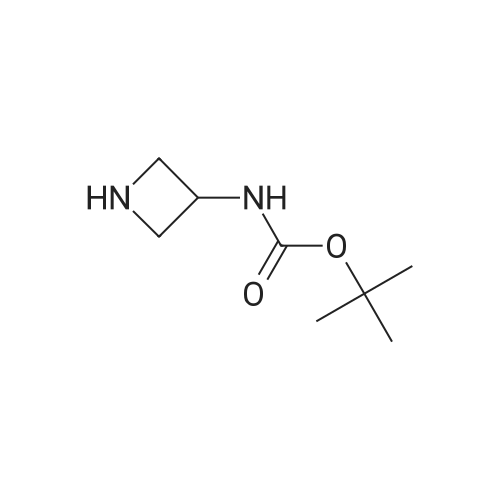
tert-butyl N-[1-(2-cyanophenyl)azetidin-3-yl]carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
20.08%
With potassium carbonate; In acetonitrile; at 100℃; for 16h;
Step 1: tert-Butyl N-[1-(2-cyanophenyl)azetidin-3-yl]carbamate A mixture of 2-fluorobenzonitrile (1.50 g, 12.39 mmol), <strong>[91188-13-5]tert-butyl N-(azetidin-3-yl)carbamate</strong> (2.35 g, 13.63 mmol) and K2CO3 (3.44 g, 24.78 mmol) in CH3CN (50.00 mL) was stirred at 100 C. for 16 h. The mixture was purified by SGC (EtOAc:PE=1:5) to afford tert-butyl N-[1-(2-cyanophenyl) azetidin-3-yl]carbamate (680.00 mg, 2.49 mmol, 20.08% yield) as product. ESI-MS (EI+, m/z): 274.1 [M+H]+.
With potassium carbonate; In N,N-dimethyl-formamide; at 85℃; for 16h;
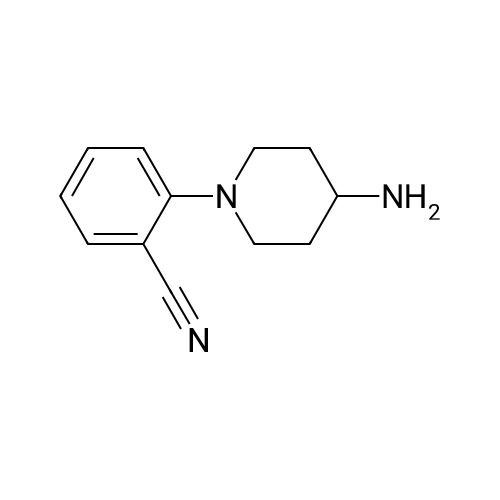
Step 1: 2-(4-Aminopiperidin-1-yl)benzonitrile To a solution of 2-fluorobenzonitrile (10.00 g, 82.57 mmol), <strong>[13035-19-3]piperidin-4-amine</strong> (10.75 g, 107.34 mmol) in DMF (100.00 mL) was added K2CO3 (28.49 g, 206.43 mmol). The reaction mixture was stirred at 85 C. for 16 h. then cooled to r.t and diluted with H2O (400 mL) and extracted with EtOAc (100 mL*3). The combined organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to afford the crude product, which was purified by chromatography (silica, dichloromethane/methanol: 1/10) to afford 2-(4-amino-1-piperidyl)benzonitrile (9.60 g, 47.70 mmol, 57.77% yield) as product. ESI-MS (EI+, m/z): 202.0 [M+H]+.
With caesium carbonate; In N,N-dimethyl-formamide; at 75℃; for 6h;Inert atmosphere;
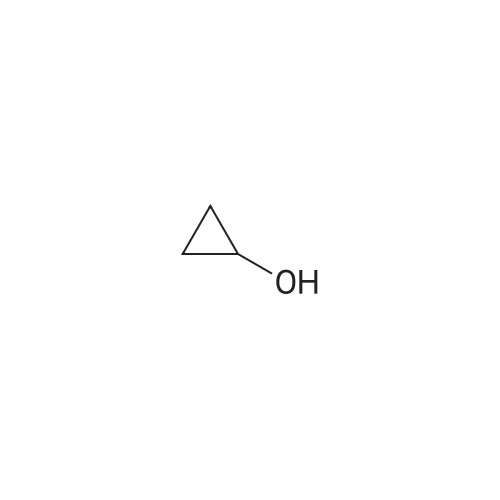
1) Under a nitrogen atmosphere, 0.30 g (2.50 mmol) of 2-fluorobenzonitrile, 3.0 mL of N,N-dimethylformamide (analytical grade) and 0.22 g of <strong>[16545-68-9]cyclopropanol</strong> (3.75 mmol) were placed in a 25 mL single-mouth flask. 2.2 g (3.70 mmol) of cesium carbonate was added in two portions under magnetic stirring, so that the temperature in the flask did not exceed 75 C. The mixture was heated to 75 C in a water bath and magnetically stirred for 6 h.2) After the reaction time is reached, the reaction liquid obtained in the step 1) is quenched with 15 mL of water, extracted with ethyl acetate, washed with saturated brine, dried over sodium sulfate and filtered, and evaporated. Have a crude product;The crude product is purified by a silica gel column. The purification is specifically: 3.0 g of column chromatography silica gel containing 200-300 mesh sieve in a column, and the crude product is dissolved in 1.0 mL of ethyl acetate and 0.5 g of the above silica gel. After steaming the mixture, the mixture was poured into a silica gel column, and a mixture of ethyl acetate and petroleum ether = 1:10 by volume was used as a detergent, and the amount was 150 mL; the eluate which satisfies the pure point of TLC detection was collected; Concentration treatment gave a pale yellow oily liquid which was found to be 2-(cyclopropoxy)benzonitrile in a yield of 95.1%.
0.15 g
With caesium carbonate; In N,N-dimethyl-formamide; at 75℃; for 6h;Inert atmosphere;
General procedure: A solution of 1-chloro-2-fluorobenzene(0.30 g, 2.3 mmol) in DMF (4 mL) was rapidly added to <strong>[16545-68-9]cyclopropanol</strong> (0.20 g, 3.6 mmol) in a 25 mLsingle-necked round-bottom flask. After that, the mixture was stirred and Cs2CO3 (1.12 g, 3.5 mmol)was put into the flask at nitrogen atmosphere. Then the mixture was refluxed under nitrogen andstirred at 135 C for 15 h. After completion of the reaction, the resulting mixture was diluted withH2O (30 mL) and extracted with AcOEt (10 mL×3). Then the combined organic phase was washedwith saturated aqueous NaCl (10 mL×3) and dried over Na2SO4 for 0.5 h. After that, the organicphase was concentrated under reduced pressure to give crude product. The crude product waspurified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 100/1) togive desired product as a light yellow liquid in 39.6% yield (0.15g)
1-(2-cyano-phenyl)-1<i>H</i>-pyrazole-3-carboxylic acid ethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
5 g
With caesium carbonate; In N,N-dimethyl-formamide; at 25℃; for 16h;
To a solution of compound B22 (5 g, 35.7 mmol) and Cs2C03 (29.1 g, 89.2 mmol) in DMF (50 mL) was added 2-fluorobenzonitrile (6.48 g, 53.5 mmol, 5.69 mL). The reaction mixture was stirred at 25C for 16 hours to give yellow mixture. TLC showed the reaction was completed. The reaction mixture was quenched by addition H20 (200 mL) and extracted with EtO Ac (150 mL x2). The combined organic layers was dried over Na2S04, filtered and concentrated under reduced pressure to give crude product. The crude product was purified by silica gel column to give compound B23 (5 g) as a white powder.
With potassium carbonate; In dimethyl sulfoxide; at 140℃; for 12h;
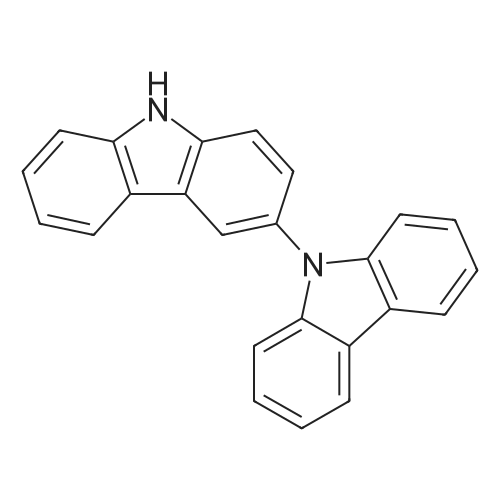
3,3?-bicarbazole (0.50 g, 1.5 mmol), potassium carbonate (0.99 g, 7.2 mmol),2-fluorobenzonitrile (0.39 g, 3.3 mmol), DMSO 6 ml, and heated at 140 C for 12 h.Cool to room temperature and pour into 200ml water to precipitate a large amount of solid and stir for 0.5h.A white solid was obtained by suction filtration and purified by column chromatography to obtain a white solid with a yield of 84%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping