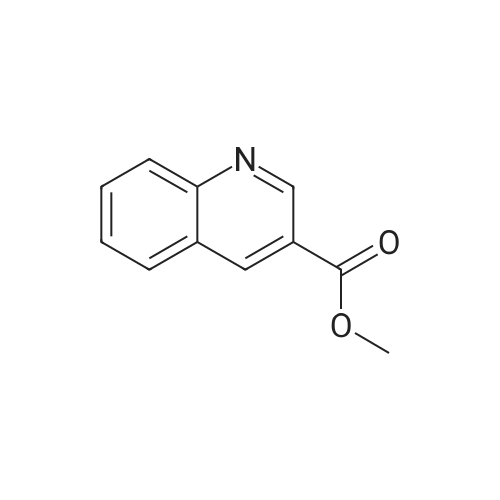
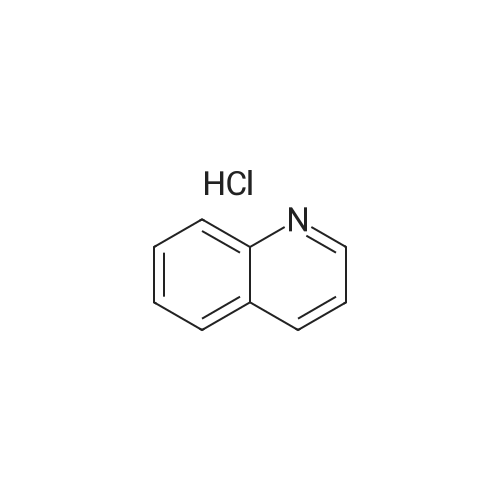
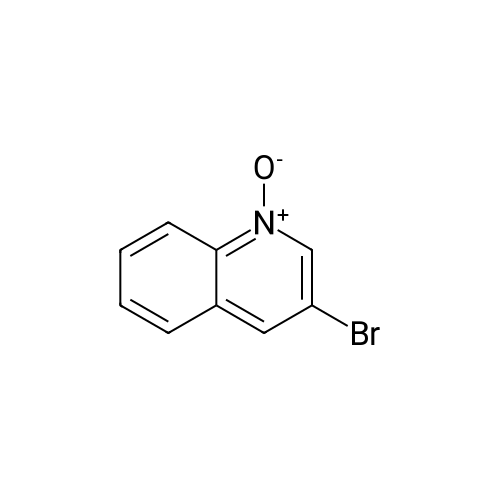
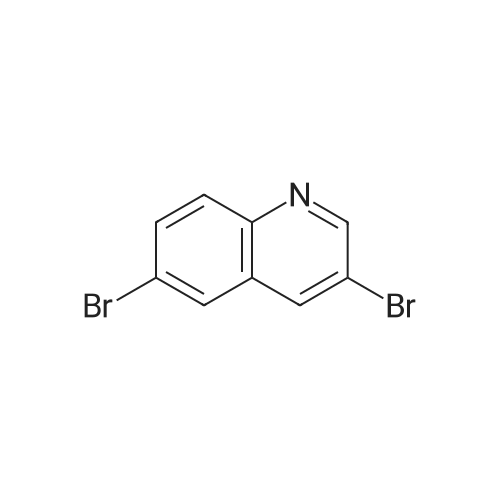
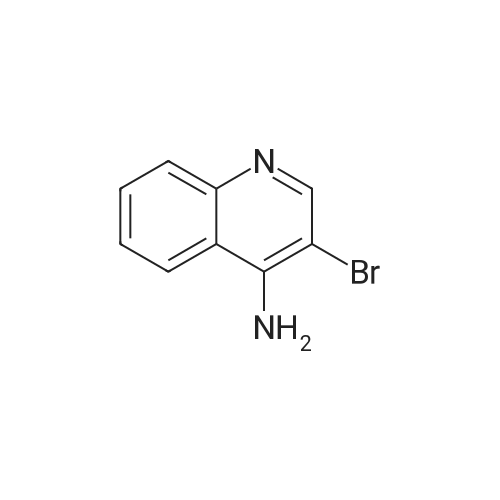
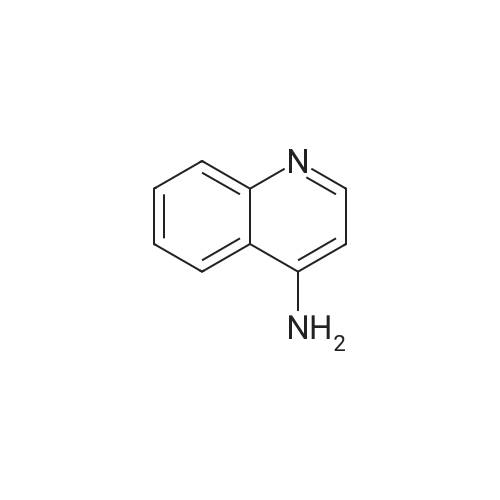
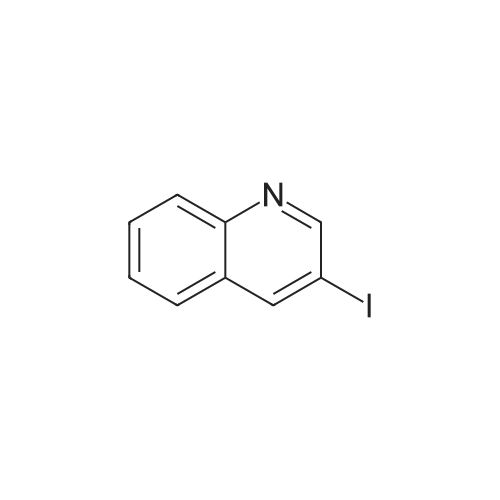
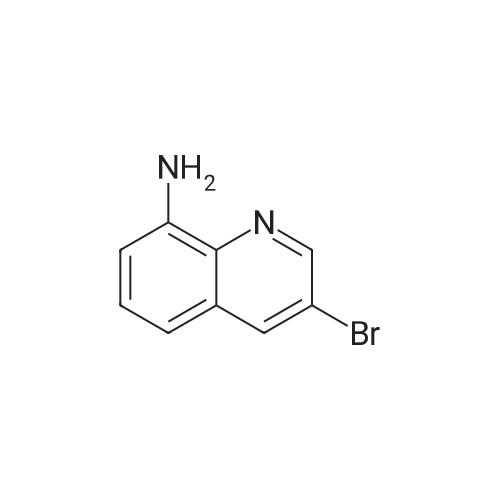
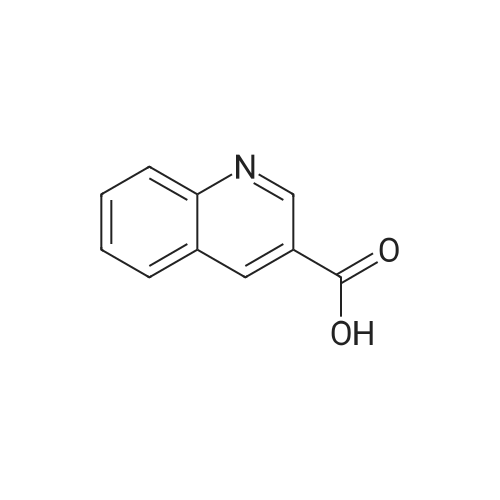
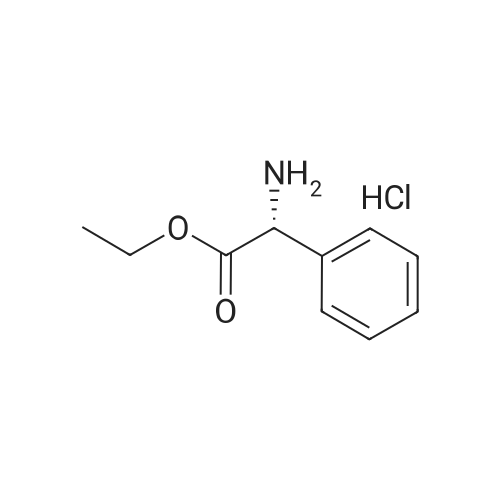
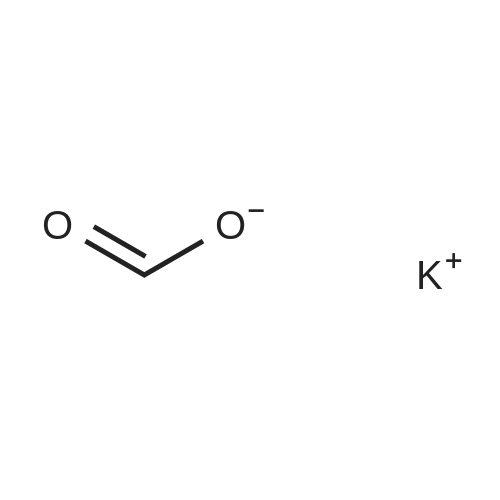
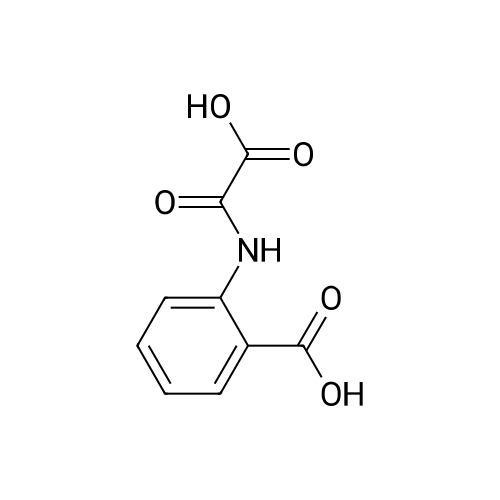
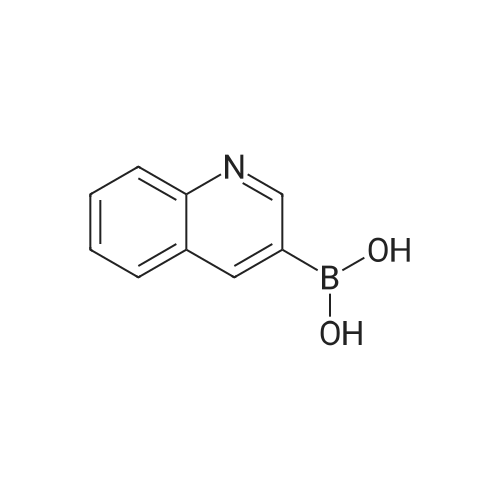
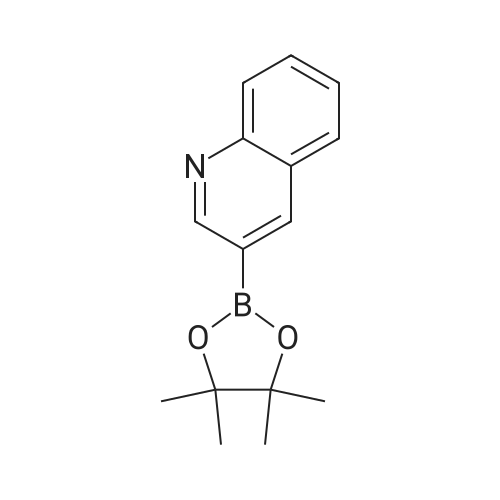
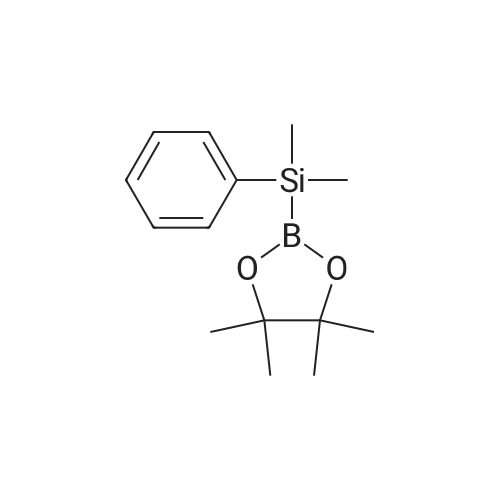
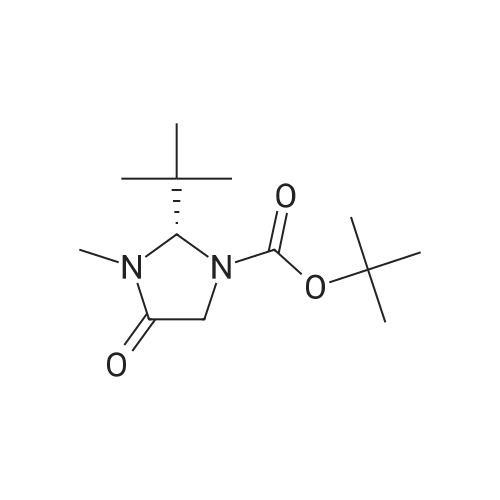
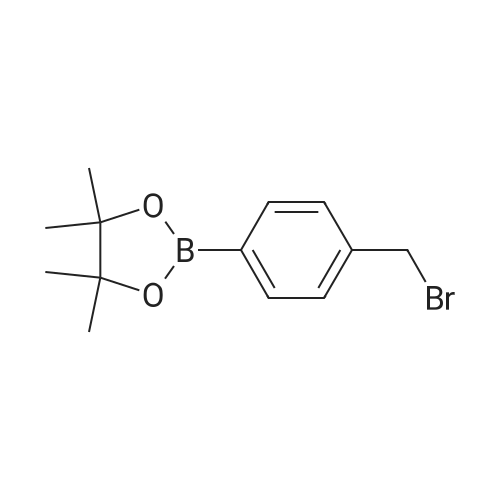
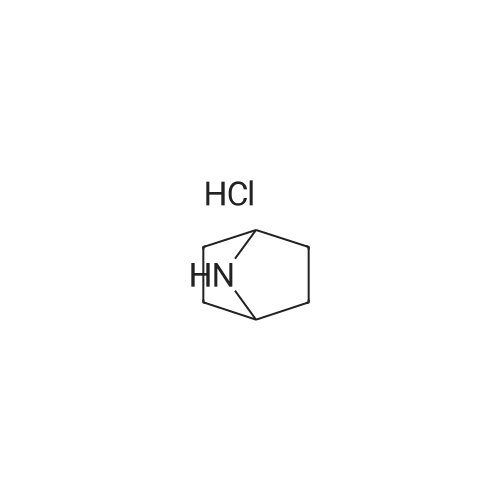
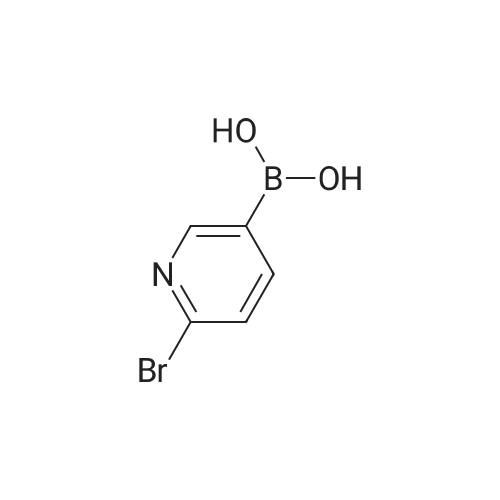
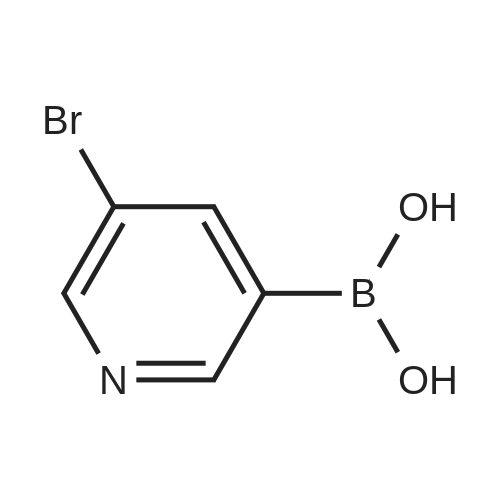
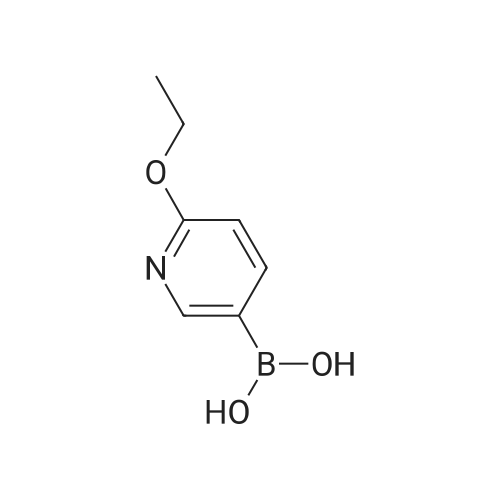
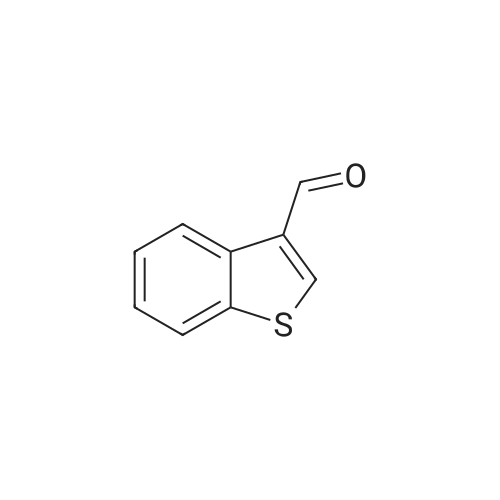
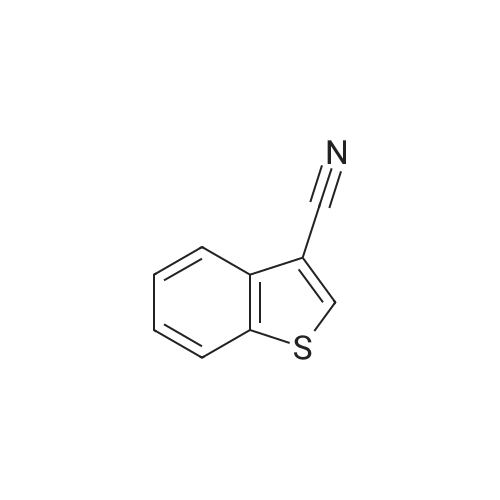
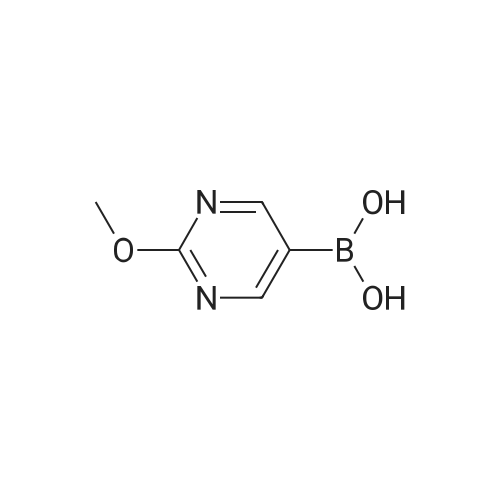
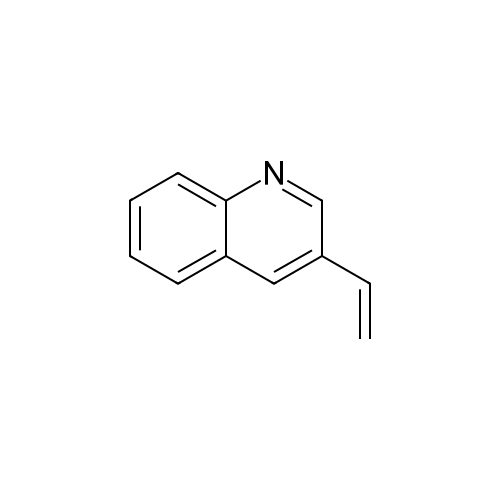
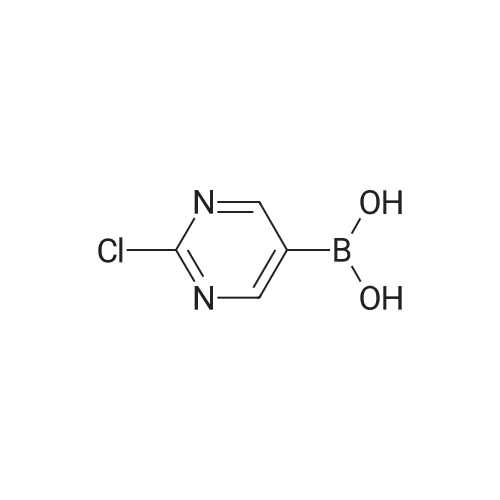
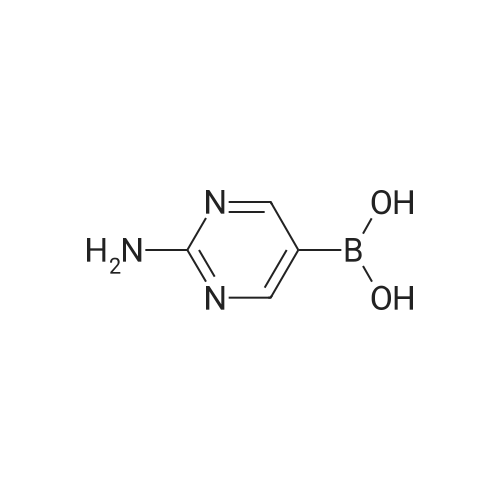
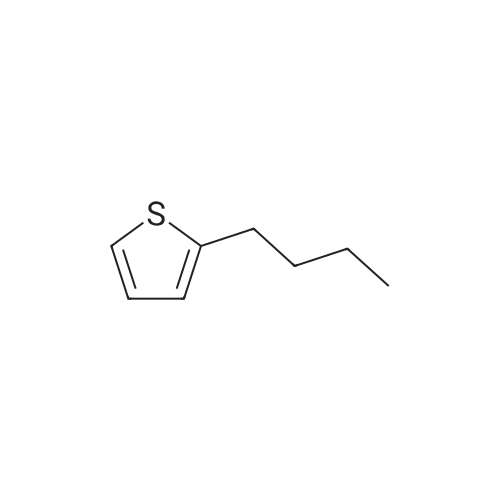
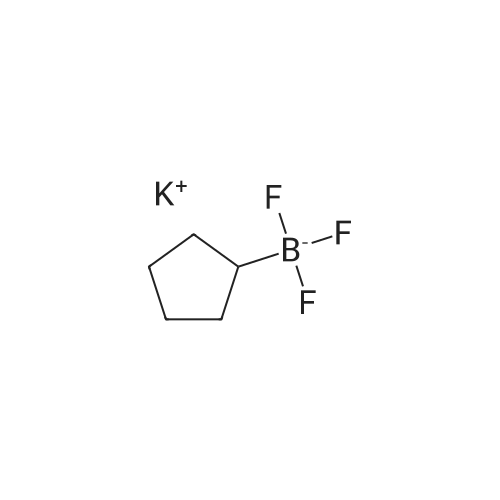
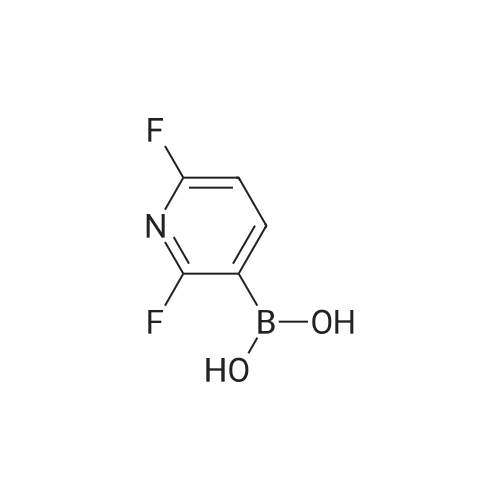
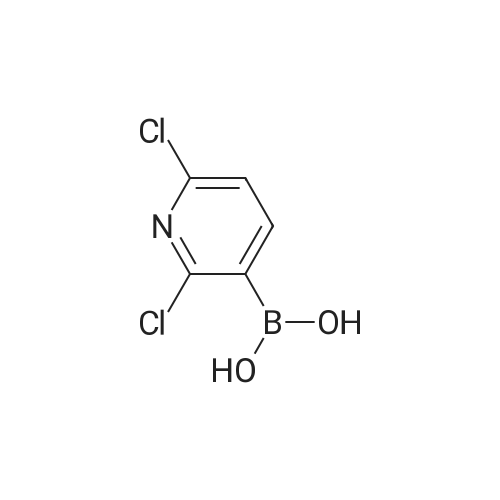
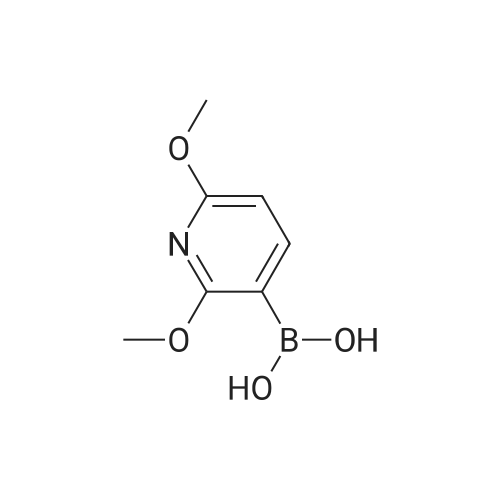
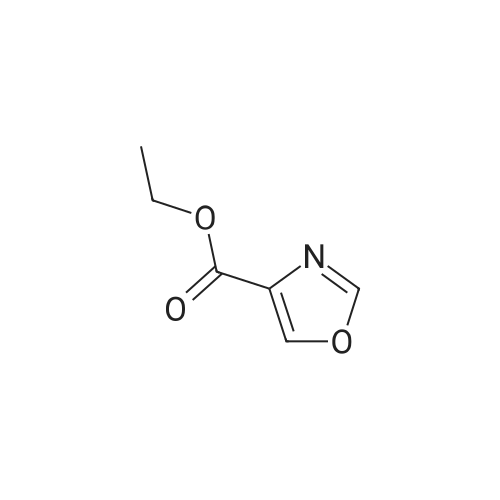
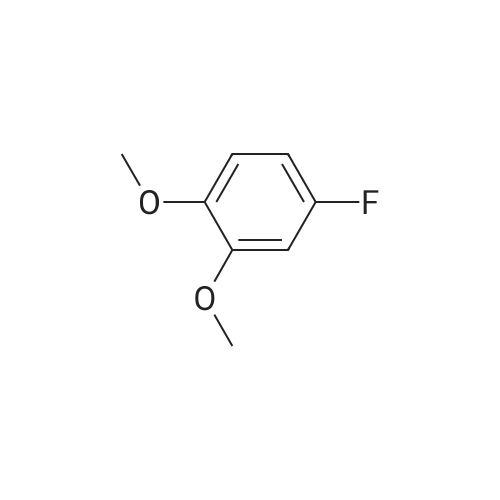
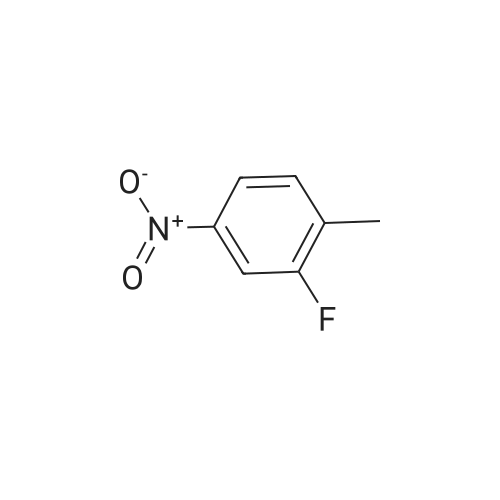
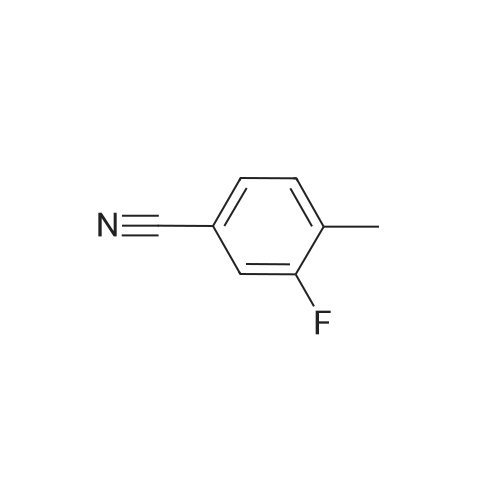
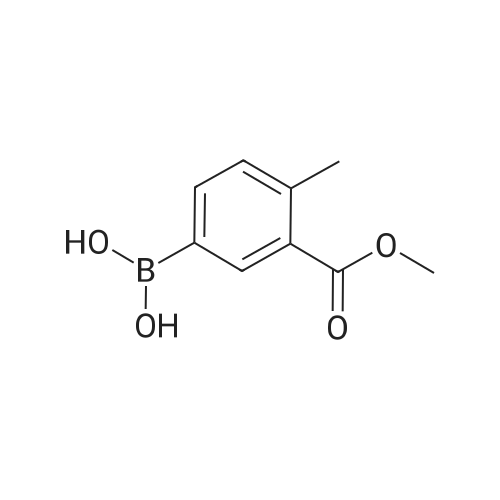
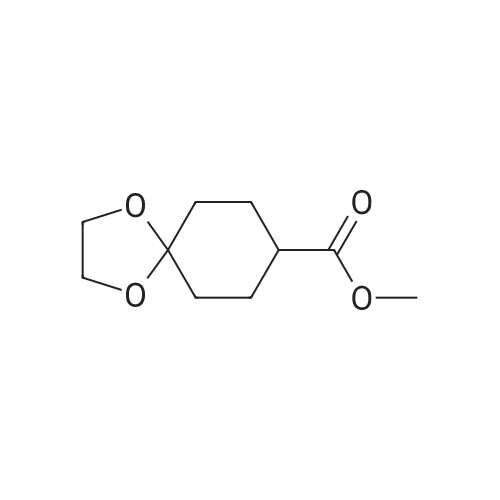
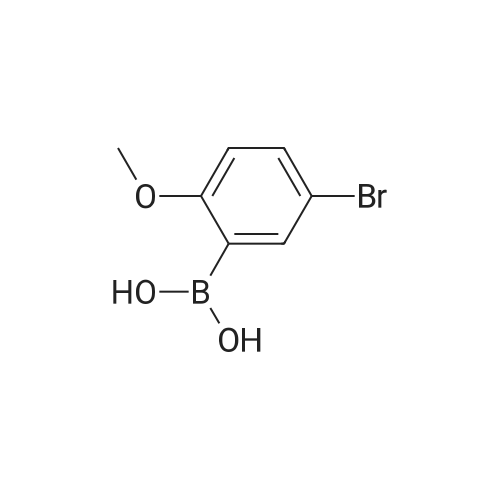
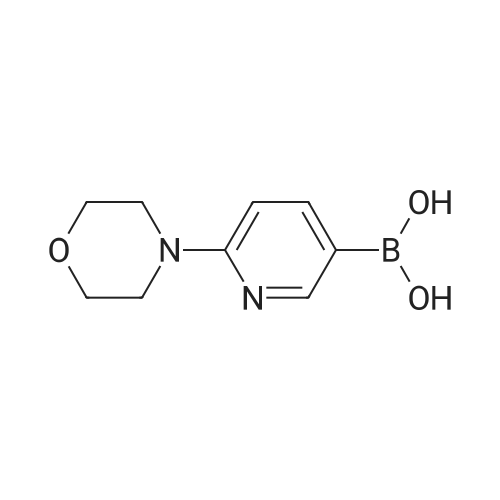
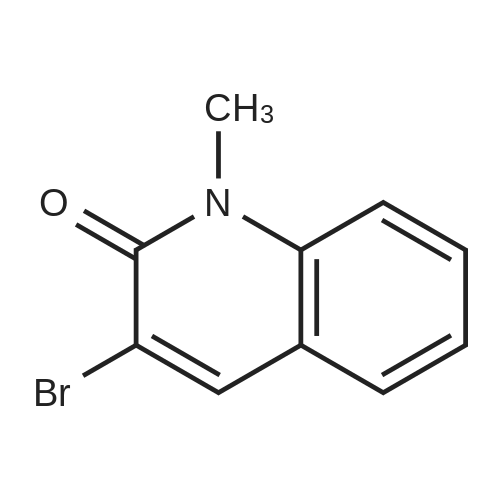
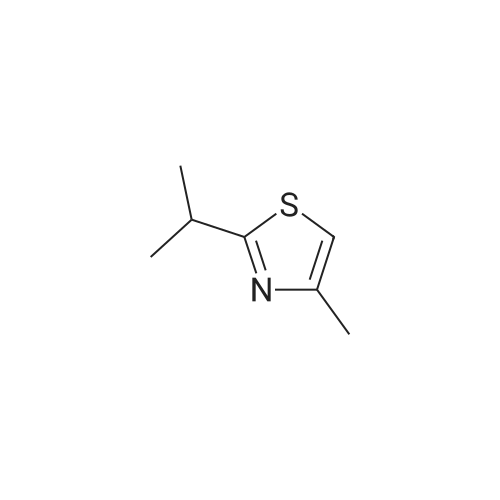
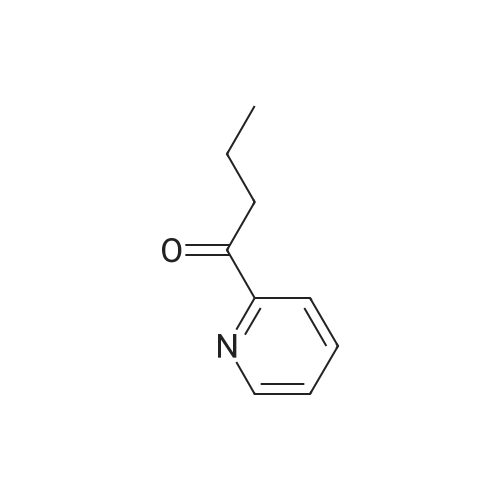
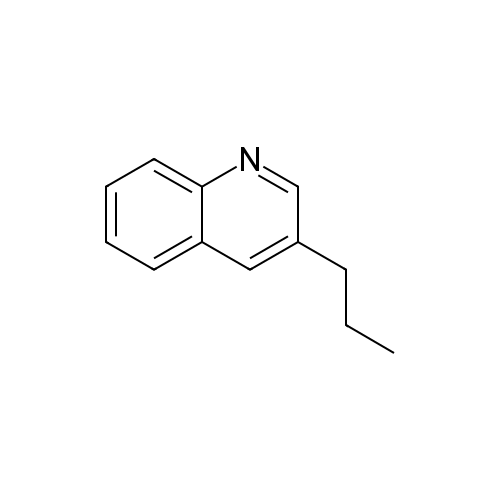
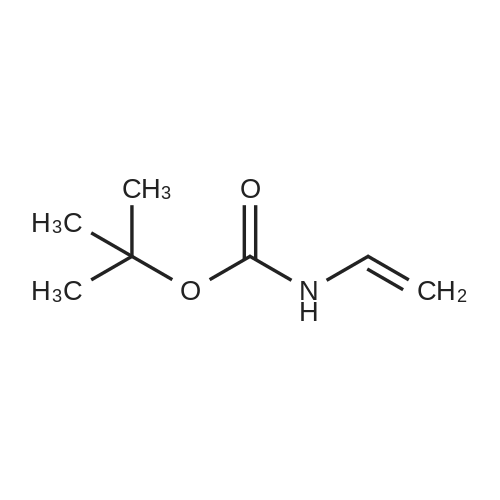
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With 1,1'-bis-(diphenylphosphino)ferrocene; palladium diacetate; triethylamine In acetonitrile at 80℃; for 23 h; Sealed tube
General procedure: An aryl bromide (1 mmol), Ph P (94 mg, 0.36 mmol), and Pd(OAc)2 (22 mg, 0.1 mmol) were weighed into screw cap vial equipped with a magnetic stir bar. Dry MeCN (5 mL) was added, followed by Et N (2.5 mL) and the corresponding amine (1.5 mmol). Neat iron pentacarbonyl (35 μL, 0.25 mmol) was added, and the vial was thoroughly closed and heated in the preheated oil bath or aluminum heating block (80 °C). The reaction mixture was stirred at the specified temperature for 23 h, then concentrated with approximately 1 g of silica, and the absorbed material was purified by gradient MPLC (hexanes–EtOAc in ratios 1:10 to 1:1). Methyl and n-butyl carboxylates, described in the article, were prepared in a similar manner; a mixture of MeCN with MeOH or n-BuOH in a ratio of 2:1 was used instead of pure MeCN, the identical amount of dppf (0.36 mmol) was used instead of Ph3P in specified cases.
Reference:
[1] Acta Chemica Scandinavica, Series B: Organic Chemistry and Biochemistry, 1981, vol. 35, # 3, p. 185 - 192
3
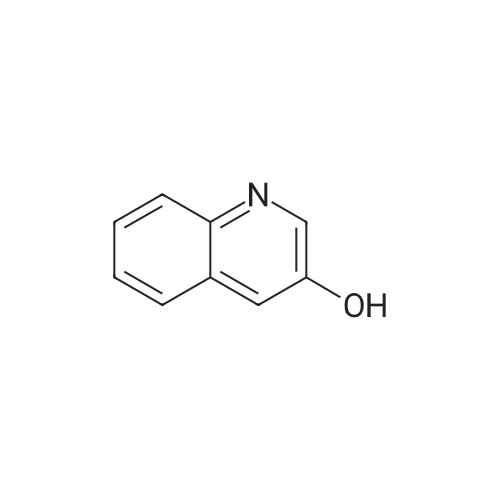
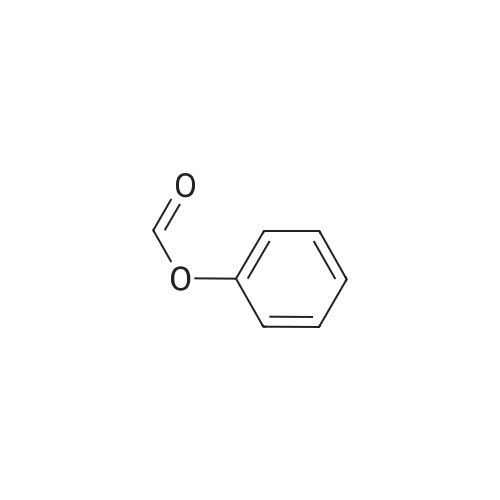
[ 530-64-3 ]
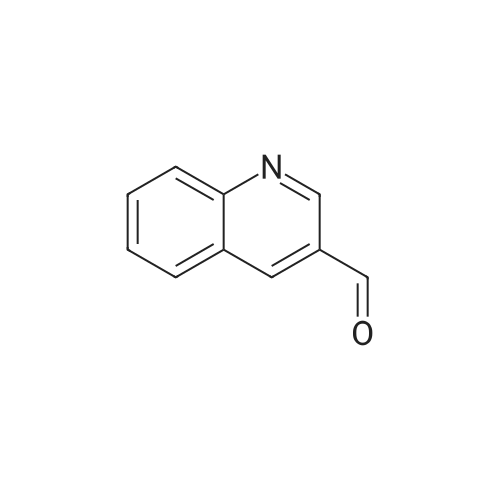
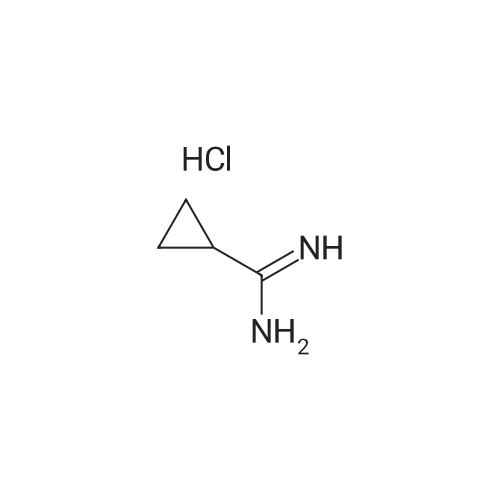
[ 5332-24-1 ]
Yield
Reaction Conditions
Operation in experiment
84.5%
With bromine In water; nitrobenzene; benzene
EXAMPLE 5 3-Bromoquinoline A slurry of 33.3 g. (0.20 mole) of quinoline hydrochloride in 50 ml. of nitrobenzene, contained in a 250 ml. three-neck round-bottom flask equipped with a paddle stirrer, condenser, dropping funnel, and thermometer, was heated to 177°-180°C., and 35.2 g. (0.22 mole) of bromine was added dropwise via the dropping funnel over a period of 11/2 hours. The temperature was maintained at about 180°C., and the mixture stirred for an additional 3 hours and 20 minutes, at which time the evolution of hydrogen chloride had ceased. Heating was stopped, the reaction product mixture was cooled to room temperature, and 200 ml. of benzene was added. The mixture was filtered, and the solid on the filter was washed with 100 ml. of benzene, and sucked dry on the filter. In this manner, 48.7 g. of crude product was obtained. This crude product was added to 200 ml. of water and the mixture made basic with saturated aqueous sodium carbonate solution. The basic mixture was extracted four times with 200 ml. portions of ether. The combined ether extracts were dried, and the solvent was removed in vacuo, leaving 35.1 g. of a pale yellow oil which solidified on standing in the refrigerator. The solid had a melting point of about 12°-13°C., and was identified as 3-bromoquinoline. Yield, 84.5 percent of theory; VPC purity, 97 percent.
Reference:
[1] Patent: US3956301, 1976, A,
[2] Patent: US5266700, 1993, A,
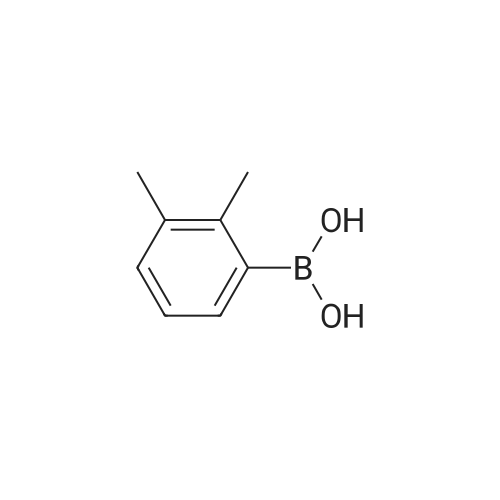
4
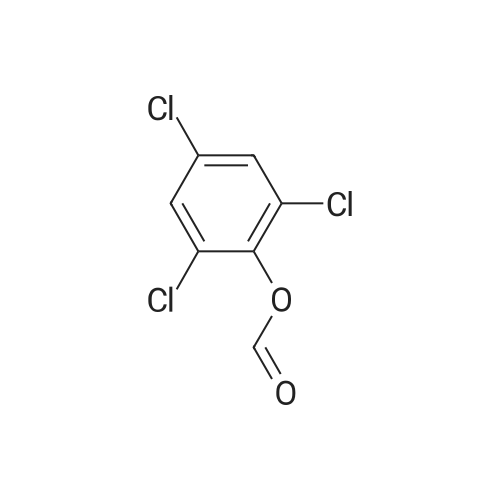
[ 22615-00-5 ]
[ 5332-24-1 ]
Reference:
[1] Tetrahedron Letters, 1997, vol. 38, # 5, p. 845 - 848
[2] Chemical Communications, 2015, vol. 51, # 32, p. 7035 - 7038
[3] Organic Letters, 2018, vol. 20, # 23, p. 7712 - 7716
5
[ 91-22-5 ]
[ 5332-24-1 ]
Reference:
[1] Bulletin of the Polish Academy of Sciences, Chemistry, 1986, vol. 34, # 7-8, p. 281 - 287
[2] Tetrahedron Letters, 1997, vol. 38, # 25, p. 4415 - 4416
[3] Chemische Berichte, 1886, vol. 19, p. 2766
[4] Recueil des Travaux Chimiques des Pays-Bas, 1937, vol. 56, p. 699,706
[5] Journal fuer Praktische Chemie (Leipzig), 1896, vol. <2> 54, p. 356[6] Chemische Berichte, 1896, vol. 29, p. 2459
[7] Chemistry and Industry (London, United Kingdom), 1959, p. 1449
6
[ 580-17-6 ]
[ 5332-24-1 ]
Reference:
[1] Canadian Journal of Chemistry, 2005, vol. 83, # 3, p. 213 - 219
With copper(l) iodide; sodium iodide; N,N`-dimethylethylenediamine In 1,4-dioxane at 110℃; Inert atmosphere
A mixture of 3-bromoquinoline (19) (9.83 g, 47.3 mmol), CuI (450 mg, 2.4 mmol), NaI (14.15 g, 94.52 mmol), N,N-dimethylethylenediamine (0.5 mL, 415 mg, 4.7 mmol) in dioxane (47 mL) was heated, stirred, and refluxed under N2 for 44-48 h at 110 °C. The reaction was monitored by GC/MS till the conversion reached 100percent. After cooling to room temperature, the mixture was diluted with 30percent aqueous ammonia (20 mL), followed by extraction with EtOAc (3 .x. 30 mL). The combined organic layers was washed with brine (100 mL), dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated by rotary evaporation under reduced pressure to yield the pure product as a pale yellow solid in almost quantitative yield. The product was used as such for the next step without further purification. 1H NMR (CDCl3): δ 9.03 (d, 1H, J = 2.4 Hz), 8.54-8.53 (m, 1H), 8.08-8.04 (m, 1H), 7.76-7.69 (m, 2H), 7.59-7.53 (m, 1H).
100%
With copper(l) iodide; sodium iodide; N,N`-dimethylethylenediamine In 1,4-dioxane at 110℃; for 48 h; Inert atmosphere
The method of Klapars was used [6]. A mixture of 3-bromoquinoline (9.8 g, 47.3 mmol), CuI (0.45 g, 2.4 mmol), NaI (14.2 g, 94.5 mmol), N, N-dimethylethylenediamine (0.5 mL) and dioxane (47.3 mL) was stirred and heated to 110 °C and allowed to reflux under N2 for 48 h. The reaction was monitored by TLC till the conversion reached 100percent. The resulting mixture was allowed to cool to rt, diluted with 30percent aqueous NH3 (20 mL), then diluted with distilled H2O and extracted with EtOAc (3 .x. 30 mL). The organic layer was washed with brine (100 mL), dried over anhydrous Na2SO4 and concentrated by rotary evaporation at reduced pressure to yield the pure product in quantitative yield as a pale yellow solid. Yield 12 g, 100percent. 1HNMR (CDCl3): δ 9.03 (d, 1H, J = 2.4 Hz), 8.54-8.53 (m, 1H), 8.08-8.04 (m, 1H), 7.76-7.69 (m, 2H), 7.59-7.53 (m, 1H).
100%
With copper(l) iodide; N,N-dimethylethylenediamine; sodium iodide In 1,4-dioxane for 48 h; Reflux; Inert atmosphere
A mixture of 3-bromoquinoline (9.8g, 4.73mmol), CuI (0.45g, 2.4mmol), NaI (14.2g, 94.5mmol), N,N-dimethylethylenediamine (0.5mL), and dioxane (47.3mL) was stirred and heated to 100°C and allowed to reflux under nitrogen for 48haccording to the method of Klapars [28]. The progress of the reaction was monitored by TLC until 100percent conversion was achieved. The reaction mixture was allowed to cool to room temperature, and then diluted with aq NH3 (20mL) followed by H2O, and extracted with EtOAc (3×30mL). The organic fraction was washed with brine (150mL), dried (Na2SO4), and concentrated by rotary evaporation at reduced pressure to yield the pure product as a pale yellow solid. Yield (12g, 100percent). 1H NMR (CDCl3): δ 9.03 (d, 1H, J=2.4Hz), 8.54–8.53 (m, 1H), 8.08–8.04 (m, 1H), 7.76–7.69 (m, 2H), 7.59–7.53 (m, 1H).
99%
With copper(l) iodide; N,N-dimethylethylenediamine; sodium iodide In 1,4-dioxane at 100℃; for 8 h; Inert atmosphere
3-Bromoquinoline (13) (8.50 g, 40.85 mmol), N,N-dimethylethylenediamine (720 mg, 8.17 mmol), copper(I) iodide (389 mg, 2.04 mmol), and sodium iodide (12.25 g, 81.71 mmol) in 1,4-dioxane (30 mL) were stirred under argon at 100 °C for 8 h. The reaction mixture was allowed to cool to room temperature, diluted with dichloromethane (40 mL), and washed with ammonia solution (25 mL) and water (25 mL). The organic layer was dried over Na2SO4, filtered, and evaporated to dryness. The title compound was obtained as a yellow solid (10.30 g, 99percent), mp 56-58 °C (lit., 36 mp 58-59 °C); 1H NMR (CDCl3, 400 MHz) δ 9.04 (br s, 1H), 8.54 (d, 1H, J=1.6 Hz) 8.06 (d, 1H, J=8.4 Hz), 7.77-7.69 (m, 2H), 7.59-7.54 (m, 1H); 13C NMR (CDCl3, 400 MHz) δ 155.6, 146.4, 143.7, 130.0, 129.9, 129.5, 127.4, 126.8, 89.8; C9H6IN (255.06); LCMS (ESI+) m/z 256 [M+H]+. Anal. Calcd for C9H6IN (255.06) C, 42.38; H, 2.37; N, 5.49. Found: C, 42.25; H, 2.36; N, 5.51.
92%
With copper(I) oxide; <i>L</i>-proline; potassium iodide In ethanol at 110℃; for 30 h; Schlenk technique; Inert atmosphere; Sealed tube
General procedure: A Schlenk tube was charged with Cu2O (7.2 mg, 10 molpercent), l-proline (11.5 mg, 20 molpercent), aryl (or heteroaryl) bromide (1 or 3,0.50 mmol), potassium iodide (KI) (249 mg, 0.75 mmol), and EtOH(1.5 mL) under nitrogen atmosphere. The Schlenk tube was sealedwith a teflon valve, and then the reaction mixture was stirred at110C for a period (the reaction progress was monitored by GCanalysis). After the reaction was completed, GC yield of high volatileproduct was determined using an appropriate internal standard(chlorobenzene or 1-chloro-4-methylbenzene) or the solvent wasremoved under reduced pressure. The residue obtained was puri-fied via silica gel chromatography (eluent: petroleum ether/ethylacetate = 10/1) to afford aryl iodides 2a–2o or heteroaryl iodides4a–4g.
90%
With copper(l) iodide; sodium iodide; N,N`-dimethylethylenediamine In 1,4-dioxane at 110℃; for 22 h; Inert atmosphere; Sealed tube
An oven dried, sealable glass tube was charged with a magnetic stir bar, 3-bromoqunoline (1.04 g, 5.0 mmol), freshly ground sodium iodide (1.52 g, 10.0 mmol), and copper (I) iodide (100 mg, 0.5 mmol). The vessel was fitted with a rubber septum, evacuated under vacuum and backfilled with argon. This process was repeated three times. The vessel was then charged with 1,4-dioxane (5 mL) followed by N, N' -dimethylethylenediamine (0.12 mL, 1.0 mmol) via syringe. The rubber septum was removed and the reaction vessel immediately sealed tightly with a Teflon screw cap and heated to 110 °C for 22 hours. After cooling to room temperature, the reaction was diluted with saturated aqueous NH4C1 (30 mli) and extracted with DCM (4 χ 25 mL) . The combined organic layers were washed with brine (30 mL) and dried over Na2S04, then concentrated to a brown residue. The residue was recrystallized in hexanes/EtOAc to provide 3-iodoqunoline (1.15g, 90percent). 3-Iodoquinoline H NMR (600 MHz, CDC13) δ 9.04 (d, J = 1.8 Hz, 1H) , 8.55-8.54 (m, 1H) , 8.08-8.06 (m, 1H) , 7.75-7.71 (m, 2H) , 7.58-7.56 (m, 1H) ; 13C NMR (150 MHz, CDC13) δ 155.57, 146.34, 143.71, 130.03, 130.01, 129.50, 127.41, 126.79, 89.77; HRMS (ESI-TOF) Calcd for C9H7IN [M+H]+: 255.9618; found: 255.9623.
87%
With copper(l) iodide; sodium iodide; N,N`-dimethylethylenediamine In 1,4-dioxane at 100℃; for 18 h;
3-Bromoquinoline (64.00 g, 308 mmol), N^-dimethylethylenediamine (13.5 ml, 127 mmol), cuprous iodide (12.00 g, 63.0 mmol) and sodium iodide (112 g, 747 mmol) in dioxane (300 ml) was placed into a preheated oil bath at 1000C. After stirring for 18 h, the heterogeneous mixture was diluted water and extracted with methylene chloride. The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was flash chromatographed with methylene chloride as the eluant to afford 68.47 g (87percent yield) of 3-iodoquinoline as a yellow solid. Method [8] retention time 6.47 min by HPLC (M+=256).
72%
Stage #1: With n-butyllithium; butyl magnesium bromide In tetrahydrofuran; hexanes at -30 - -10℃; for 3.5 h; Stage #2: With iodine In tetrahydrofuran; hexanes at -10 - 20℃;
Example 2. Synthesis of 3-f 18r Fifluoroquinoline; Example 2(i) : 3-lodoquinoline; n-Butyl lithium (20 ml_ of a 1.9M solution in hexanes, 38 mmol) was added 5 dropwise to a stirred solution of butylmagnesium chloride (0.93 ml_ of a 2M solution in hexanes, 19 mmol) in THF at -10 0C and stirred for 1 h when the solution was cooled to -30 0C and 3-bromoquinoline (6.8 ml_, 50 mmol) added dropwise. The deep red solution was stirred at -10 °C for 2.5 h when iodine (12.7 g, 50 mmol) was added and the reaction mixture allowed to warm to room i o temperature overnight. Water (100 mL) was added and the mixture was extracted with ethyl acetate (3 * 100 mL), dried (MgSO4) and concentrated in vacuo to give a brown oil. Purification by flash chromatography (SiO2; DCM) gave the title compound as an off white solid. (9.20 g, 36 mmol, 72percent); mp 48-^49 °C (from ether-petrol); silica gel TLC Rf 0.36 (DCM); (Found C, 42.24; H, 2.29; N, 5.40.15 C9H6IN requires C, 42.38; H, 2.37; N, 5.49percent.); iWcrrT1 (neat) 2083, 1654, 1489,1275, 1070; δH (300 MHz; CDCI3) 9.05 (1 H, s, H2), 8.55 (1 H, d, H4 J 2 Hz), 8.08(1 H, d, H8 J 8 Hz), 7.78-7.51 (3H, m, H5-H7); δc (75 MHz; CDCI3) 155.91 (C2),146.89 (C9), 143.85 (C4), 130.31 (C7), 130.15 (C10), 130.00 (C8), 127.60 (C5),.. 127.02 (C6), 90.04 (C3); m/z (El) 255(M+H+, 40percent), 127(100), 101(95), 75(70). 0>~r[Found: * "-
Reference:
[1] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 1, p. 458 - 470
[2] European Journal of Medicinal Chemistry, 2011, vol. 46, # 5, p. 1789 - 1797
[3] European Journal of Medicinal Chemistry, 2013, vol. 70, p. 130 - 142
[4] Tetrahedron, 2013, vol. 69, # 45, p. 9512 - 9519
[5] Journal of the American Chemical Society, 2002, vol. 124, # 50, p. 14844 - 14845
[6] Catalysis Today, 2016, vol. 274, p. 129 - 132
[7] Journal of the American Chemical Society, 2015, vol. 137, # 9, p. 3338 - 3351
[8] Patent: WO2015/131100, 2015, A1, . Location in patent: Page/Page column 166; 167
[9] Angewandte Chemie - International Edition, 2015, vol. 54, # 1, p. 263 - 266[10] Angew. Chem., 2015, vol. 127, # 01, p. 265 - 268,4
[11] Patent: WO2010/91310, 2010, A1, . Location in patent: Page/Page column 147
[12] Journal of the American Chemical Society, 2006, vol. 128, # 26, p. 8404 - 8405
[13] Chemistry - A European Journal, 2008, vol. 14, # 33, p. 10348 - 10356
[14] Tetrahedron, 2003, vol. 59, # 43, p. 8629 - 8640
[15] Tetrahedron Letters, 2003, vol. 44, # 10, p. 2033 - 2035
[16] Patent: WO2007/141529, 2007, A1, . Location in patent: Page/Page column 12-13
[17] Journal of the American Chemical Society, 2015, vol. 137, # 26, p. 8328 - 8331
[18] Chemical & Pharmaceutical Bulletin, 1982, vol. 30, # 5, p. 1731 - 1737
[19] Journal of Organic Chemistry, 2011, vol. 76, # 18, p. 7563 - 7568
[20] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 22, p. 7194 - 7201
17
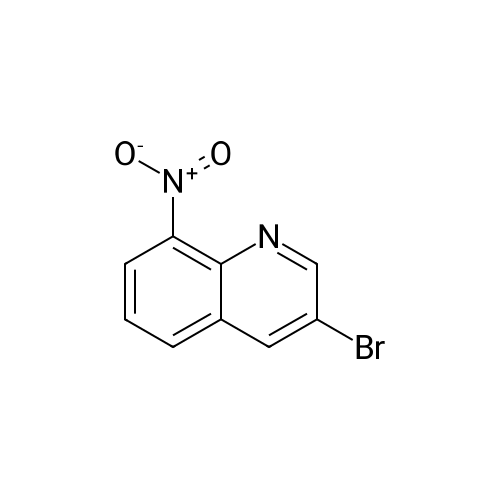
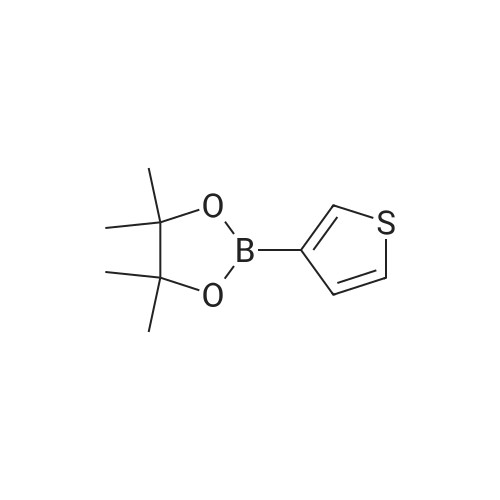
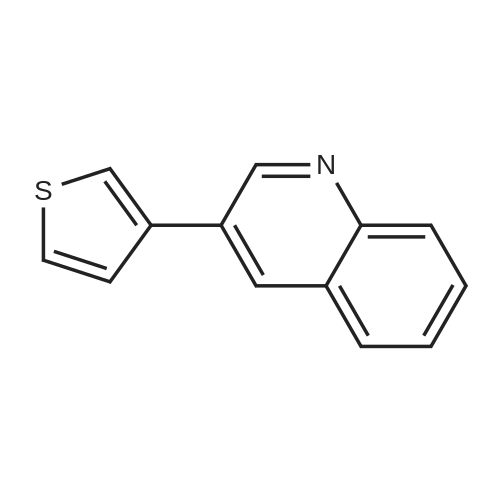
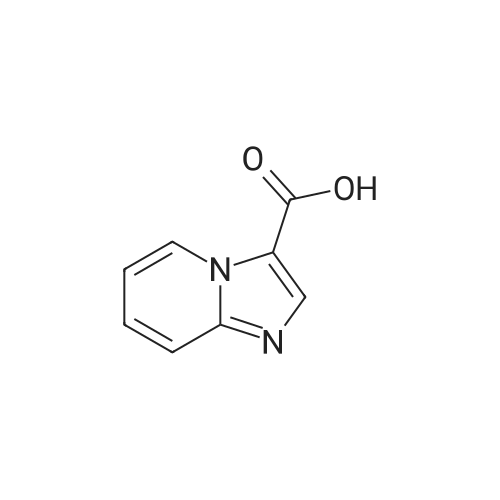
[ 5332-24-1 ]
[ 79476-07-6 ]
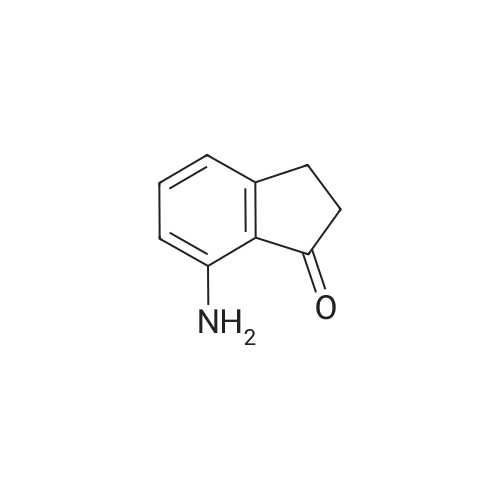
[ 24844-91-5 ]
Reference:
[1] Chemistry - A European Journal, 2010, vol. 16, # 41, p. 12425 - 12433
18
[ 5332-24-1 ]
[ 580-18-7 ]
Yield
Reaction Conditions
Operation in experiment
41%
Stage #1: With n-butyllithium In hexanes; diethyl ether at -78℃; for 1 h; Stage #2: With Trimethyl borate In hexanes; diethyl ether at 0 - 20℃; for 1.16667 - 1.25 h; Stage #3: With peracetic acid In hexanes; diethyl ether at -78 - 20℃; for 20 h;
3-Bromoquinoline (5.0 g, 24.03 mmoL) was dissolved in anhydrous ether and cooled to 780° C. It was then treated with n-butyllithium (10.57 mL, 26.43 mmoL 2.5M in hexanes) by slow addition over 30 min. The reaction was stirred at -78° C. for 30 min then it was treated with trimethylborate (2.50 g, 24.03 mmol) by slow addition over 10-15 min. The reaction was then allowed to stir at 0° C. for 45 min then at room temperature for 15 min. The reaction was then cooled back to -78° C. and treated with peracetic acid (5.49 g, 26.44 mmoL) and stirred at room temperature for 20 hr. The reaction was diluted with H2O (40 mL) and solid sodium bisulfite was added until the peroxides were destroyed as indicated with peroxide test strips. The layers were separated and the aqueous layer extracted with ethyl acetate (2*50 mL). The organic layer was concentrated in vacuo and the residue was azeodried with toluene (4*50 mL). The residue was triturated with toluene and the solids were collected by vacuum filtration to give 1.43 g (41percent) of the title compound. 1H NMR (300 MHz, DMSO-d6) δ ppm 10.29 (s, 1H) 8.58 (d, J=2.57 Hz, 1H) 7.85-7.95 (m, 1H) 7.72-7.83 (m, 1H) 7.40-7.57 (m, 3H). MS m/z (DCI) 146.0 (M+H)+.
Reference:
[1] Journal of the American Chemical Society, 2016, vol. 138, # 41, p. 13493 - 13496
[2] Tetrahedron, 2013, vol. 69, # 31, p. 6409 - 6414
[3] Tetrahedron, 2002, vol. 58, # 6, p. 1125 - 1129
[4] Angewandte Chemie - International Edition, 2014, vol. 53, # 13, p. 3353 - 3357[5] Angew. Chem., 2014, vol. 126, # 13, p. 3421 - 3425,5
[6] Patent: US2007/219258, 2007, A1, . Location in patent: Page/Page column 24
19
[ 5332-24-1 ]
[ 68-12-2 ]
[ 13669-42-6 ]
Yield
Reaction Conditions
Operation in experiment
45%
With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.166667 h;
General procedure: To a solution of 13a (100mg, 0.48mmol) in dry THF (1.5mL) at -78°C nBuLi (2.5M in n-hexane, 300μL, 0.72mmol) was added dropwise. The resulting solution turned to red and DMF (192μL, 2.49mmol) was added. After 10minat -78°C the mixture was quenched with water. The reaction was poured into a saturated aqueous solution of NaHCO3 (10mL) and extracted with EtOAc (3*10mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated. The residue was purified by flash chromatography on silica gel (10percent EtOAc in n-hexane) to afford the title compound as a yellow solid (53percent yield).
Reference:
[1] Tetrahedron Letters, 2003, vol. 44, # 10, p. 2033 - 2035
[2] Tetrahedron, 2003, vol. 59, # 43, p. 8629 - 8640
[3] Tetrahedron, 2002, vol. 58, # 17, p. 3387 - 3400
[4] European Journal of Medicinal Chemistry, 2019, p. 290 - 320
Reference:
[1] Angewandte Chemie - International Edition, 2013, vol. 52, # 33, p. 8611 - 8615[2] Angew. Chem., 2013, vol. 125, # 33, p. 8773 - 8777,5
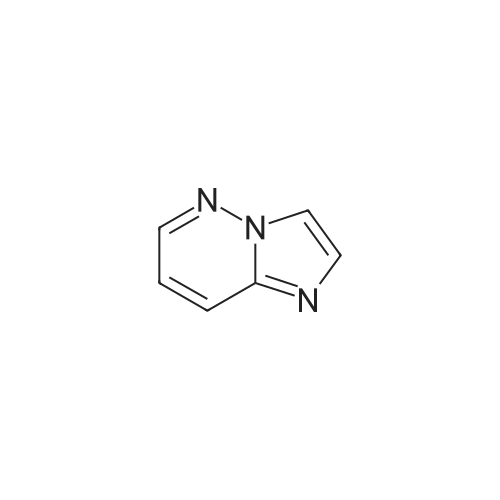
22
[ 5332-24-1 ]
[ 20461-86-3 ]
[ 13669-42-6 ]
Reference:
[1] Angewandte Chemie - International Edition, 2017, vol. 56, # 6, p. 1500 - 1505[2] Angew. Chem., 2017, vol. 129, # 6, p. 1522 - 1527,6
23
[ 5332-24-1 ]
[ 13669-42-6 ]
Reference:
[1] Bulletin of the Polish Academy of Sciences, Chemistry, 1986, vol. 34, # 7-8, p. 281 - 287
[2] RSC Advances, 2015, vol. 5, # 22, p. 17060 - 17063
24
[ 5332-24-1 ]
[ 124-41-4 ]
[ 6931-17-5 ]
Yield
Reaction Conditions
Operation in experiment
94%
With copper(l) iodide In N,N-dimethyl-formamide for 16 h; Reflux
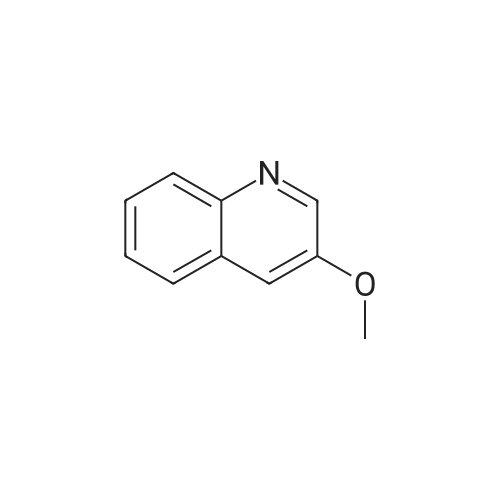
Step 1: To a solution of 3-bromoquinoline (3) (6.5 mL, 48 mmol)in dimethylformamide (80 mL) were added sodium methanolate30percent (16 mL, 54 mmol) and copper iodide (458 mg,2.4 mmol). The mixture was refluxed for 16 h, hydrolyzed andextracted with Et2O. The organic layer was washed with waterand brine, dried over MgSO4 and evaporated under vacuum toafford 3-methoxyquinoline in 94percent yield as a colorless oil; 1HNMR (300 MHz, CDCl3) δ: 8.45 (d, 1H, 2.9 Hz), 7,85 (d, 1H, 8.1 Hzand 1.2 Hz), 7.49 (d, 1H, 8.1 Hz and 1.2 Hz), 7.36-7.25 (m, 2H),7.12 (d, 1H, 2.9 Hz), 3.67 (s, 3H); MS (APCI, pos. 30 V) m/z:[M+H]+, 160.
Reference:
[1] European Journal of Medicinal Chemistry, 2017, vol. 127, p. 621 - 631
[2] Tetrahedron, 2002, vol. 58, # 6, p. 1125 - 1129
25
[ 67-56-1 ]
[ 5332-24-1 ]
[ 6931-17-5 ]
Reference:
[1] European Journal of Organic Chemistry, 2012, # 26, p. 4914 - 4917,4
[2] European Journal of Organic Chemistry, 2012, # 26, p. 4914 - 4917
[3] Chemistry - A European Journal, 2012, vol. 18, # 9, p. 2498 - 2502
Reference:
[1] Journal of the American Chemical Society, 1950, vol. 72, p. 393
28
[ 5332-24-1 ]
[ 101870-60-4 ]
Reference:
[1] Tetrahedron, 2013, vol. 69, # 45, p. 9512 - 9519
[2] European Journal of Organic Chemistry, 2016, vol. 2016, # 8, p. 1606 - 1611
29
[ 5332-24-1 ]
[ 13721-00-1 ]
Yield
Reaction Conditions
Operation in experiment
79%
Stage #1: With 3-chloro-benzenecarboperoxoic acid In dichloromethane at 0 - 20℃; Inert atmosphere Stage #2: With N,N-dimethyl-formamide; phosphorus(V) oxybromide In dichloromethane at 0 - 25℃; Inert atmosphere
To a stirred solution of 2-bromoquinoline (306 mg, 1.47 mmol) in CH2Cl2 (3 ml) at 0 °C was added m-CPBA (358 mg, 85percent max., 1.76 mmol) and the reaction is allowed to stir at room temperature overnight. Extra CH2Cl2 (11.6 ml) was added to the reaction mixture. To the resulting solution at 0 °C was added POBr3 (516 mg, 1.76 mmol) followed by dropwise addition of DMF (57 ul, 0.74 mmol) under argon. The resulting reaction mixture was warmed to 25 °C and stirred for two hours before extra CH2Cl2 (10 ml) was added. Saturated aqueous sodium carbonate solution is added to the reaction mixture slowly to adjust the pH to 7–8. The resulting mixture is separated and the aqueous phase is extracted with CH2Cl2 thoroughly. The organic phase is combined and washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (PE:EA=100:1) to afford the title compound (335 mg, 79percent yield) as a white solid.
Reference:
[1] Journal of the American Chemical Society, 2013, vol. 135, # 8, p. 2891 - 2894
34
[ 5332-24-1 ]
[ 6480-68-8 ]
Reference:
[1] Journal of Organic Chemistry, 1957, vol. 22, p. 565
[2] Journal of the American Chemical Society, 1941, vol. 63, p. 1553,1554
35
[ 5332-24-1 ]
[ 124-38-9 ]
[ 584-08-7 ]
[ 6480-68-8 ]
Reference:
[1] Angewandte Chemie - International Edition, 2017, vol. 56, # 43, p. 13426 - 13430[2] Angew. Chem., 2017, vol. 129, p. 13611 - 13615,5
36
[ 5332-24-1 ]
[ 5341-07-1 ]
[ 116632-33-8 ]
Reference:
[1] Chemistry - A European Journal, 2006, vol. 12, # 35, p. 8935 - 8951
[2] Journal fuer Praktische Chemie (Leipzig), 1889, vol. <2> 39, p. 301
[3] Chemische Berichte, 1905, vol. 38, p. 1279
[4] Journal fuer Praktische Chemie (Leipzig), 1893, vol. <2> 48, p. 158[5] Journal fuer Praktische Chemie (Leipzig), 1894, vol. <2> 50, p. 239
[6] Tetrahedron Asymmetry, 2003, vol. 14, # 11, p. 1517 - 1527
37
[ 5332-24-1 ]
[ 116632-33-8 ]
Reference:
[1] New Journal of Chemistry, 2012, vol. 36, # 3, p. 570 - 574
38
[ 5332-24-1 ]
[ 98555-51-2 ]
Yield
Reaction Conditions
Operation in experiment
48%
Stage #1: With ruthenium(IV) oxide; sodium hypochlorite In tetrachloromethane for 24 h; Stage #2: With hydrogenchloride; water In tetrachloromethane
Example 12 2-Bromo-7-(4-fluoro-benzyl)-5,9-dihydroxy-pyrrolo[3,4-g]quinoline-6,8-dione 1008 Following the literature procedure of M.-D. Le Bas et al. (Synthesis 2001, 16, p. 2495), 100 ml CCl4 was mixed with 250 ml of an aqueous NaOCl solution. To this mixture was added 40 mg of RuO2, followed by 3 g 3-bromoquinoline dissolved in 50 ml CCl4. Additional 30 ml portions of bleach were added at 2, 4, and 6 h. After 24 h, the aqueous layer was collected and acidified to pH 1 with 3N HCl. The aqueous layer was then extracted with ethyl acetate, dried over Na2SO4 and volatiles removed by evaporation to give the 1.7 g product as a yellow resin, (48percent yield). 1H NMR and MS data matched that reported in the literature. The resulting anhydride, 1 g, was then carried through the previously reported multistep sequence to afford the corresponding cyano-ester. Dieckmann condensation between 80 mg (0.3 mmol) of the ester and 80 mg (3.6 mmol) of the imide utilizing 900 uL LiHMDS in 2 ml dry THF gave the crude product. After the typical work-up, approximately 60 mg (30percent) of unpurified product was obtained as a yellow solid which was further refined by trituration with diethyl ether to provide 2 mg highly pure product 1008. 1H NMR (300 MHz, d6-DMSO) δ 9.20 (d, 1H), 9.05 (d, 1H) and 4.85 (s, 2H) ppm, MS=416.1 (M+H).
25%
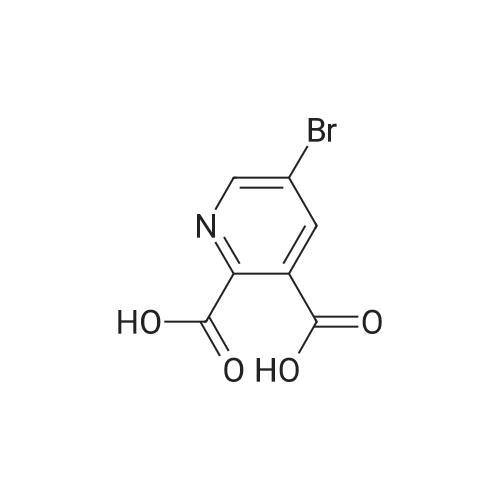
With potassium permanganate In methanol; water at 20 - 80℃; for 1.5 h;
To a mixture of 3-bromoquinoline (10 ml, 72.7 mmol) and water (20OmL) was added KMnO4 (69.0 g, 436 mmol) at 6 portion each 15 min at 80°C with stirring. After allowing the reaction to cool to rt, MeOH (20 mL) was added to the solution. The resulting mixture was washed with toluene (100 mL)5 and the aqueous layer was adjusted to pH 1 with cone. HCl. The mixture was extracted twice with EtOAc/THF (10OmL / 50 mL). The combined extracts were washed with brine, dried over Na2SO4, and the solvent was removed in vacuo. Water was added to the residue, the resulting insoluble materials were filtered off. Isobutyl acetate was added to the filtrate, and then water was removed in vacuo. The resulting precipitates were collected by filtration, and dried to give 4.45 g of compound B-2 (yield = 25 percent) as a colorless crystal. EPO <DP n="48"/>1H NMR (DMSO-(IO) δ 12.5O(1H, s), 8.9O(1H, d, J = 2.1 Hz), 8.43(1H, d, J = 2.1 Hz).
Reference:
[1] Journal of Labelled Compounds and Radiopharmaceuticals, 2001, vol. 44, p. S283 - S285
[2] Synthesis, 2001, # 16, p. 2495 - 2499
[3] Patent: US2008/58315, 2008, A1, . Location in patent: Page/Page column 25-26
[4] Patent: WO2007/19098, 2007, A2, . Location in patent: Page/Page column 46-47
39
[ 5332-24-1 ]
[ 110-19-0 ]
[ 98555-51-2 ]
Yield
Reaction Conditions
Operation in experiment
25%
With potassium permanganate In water
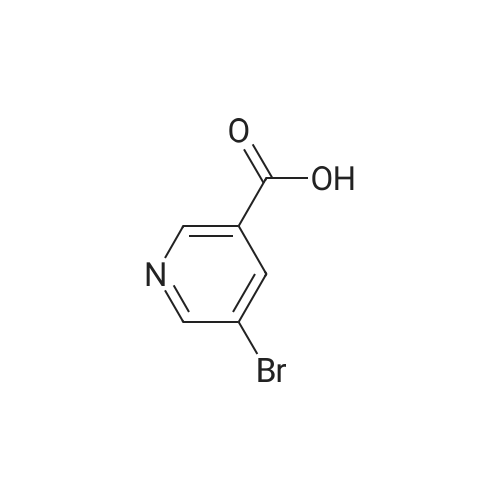
Compound B-2: 5-Bromo-pyridine-2,3-dicarboxylic acid To a mixture of 3-bromoquinoline (10 ml, 72.7 mmol) and water (200 mL) was added KMnO4 (69.0 g, 436 mmol) at 6 portion each 15 min at 80° C. with stirring. After allowing the reaction to cool to rt, MeOH (20 mL) was added to the solution. The resulting mixture was washed with toluene (100 mL), and the aqueous layer was adjusted to pH 1 with conc. HCl. The mixture was extracted twice with EtOAc/THF (100 mL/50 mL). The combined extracts were washed with brine, dried over Na2SO4, and the solvent was removed in vacuo. Water was added to the residue, the resulting insoluble materials were filtered off. Isobutyl acetate was added to the filtrate, and then water was removed in vacuo. The resulting precipitates were collected by filtration, and dried to give 4.45 g of compound B-2 (yield=25percent) as a colorless crystal. 1H NMR (DMSO-d6) δ 12.50 (1H, s), 8.90 (1H, d, J=2.1 Hz), 8.43 (1H, d, J=2.1 Hz).
Reference:
[1] Patent: US2015/225399, 2015, A1,
40
[ 5332-24-1 ]
[ 98555-51-2 ]
[ 5651-01-4 ]
Reference:
[1] Chemische Berichte, 1886, vol. 19, p. 2766
41
[ 5332-24-1 ]
[ 191162-39-7 ]
Yield
Reaction Conditions
Operation in experiment
90%
With n-butyllithium; Triisopropyl borate In tetrahydrofuran; hexane at -78℃; for 1 h; Inert atmosphere; Schlenk technique
General procedure: To a two-neck 250mL round bottom flask, triisopropyl borate (3.30mL, 29.06mmol) and 3-bromoquinoline (3.00g, 14.49mmol) was dissolved in dry THF (100mL), then n-butyllithium (14.50mL of a 2M solution in hexane, 29.00mmol) was added dropwise via a dropping funnel over 1h under N2 at−78°C. After 2h, the acetone/dry ice bath was removed, and the reaction solution was allowed to warm to 0°C. The reaction was then quenched with a 2M HCl solution, and the pH value was adjusted to 7 with a solution of 2M NaHCO3. The resulting solution was extracted with ethyl acetate (EA) (3×100mL). The combined organic layers were dried with MgSO4 and evaporated to dryness. n-Hexane was then added to precipitated the product as a white solid (80percent yield).
Reference:
[1] Dyes and Pigments, 2013, vol. 99, # 1, p. 105 - 115
[2] Synthetic Communications, 2003, vol. 33, # 5, p. 795 - 800
[3] Journal of Organic Chemistry, 2002, vol. 67, # 15, p. 5394 - 5397
[4] Patent: US2009/181941, 2009, A1, . Location in patent: Page/Page column 49-50
[5] Journal of the American Chemical Society, 2012, vol. 134, # 28, p. 11667 - 11673
[6] Organic Letters, 2012, vol. 14, # 18, p. 4814 - 4817,4
42
[ 5332-24-1 ]
[ 5419-55-6 ]
[ 191162-39-7 ]
Yield
Reaction Conditions
Operation in experiment
72%
Stage #1: With n-butyllithium In tetrahydrofuran at -75 - 0℃; for 0.333333 h; Stage #2: With hydrogenchloride; water In tetrahydrofuran
Compound 3-bromo-quinoline (3-bromoquinoline) 10g (48.06mmol) and triisopropylborate (triisopropylborate) 22mL (96.13mmol) dissolved in 200mL of THF and then, at -75 ° C n-BuLi 53mL (96.13mmol) of It was added slowly. After stirring at ° C or less for 20 minutes into the 2N HCl, it was adjusted to pH 7 neoteumyeonseo a 5NNaOH slowly. Ethyl acetate (Ethyl acetate) and was extracted with distilled water,methanol and recrystallization conducted by removing water to give the title compound21-45.95g (72percent).
56%
With n-butyllithium In tetrahydrofuran; hexane at -78 - -20℃;
To a solution of compound 3-bromoquinoline (2.08 g, 10 mmol), triisopropyl borate (2.3g, 12 mmol) in THF (20 mL) was added n-BuLi (4 mL, 2.5 M in hexane) at -780C. The reaction mixture was stirred for 1 h at -780C, then warmed to -200C and quenched with 2 N HCl (50 mL, 100 mmol). The mixture was concentrated and purification by chromatography (MeOH:DCM=l : 10) to give quinolin-3-ylboronic acid (0.97g, 56percent) as light yellow solid. MS (M/Z) M*+H): M"+H=174
Reference:
[1] Patent: KR2016/52399, 2016, A, . Location in patent: Paragraph 0151-0154
[2] Patent: WO2009/155527, 2009, A2, . Location in patent: Page/Page column 134
[3] Chemistry - A European Journal, 2014, vol. 20, # 1, p. 263 - 271
43
[ 5332-24-1 ]
[ 121-43-7 ]
[ 191162-39-7 ]
Yield
Reaction Conditions
Operation in experiment
34%
With sodium hydroxide; n-butyllithium In hexane; di-isopropyl ether; acetic acid; ethyl acetate
PREPARATION 98 Quinolin-3-ylboronic Acid A 2.5M solution of n-butyllithium in hexane (4.4 ml, 11 mmol) was slowly added to a stirred solution of 3-bromoquinoline (2.08 g, 10 mmol) in anhydrous ether (20 ml), under nitrogen, at -75° C. After a further 20 minutes at -75° C., trimethylborate (1.46 ml, 13 mmol) was added, whereupon the red colour changed to yellow. The reaction mixture was allowed to warm to room temperature and quenched with water, followed by 1M aqueous sodium hydroxide solution (10 ml). The resulting mixture was stirred for 30 minutes and then glacial acetic acid added until a pH ~5-6 was attained, which generated a gummy precipitate. Diisopropyl ether was added to this mixture, stirring continued for 1 hour and then the clear aqueous and organic phases were decanted from the solid and discarded. The solid residue was dissolved in ethyl acetate and the solution washed with water, dried (MgSO4) and evaporated under reduced pressure to give the title compound as a pale yellow solid (580 mg, 34percent). Found: C, 62.74; H, 4.11; N, 7.92. C9H8BNO2 requires C, 62.49; H, 4.66; N, 8.10percent. δ(DMSOd6): 7.59 (t,1H), 7.76 (t,1H), 7.98 (m,2H), 8.42 (brs,2H,exchangeable), 8.70 (s,1H), 9.18 (s,1H).
Example 12 2-Bromo-7-(4-fluoro-benzyl)-5,9-dihydroxy-pyrrolo[3,4-g]quinoline-6,8-dione 1008 Following the literature procedure of M.-D. Le Bas et al. (Synthesis 2001, 16, p. 2495), 100 ml CCl4 was mixed with 250 ml of an aqueous NaOCl solution. To this mixture was added 40 mg of RuO2, followed by 3 g 3-bromoquinoline dissolved in 50 ml CCl4. Additional 30 ml portions of bleach were added at 2, 4, and 6 h. After 24 h, the aqueous layer was collected and acidified to pH 1 with 3N HCl. The aqueous layer was then extracted with ethyl acetate, dried over Na2SO4 and volatiles removed by evaporation to give the 1.7 g product as a yellow resin, (48% yield). 1H NMR and MS data matched that reported in the literature. The resulting anhydride, 1 g, was then carried through the previously reported multistep sequence to afford the corresponding cyano-ester. Dieckmann condensation between 80 mg (0.3 mmol) of the ester and 80 mg (3.6 mmol) of the imide utilizing 900 uL LiHMDS in 2 ml dry THF gave the crude product. After the typical work-up, approximately 60 mg (30%) of unpurified product was obtained as a yellow solid which was further refined by trituration with diethyl ether to provide 2 mg highly pure product 1008. 1H NMR (300 MHz, d6-DMSO) delta 9.20 (d, 1H), 9.05 (d, 1H) and 4.85 (s, 2H) ppm, MS=416.1 (M+H).
25%
With potassium permanganate; In methanol; water; at 20 - 80℃; for 1.5h;
To a mixture of 3-bromoquinoline (10 ml, 72.7 mmol) and water (20OmL) was added KMnO4 (69.0 g, 436 mmol) at 6 portion each 15 min at 80C with stirring. After allowing the reaction to cool to rt, MeOH (20 mL) was added to the solution. The resulting mixture was washed with toluene (100 mL)5 and the aqueous layer was adjusted to pH 1 with cone. HCl. The mixture was extracted twice with EtOAc/THF (10OmL / 50 mL). The combined extracts were washed with brine, dried over Na2SO4, and the solvent was removed in vacuo. Water was added to the residue, the resulting insoluble materials were filtered off. Isobutyl acetate was added to the filtrate, and then water was removed in vacuo. The resulting precipitates were collected by filtration, and dried to give 4.45 g of compound B-2 (yield = 25 %) as a colorless crystal. EPO <DP n="48"/>1H NMR (DMSO-(IO) delta 12.5O(1H, s), 8.9O(1H, d, J = 2.1 Hz), 8.43(1H, d, J = 2.1 Hz).
3-Bromoquinoline (5.0 g, 24.03 mmoL) was dissolved in anhydrous ether and cooled to 780 C. It was then treated with n-butyllithium (10.57 mL, 26.43 mmoL 2.5M in hexanes) by slow addition over 30 min. The reaction was stirred at -78 C. for 30 min then it was treated with trimethylborate (2.50 g, 24.03 mmol) by slow addition over 10-15 min. The reaction was then allowed to stir at 0 C. for 45 min then at room temperature for 15 min. The reaction was then cooled back to -78 C. and treated with peracetic acid (5.49 g, 26.44 mmoL) and stirred at room temperature for 20 hr. The reaction was diluted with H2O (40 mL) and solid sodium bisulfite was added until the peroxides were destroyed as indicated with peroxide test strips. The layers were separated and the aqueous layer extracted with ethyl acetate (2*50 mL). The organic layer was concentrated in vacuo and the residue was azeodried with toluene (4*50 mL). The residue was triturated with toluene and the solids were collected by vacuum filtration to give 1.43 g (41%) of the title compound. 1H NMR (300 MHz, DMSO-d6) delta ppm 10.29 (s, 1H) 8.58 (d, J=2.57 Hz, 1H) 7.85-7.95 (m, 1H) 7.72-7.83 (m, 1H) 7.40-7.57 (m, 3H). MS m/z (DCI) 146.0 (M+H)+.
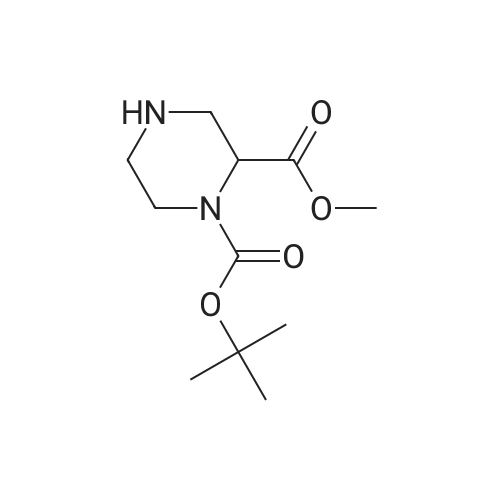
1-tert-butyl 2-methyl 4-quinolin-3-ylpiperazine-1,2-dicarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
To a solution of 3.50 g (14.3 mmol) 1-tert-butyl 2-methyl piperazine-l,2-dicarboxylate in 150 mL anhydrous toluene was added 1.62 mL (11.9 mmol) of 3-bromoquinoline. The resulting solution was purged with nitrogen gas for 15 min, and then purged under vacuum for 5 min. Next, 1.59 g (17.6 mmol) of sodium tert-butoxide was added, and the system was again purged for 2 min under vacuum. To this solution were added 444 mg (0.714 mmol) of (+/-)-2,2'-bis(diphenylphosphino)-l,l'- binaphthalene and 342 mg (0.595 mmol) of bis(dibenzylideneacetone)palladium, and the system was purged one last time for 2 min under vacuum. The mixture was then heated under nitrogen to 95 "C for 3.5 h, taken up in anhydrous diethyl ether, and filtered through a plug of Celite. The filtrate was concentrated in vacuo to yield a red solid, which was purified using a Biotage Horizon system (30% ethyl acetate/ hexanes mixture) to give the title compound as a racemic mixture. Chiral HPLC separation (Chiralcel AD, 60% 2-propanol/heptane) afforded the R enantiomer (first eluting) and the S enantiomer (second eluting), each in ^>9% ee. For the S enantiomer, 1H NMR (CDCl3): delta 8.75 (d, J = 2.1 Hz, IH), 7.99 (d, J = 8.2 Hz, 1 H), 7.68 (dd, J= 6.9, 1.1 Hz, 1 H), 7.53 (ddd, J = 8.7, 7.1, 1.6 Hz, 1 H), 7.48 (td, J = 8.2,1.3 Hz5 1 H) 7.36 (d, J = 2.5 Hz, 1 H), 4.96 (s, 0.55 H) 4.78 (s, 0.45 H), 4.11 (d, J = 7.1, 0.55 H), 4.01 (d, J = 13.0 Hz, 0.45 H), 3.80 (d, J = 8.0 Hz, 3 H), 3.57 (m, 1 H), 3.44 (t, J = 9.4 Hz, 0.55 H), 3.32 (t, J = 9.6 Hz, 0.45 H), 3.04 (m, 1 H), 2.89 (q, J = 8.6 Hz, IH) 1.52 (s, 5 H), 1.48 (s, 4 H). LC/MS 372.3 (M+l).
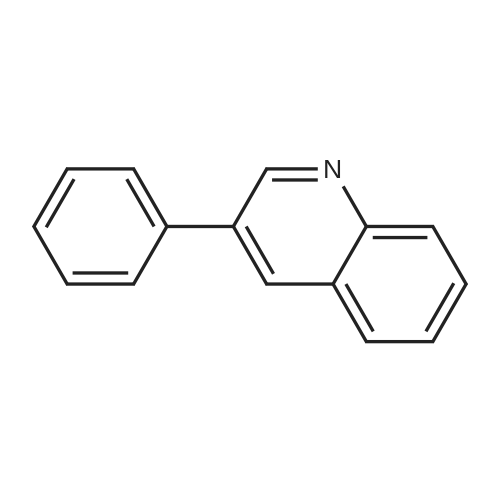
With potassium carbonate;palladium diacetate; In N,N-dimethyl-formamide; at 90℃; for 1.5 - 12h;
To a degassed solution of the aryl bromide (68, 74 or 77, Schemes 8 and 9) in DMF (4.0 niL) was added aryl boronic acid (53, 55, 63 or 71, 1.2 equiv), EPO <DP n="103"/>Pd(OAc>2 (0.05 equiv) and K2CO3 (2 equiv) at room temperature. After degassing and purging with argon (repeated thrice), the reaction mixture was stirred at 9O0C. Reaction times vary from 1.5 hours to 12 hours. The mixture was allowed to cool to room temperature and diluted with H2O (15 mL). The aqueous solution was extracted with ethyl acetate (5 x 15 mL) and the combined organic layer was concentrated under reduced pressure. The residue was purified by flash column chromatography.6-(Quinolin-3-yl)-naphthalen-2-ol (69)[00370] Light yellow solid (0.107 g, 74%). 1H NMR (DMSO): 7.18 (d, IH,J=8.7 ), 7.21 (s, IH), 7.67 (t, IH, J=7.3), 7.78 (t, IH, J=7.5), 7.86-7.95 (m, 3H), 8.07 (s, IH), 8.36 (s, IH), 8.74 (s, IH), 9.39 (s, IH), 9.90 (s, IH); 13C NMR (DMSO): 109.1, 119.8, 125.7, 126.4, 127.5, 127.6, 128.3, 128.5, 128.8, 129.2, 129.8, 130.5, 131.6, 132.8, 133.4, 134.7, 147.1, 150.2, 156.5.
To a stirred solution of 2-bromoquinoline (306 mg, 1.47 mmol) in CH2Cl2 (3 ml) at 0 C was added m-CPBA (358 mg, 85% max., 1.76 mmol) and the reaction is allowed to stir at room temperature overnight. Extra CH2Cl2 (11.6 ml) was added to the reaction mixture. To the resulting solution at 0 C was added POBr3 (516 mg, 1.76 mmol) followed by dropwise addition of DMF (57 ul, 0.74 mmol) under argon. The resulting reaction mixture was warmed to 25 C and stirred for two hours before extra CH2Cl2 (10 ml) was added. Saturated aqueous sodium carbonate solution is added to the reaction mixture slowly to adjust the pH to 7-8. The resulting mixture is separated and the aqueous phase is extracted with CH2Cl2 thoroughly. The organic phase is combined and washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (PE:EA=100:1) to afford the title compound (335 mg, 79% yield) as a white solid.
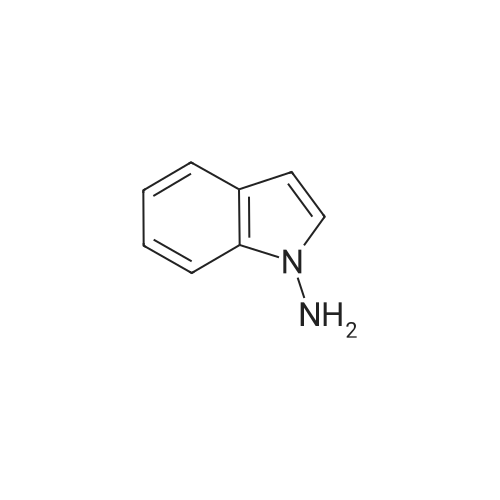
With tris-(dibenzylideneacetone)dipalladium(0); (R)-(-)-1-[(SP)-2-(dicyclohexylphosphino)ferrocenyl]ethyldi-tert-butylphosphine; potassium tert-butylate; lithium chloride; In toluene; at 130℃; for 3h;Inert atmosphere;
General procedure: A flame-dried resealable Schlenk tube was charged with Pd2(dba)3 (0.025 mmol, 2.5 mol %), Josiphos (0.05 mmol, 5 mol %), the solid reactant(s) (1.0 mmol of the <strong>[53406-38-5]1-aminoindole</strong>, 2.0 mmol of the aryl halide), LiCl (2.0 mmol) and KOtBu (1.4 mmol). The Schlenk tube was capped with a rubber septum, evacuated, and backfilled with argon; this evacuation/backfill sequence was repeated one additional time. The liquid reactant(s) and toluene (2 mL/mmol) were added through the septum. The septum was replaced with a teflon screwcap. The Schlenk tube was sealed, and the mixture was stirred at 130 C for 3 h. The resulting suspension was cooled to room temperature and filtered through a pad of celite eluting with ethyl acetate, and the inorganic salts were removed. The filtrate was concentrated and purification of the residue by silica gel column chromatography gave the desired product. All the compounds gave satisfactory spectroscopic data. Data for the selected compounds are given below:Compound 3a: Yield: 94%; TLC : Rf 0.39 (c-hexane/AcOEt 8:2). IR (neat): 3306, 1604, 1508, 1446, 1359, 1246, 1220, 1112, 1063, 832, 754 cm-1; 1H NMR (CDCl3, 300 MHz) : delta 7.55 (dd, 1H, J = 6.5, 2.3 Hz), 7.25-6.95 (m, 4H), 6.65 (d, 2H, J = 8.9 Hz), 6.42 (d, 1H, J = 3.3 Hz), 6.37 (d, 2H, J = 8.9 Hz), 6.32 (s, 1H), 3.62 (s, 3H).13C NMR (75 MHz, CDCl3) delta 154.5, 141.0, 135.9, 128.7, 126.6, 122.3, 121.1, 120.3, 114.7 (2C), 114.2 (2C), 109.5, 100.6, 55.6. m/z MS (ES+) 239.0 (M+H+).Compound 3i: Yield: 63%; TLC : Rf 0.40 (c-hexane/AcOEt 8:2). IR (neat): 3326, 1521, 1466, 1236, 1220, 1125, 1103, 832, 754 cm-1; 1H NMR (CDCl3, 300 MHz) : delta 7.79-7.61 (m, 1H), 7.33-7.10 (m, 4H), 6.99-6.83 (m, 2H), 6.55 (dd, 1H, J = 3.3, 0.7 Hz), 6.54, (br s, 1H), 6.46 (dd, 2H, J = 9.0, 4.4 Hz). 13C NMR (75 MHz, CDCl3) delta 159.4 (d, 1C, JC-F = 237.0 Hz), 143.4, 135.6, 128.5, 126.7, 122.5, 121.2, 120.4, 116.0 (d, 2C, JC-F = 23.5 Hz), 114.0 (d, 2C, JC-F = 8.3 Hz), 109.3, 100.9. m/z MS (ES+) 227.0 (M+H+).
With potassium carbonate;copper(l) iodide; In 1-methyl-pyrrolidin-2-one; at 200℃; for 2h;
Example 11 Synthesis of 7-(quinolin-3-ylamino)-indan-1-one 0.43 g of 3-bromoquinoline, 0.29 g of <strong>[628732-03-6]7-aminoindan-1-one</strong>, 0.08 g of copper(I) iodide, 0.41 g of potassium carbonate, and 4 ml of N-methylpyrrolidone were mixed, and then the mixture was stirred for 2 hours at 200 C. The resultant mixture was purified by silica gel column chromatography to obtain 0.15 g of 7-(quinolin-3-ylamino)-indan-1-one (Compound Number 50).
With C37H45ClN2O3PPd; potassium carbonate; In ethanol; water; at 80℃; for 6h;Inert atmosphere; Schlenk technique;
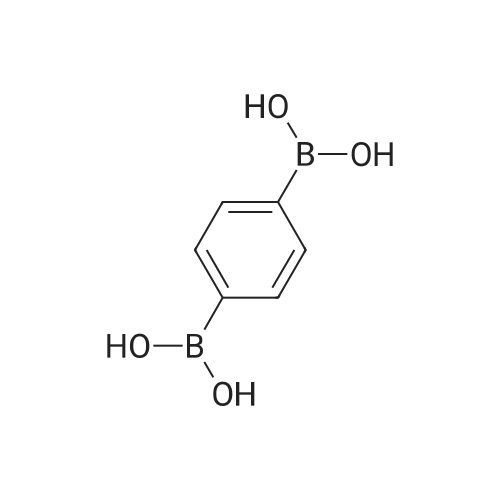
A 10-mL round-bottom flask was charged with the prescribe damount of catalyst, 1,4-benzenediboronic acid (0.5 mmol), N-heteroaryl halides (1.5 mmol), the selected base (1.5 mmol) and solvent (4 mL). The flask was placed in an oil bath and heated at 80 °C for 6 h, then cooled to room temperature and extracted with CH2Cl2. The crude products obtained from evaporation were purified by flash chromatography on silica gel. The products 5b?c, 5f, 5m [21], 5d [22], 5e [23], 5l [24] were known compounds and characterized by the comparison of data with those in the literature. The products 5a, 5g?k, 5n?o were new compounds and characterized by elemental analysis, IR, MS,1H and 13C NMR.
52%
With palladium; potassium carbonate; In methanol; acetonitrile; at 20℃; for 4h;
General procedure: To a freshly prepared solution of PdNPs (10 mL, 0.02 mmol), required amount of K2CO3 (2 mmol) was added followed by aryldihalides/ arylhalide (1 mmol) and arylboronic acid (3 mmol)/diboronic acid (0.75 mmol). Then, the reaction mixture was stirred at room temperature in open atmosphere. The reaction was monitored by TLC and was stopped after the complete consumption of starting material. The desired product got precipitated out which was separated by filtration and extracted with chloroform. The chloroform layer was evaporated to get the terphenyl in pure state.
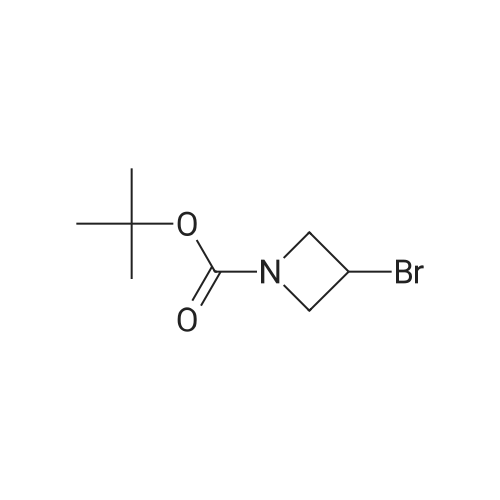
With 1,1'-bis-(diphenylphosphino)ferrocene; palladium diacetate; triethylamine; In acetonitrile; at 80℃; for 23h;Sealed tube;
General procedure: An aryl bromide (1 mmol), Ph P (94 mg, 0.36 mmol), and Pd(OAc)2 (22 mg, 0.1 mmol) were weighed into screw cap vial equipped with a magnetic stir bar. Dry MeCN (5 mL) was added, followed by Et N (2.5 mL) and the corresponding amine (1.5 mmol). Neat iron pentacarbonyl (35 muL, 0.25 mmol) was added, and the vial was thoroughly closed and heated in the preheated oil bath or aluminum heating block (80 C). The reaction mixture was stirred at the specified temperature for 23 h, then concentrated with approximately 1 g of silica, and the absorbed material was purified by gradient MPLC (hexanes-EtOAc in ratios 1:10 to 1:1). Methyl and n-butyl carboxylates, described in the article, were prepared in a similar manner; a mixture of MeCN with MeOH or n-BuOH in a ratio of 2:1 was used instead of pure MeCN, the identical amount of dppf (0.36 mmol) was used instead of Ph3P in specified cases.
Compound B-2: 5-Bromo-pyridine-2,3-dicarboxylic acid To a mixture of 3-bromoquinoline (10 ml, 72.7 mmol) and water (200 mL) was added KMnO4 (69.0 g, 436 mmol) at 6 portion each 15 min at 80 C. with stirring. After allowing the reaction to cool to rt, MeOH (20 mL) was added to the solution. The resulting mixture was washed with toluene (100 mL), and the aqueous layer was adjusted to pH 1 with conc. HCl. The mixture was extracted twice with EtOAc/THF (100 mL/50 mL). The combined extracts were washed with brine, dried over Na2SO4, and the solvent was removed in vacuo. Water was added to the residue, the resulting insoluble materials were filtered off. Isobutyl acetate was added to the filtrate, and then water was removed in vacuo. The resulting precipitates were collected by filtration, and dried to give 4.45 g of compound B-2 (yield=25%) as a colorless crystal. 1H NMR (DMSO-d6) delta 12.50 (1H, s), 8.90 (1H, d, J=2.1 Hz), 8.43 (1H, d, J=2.1 Hz).
General procedure: Under a nitrogen atmosphere 0.075 g (0.2 mmol) of2,5-dibromo-3,4-dinitrothiophene were dissolved in 8 mLof anhydrous toluene and treated with 10 mol% of the catalyst(catalysts I-IV). The mixture was then subjected toultrasound irradiation for 5 min and then stirred at roomtemperature for additional 25 min. Then, 0.5 mmol of theboronic acid, 1.6 mmol of potassium phosphate, and 2 mLof water were added, and the mixture was heated underreflux at 100C for 48 h. After cooling to room temperature,the mixture was dried over magnesium sulfate andchromatographed (petroleum ether/dichloromethane 1:3).
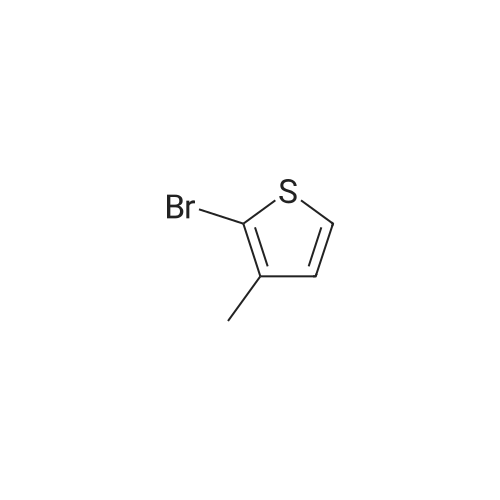
3-(5-bromo-4-methylthiophen-2-yl)quinoline[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
63%
With potassium acetate; palladium diacetate; In N,N-dimethyl acetamide; at 80℃; for 10h;Inert atmosphere; Schlenk technique; Green chemistry;
General procedure: In a similar manner as described in [1], as a typical experiment, the 2-bromothiophene derivative (2 mmol), aryl bromide derivative (1 mmol), KOAc (0.196 g, 2 mmol) and Pd(OAc)2 (2.2 mg, 0.01 mmol) were dissolved in DMA (5 mL) under an argon atmosphere. The reaction mixture was stirred at 80 C for 2 h. After evaporation of the solvent, the product was purified by silica gel column chromatography.
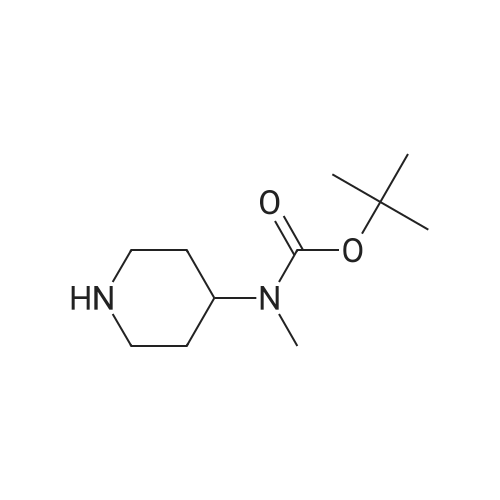
1-(3'-quinolyl)-4-(N-Boc-N-methyl)aminopiperidine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
With potassium tert-butylate; palladium diacetate; DavePhos; In toluene; at 120℃; for 12h;Inert atmosphere;
General procedure: An oven-dried reaction flask, equipped with a reflux condenser was charged with heteroaryl chloride (1.0 mmol), 4-(N-Boc-N-methyl)aminopiperidine (1.3 equiv.), Pd(OAc)2 (3 mol%), DavePhos (3 mol%), t-BuONa (1.2 equiv.). The flask was sealed, and was evacuated and backfilled with argon for three times. Then 3 mL toluene was added to the system with a syringe. The reaction mixture was stirred at 120 C for 12 h. After cooling to the room temperature, the resulting residue was filtered through a plug of silica gel and washed with ethyl acetate. The mixture was then poured into water and extracted. The combined organic layers were washed with brine, dried over MgSO4, and filtered. The solvent was removed under vacuum. The residue was purified by flash column chromatography to afford the desired product.
With potassium phosphate; tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; at 90 - 100℃; for 8h;Inert atmosphere;
General procedure: Compounds 3a-h were prepared according to the followinggeneral procedure [2,3]: To a 3-bromoquinoline (100 mg,0.48 mmol) suspension in dioxane (3.0 mL), placed in ascrew capped reaction tube, was added Pd(PPh3)4 (1.5 mol%, 2 mmol), aryl boronic acid (1.2 mmol), and K3PO4 (153mg, 1.5 mmole), and the mixture was flushed with dry nitrogengas for few minutes. The reaction mixture was thenheated at 90-100 C for 8 h. After completion of reaction, 20mL of water was added and the mixture was cooled to roomtemperature. The organic and aqueous layers were separatedand the latter was extracted three times with ethyl acetate (3 15 mL). Solvent was removed under reduced pressure by meansof rotary evaporation and the residue obtained was purifiedthrough column chromatography. Using the aforementionedprocedure, the following compounds were prepared:
4-(5-(quinolin-3-yl)pyridin-2-yl)morpholine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
88%
With (DPEPhos)Ni(mesityl)Br; potassium tert-butylate; In 1,4-dioxane; ethanol; at 60℃; for 16h;Inert atmosphere; Sealed tube;
Unless otherwise specified, under an inert atmosphere C1 (31.9mg, 0.04 mmol, 10 mol %), aryl halide (0.4 mmol), boronic acid(1.2 mmol), and KOtBu (157.1 mg, 1.4 mmol), were added to anoven-dried 4 dram vial containing a magnetic stir bar. 1,4-Dioxane (4 mL) and EtOH (101.7 muL) were added. The vial wassealed with a screwcap featuring a PTFE/silicone septum andwas removed from the glovebox. The reaction mixture wasmagnetically stirred for 16 h in a temperature-controlled aluminumheating block set to 60 C. After 16 h, the reactionmixture was cooled to room temperature, taken up in EtOAc (ca.10 mL), and extracted with distilled water (3 × 10 mL). Theorganic layer was dried over anhydrous Na2SO4, filtered, andconcentrated with the aid of a rotary evaporator.
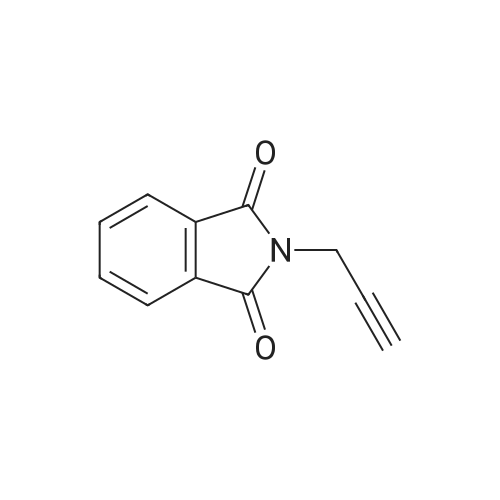
2-[3-(quinolin-3-yl)prop-2-yn-1-yl]isoindolin-1,3-dione[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
62.2%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In acetonitrile; at 80℃; for 4h;Inert atmosphere;
General procedure: To a solution of compound 2 or 3 (1.2 eq) in acetonitrile in a high pressure vessel, CuI (0.1 eq), bis(triphenylphosphine)palladium(II) dichloride (0.05 eq), the corresponding halogenated aryl (1 eq) and triethylamine (1.5 eq) were added. The reaction mixture was stirred at 80C for 4h under Ar. Water was added and the resulting mixture was extracted with EtOAc. The organic layer was washed with water and brine, and concentrated by evaporation. The residue was dissolved with EtOAc and treated with petroleum ether. The product was precipitated and filtered to yield the compounds 6a, 7a, 7b and 7d-7i.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping