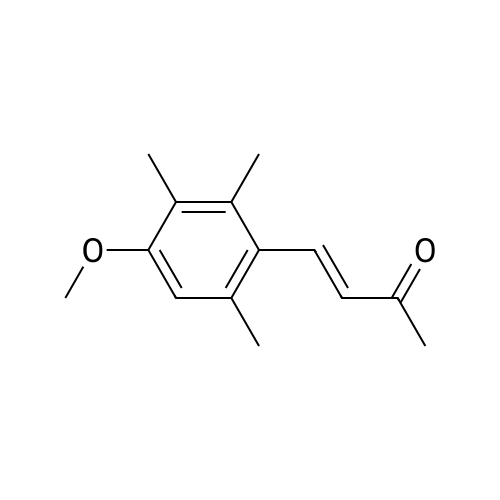
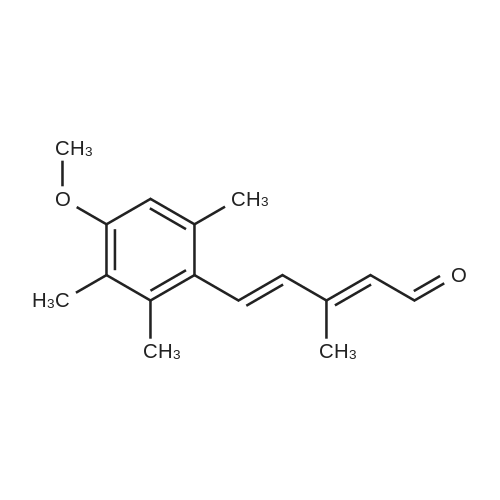
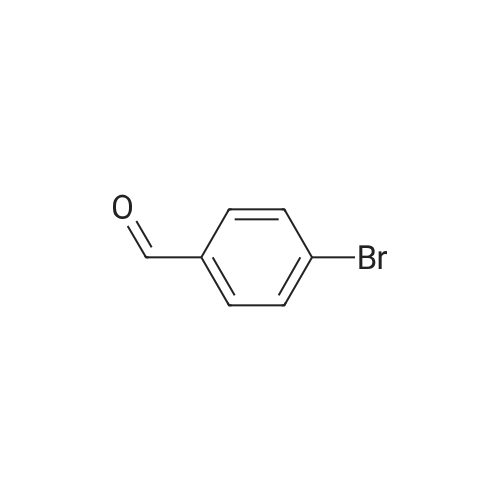
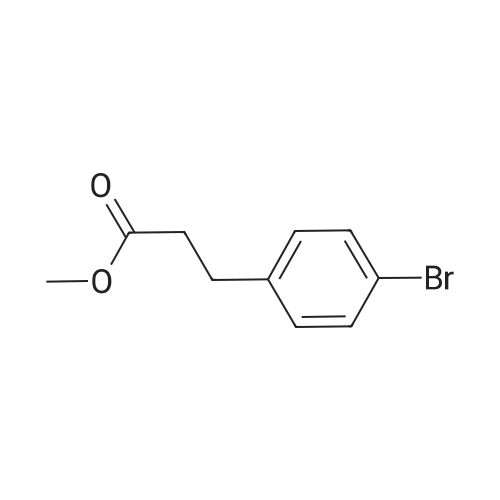
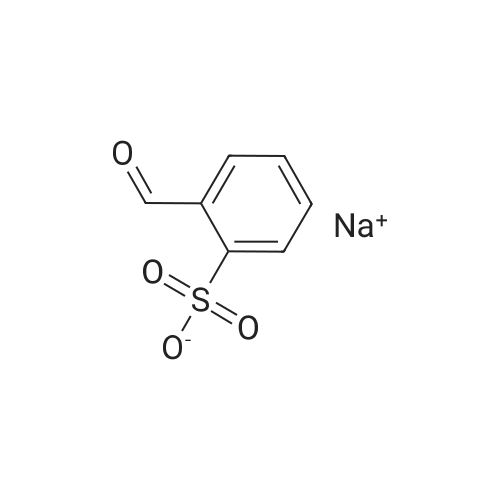
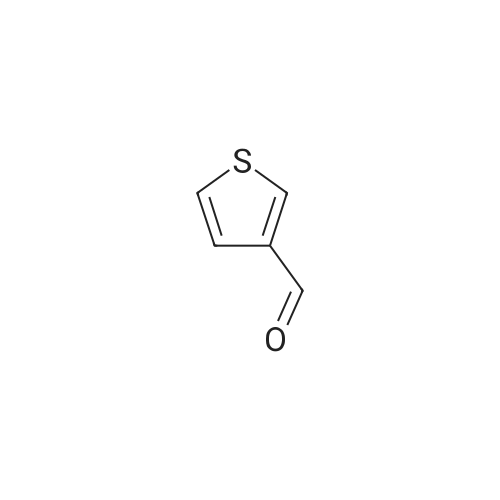
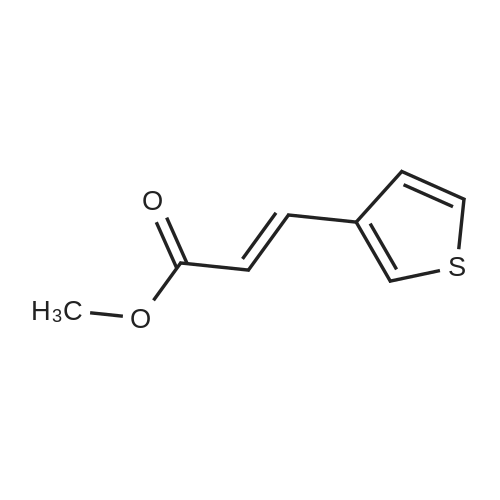
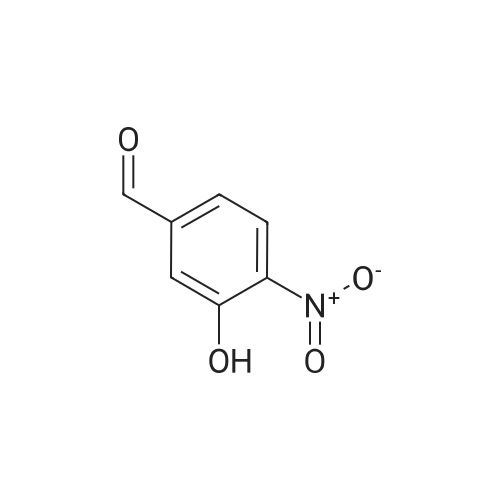
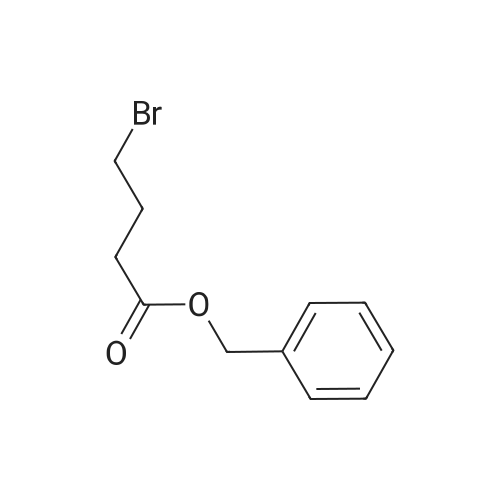
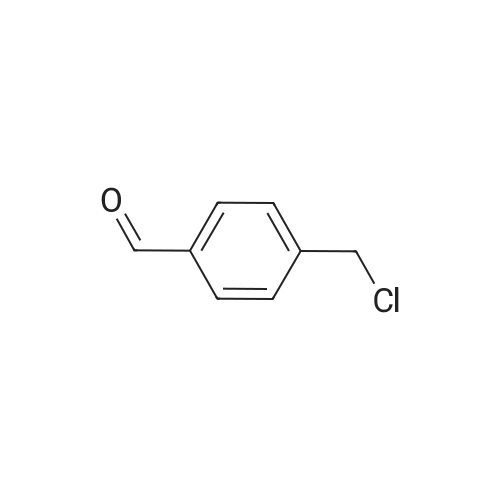
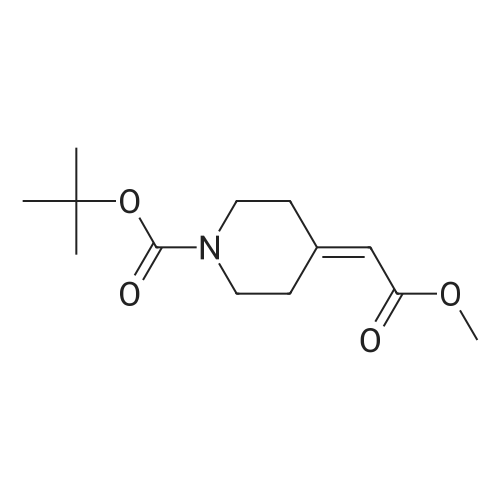
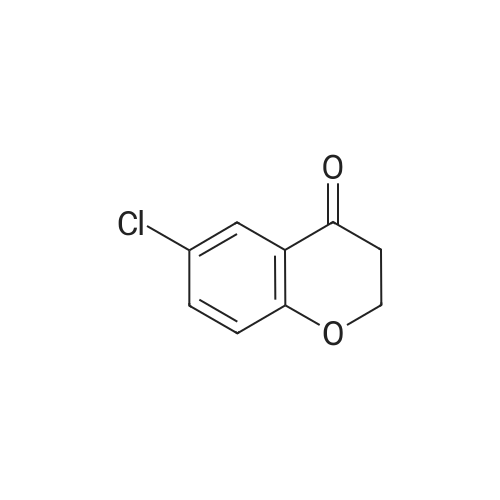
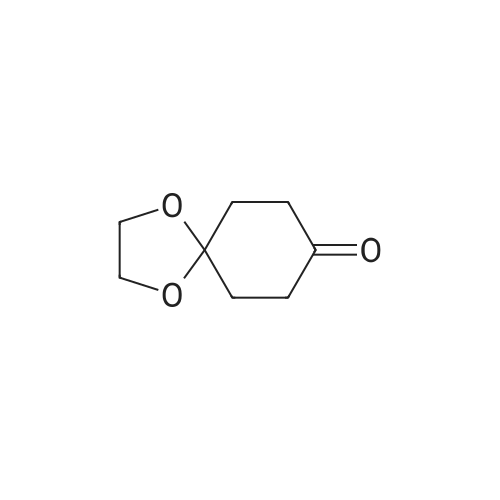
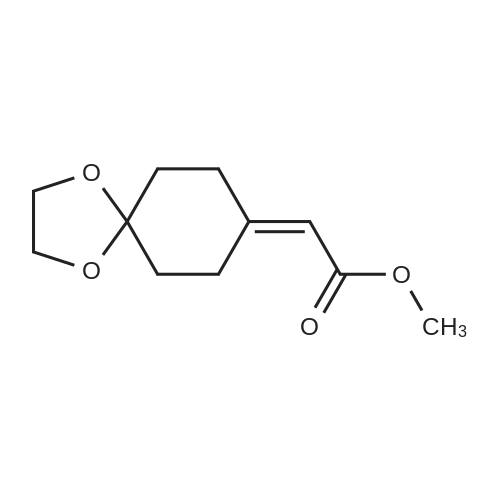
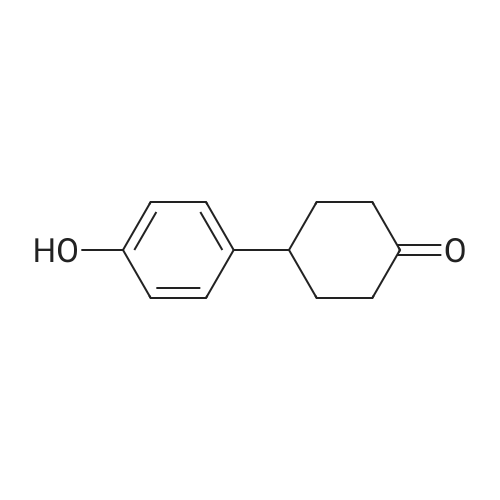
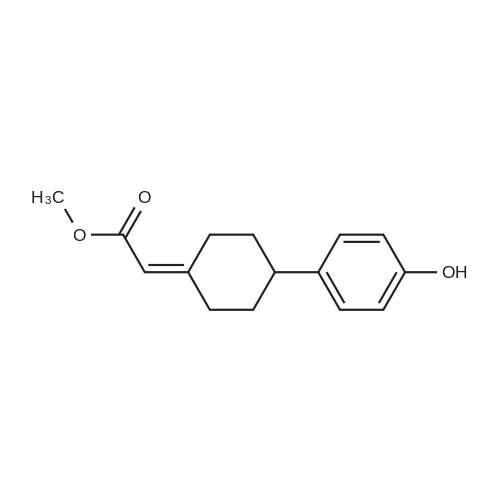
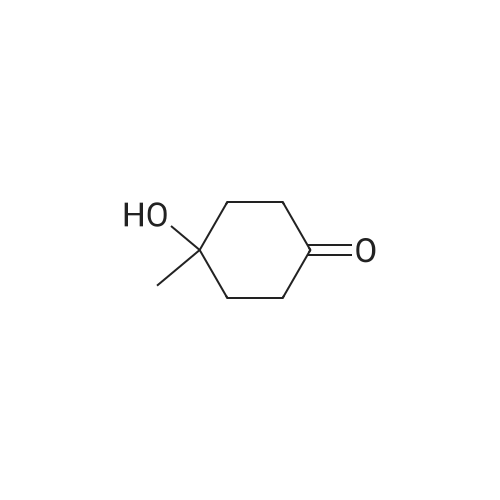
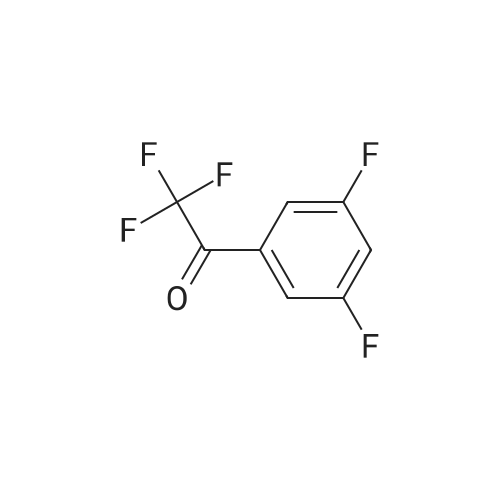
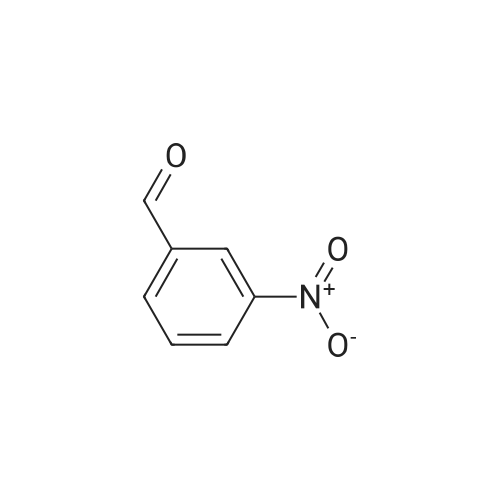
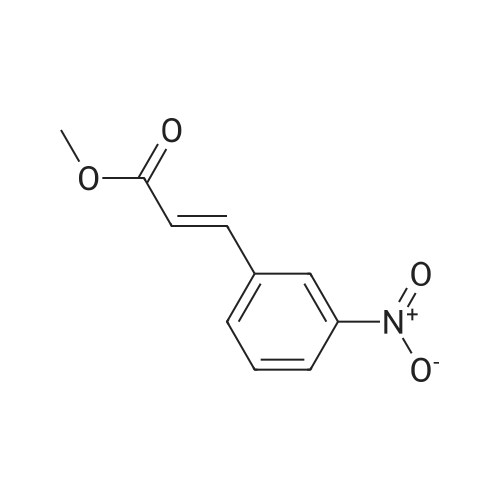
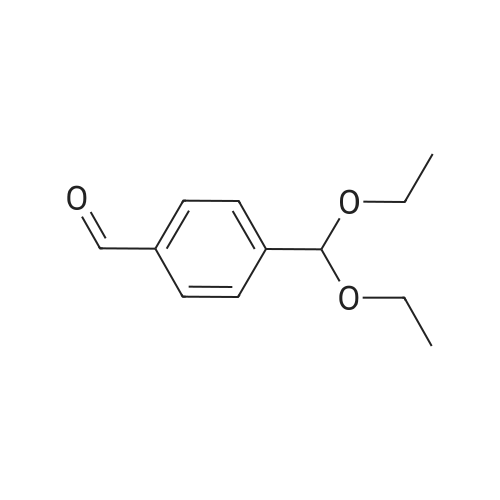
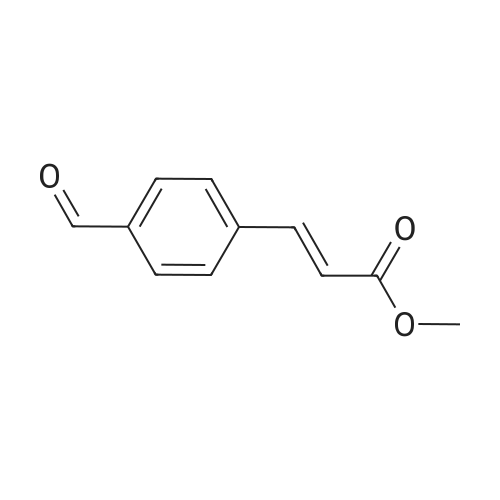
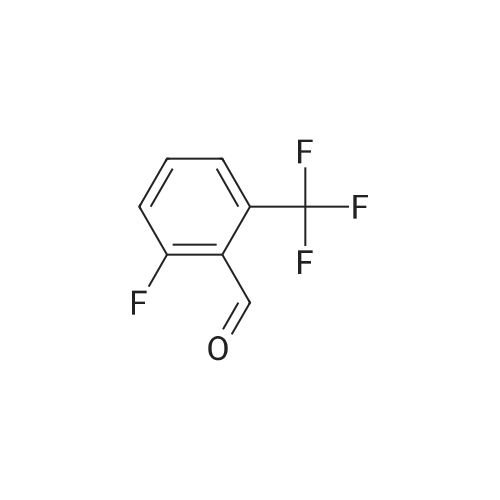
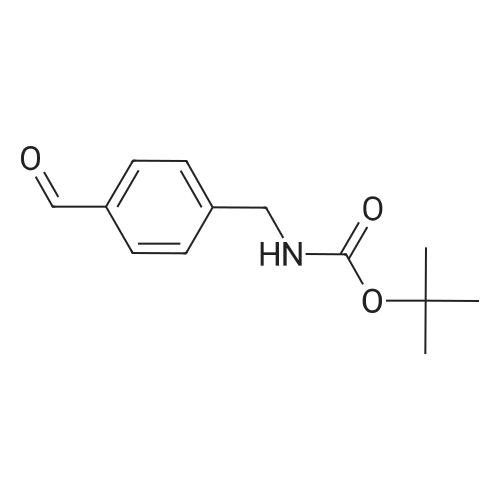
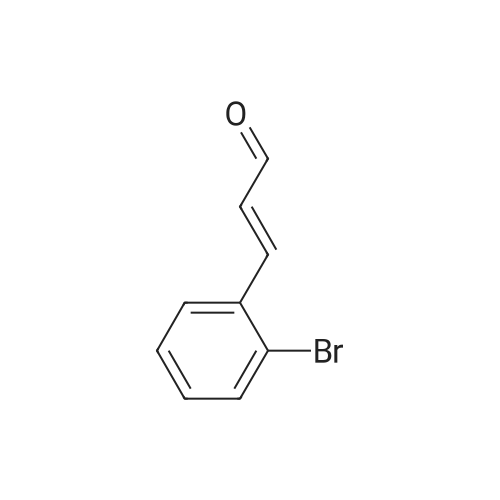
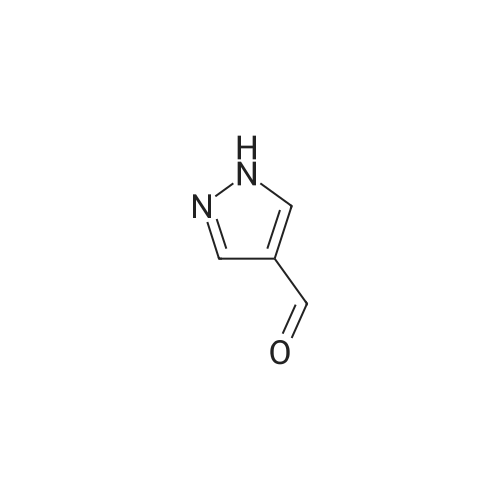
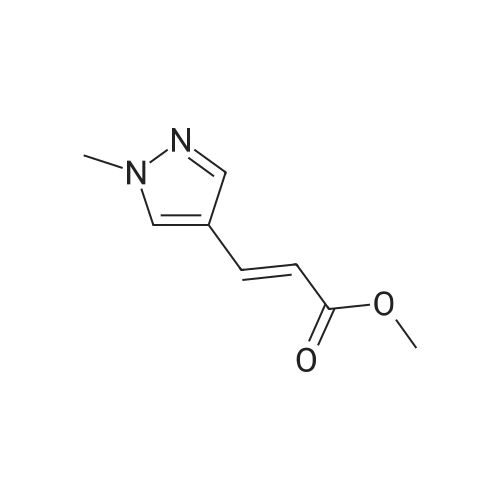
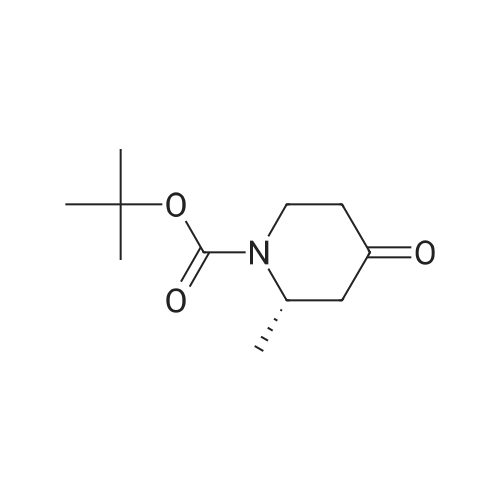
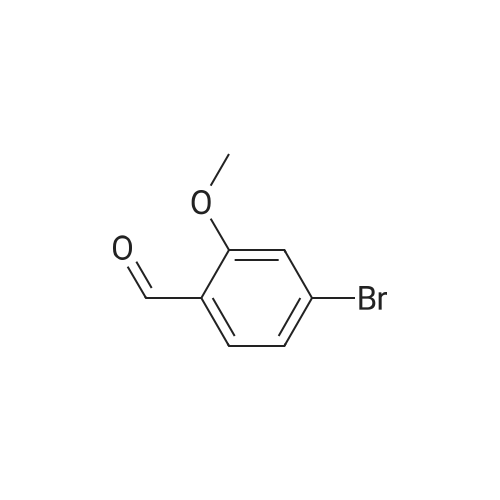
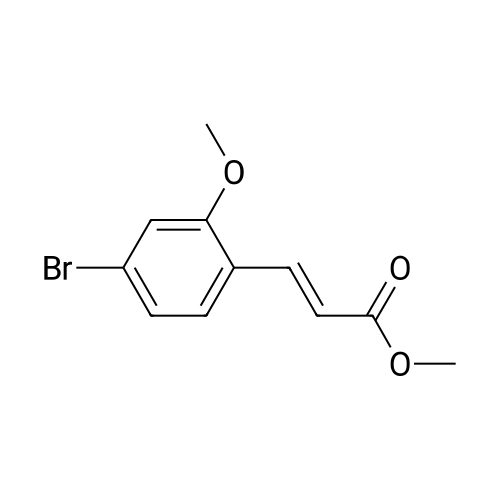
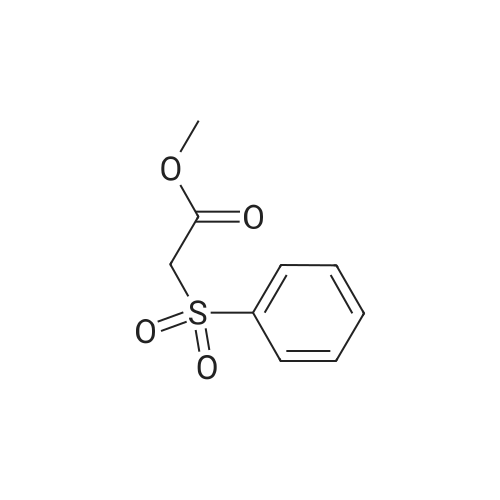
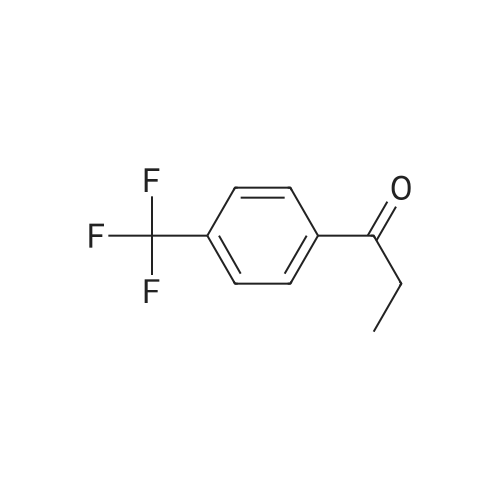
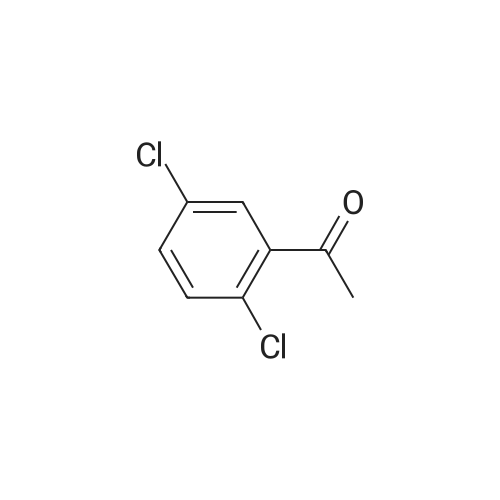
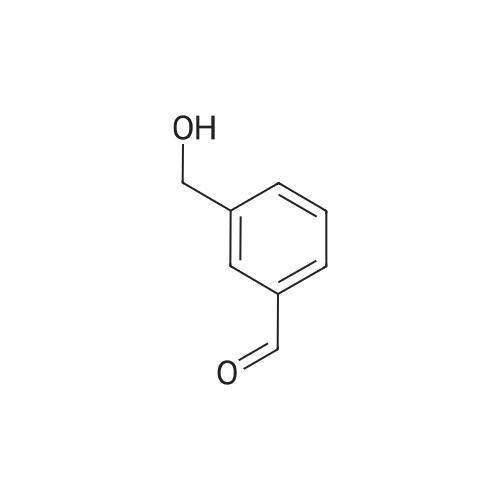
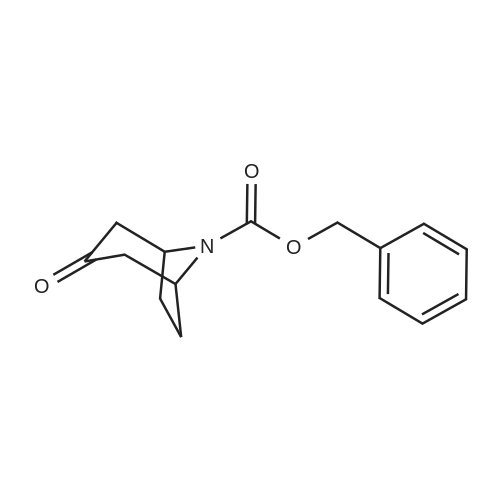
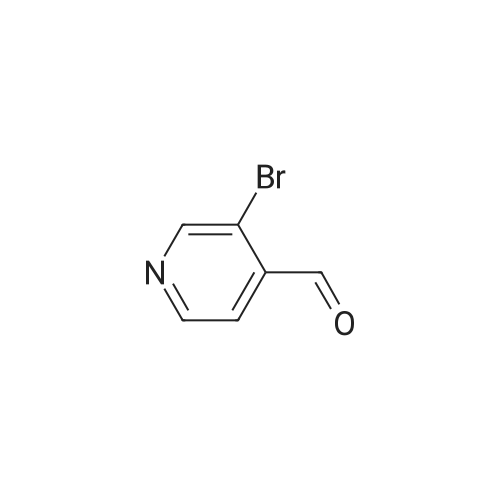
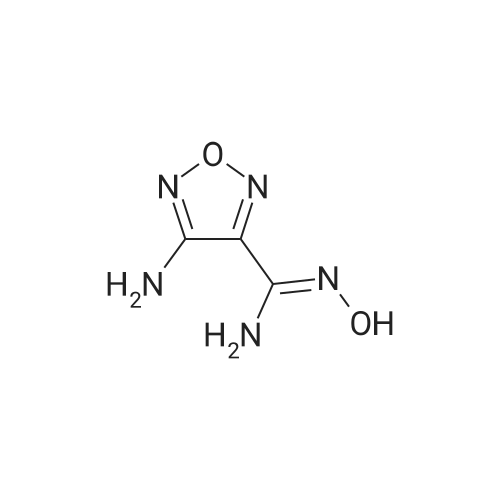
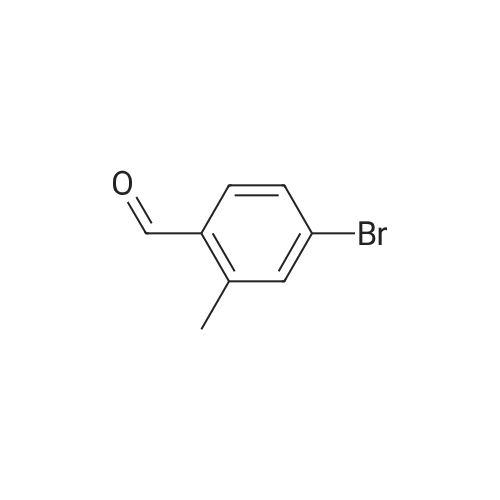
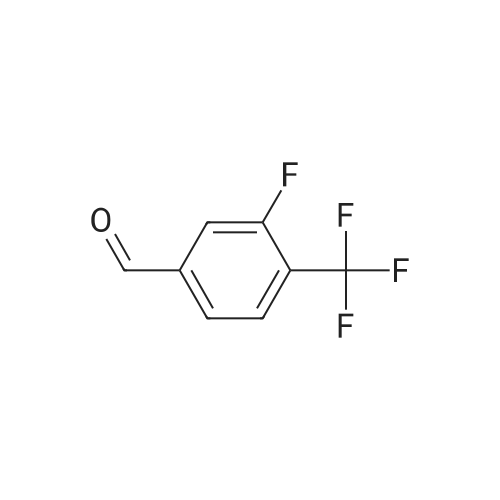
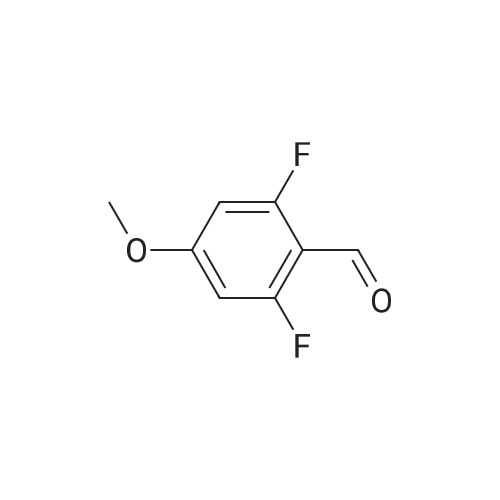
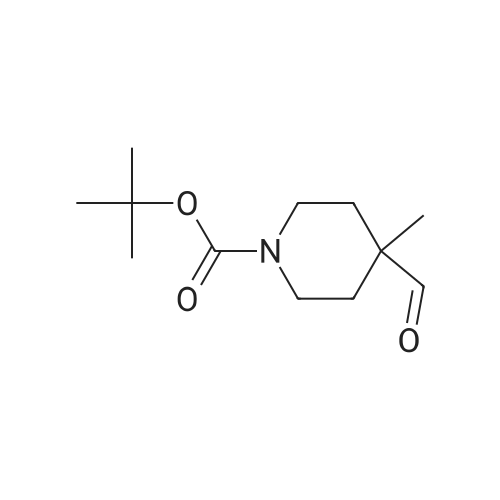
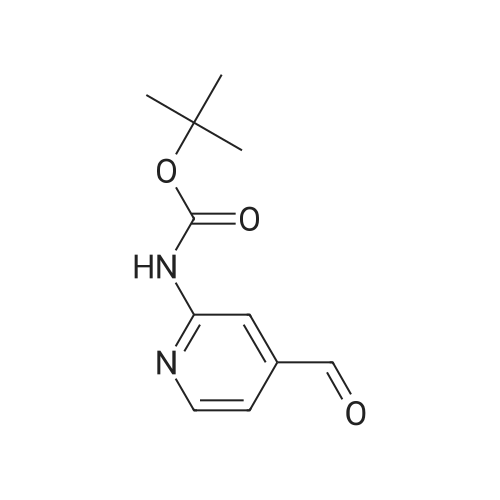
| 82% |
|
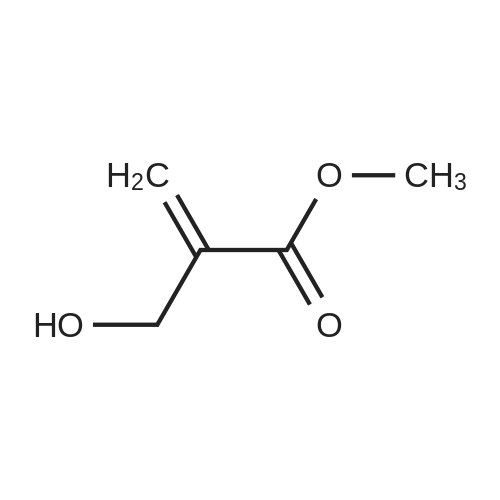
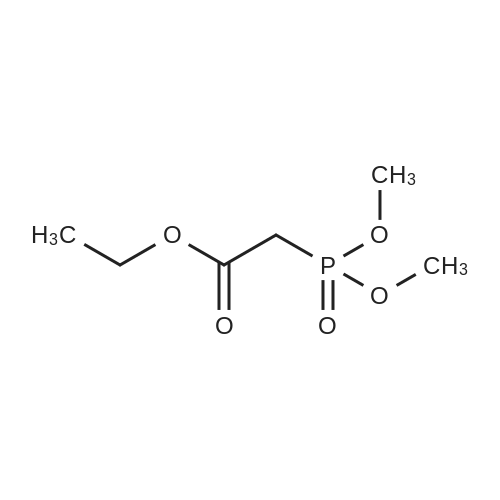
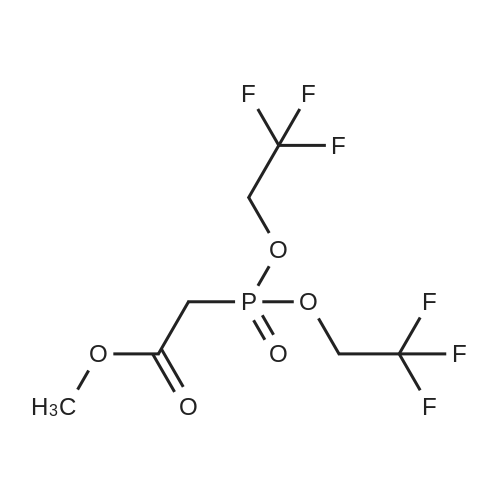
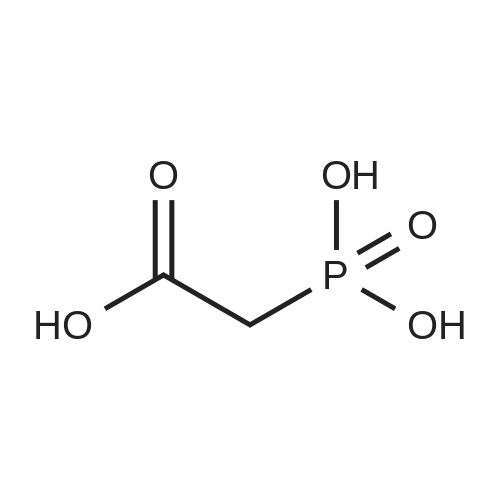
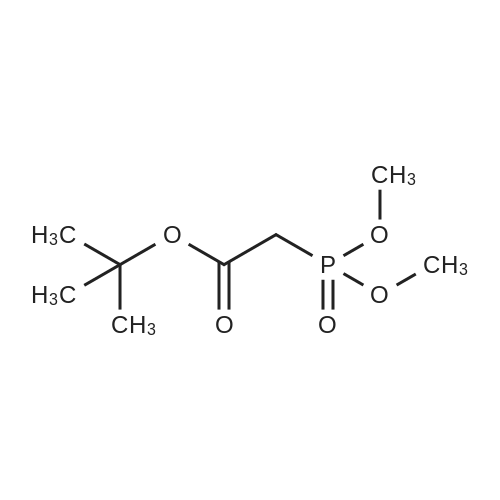
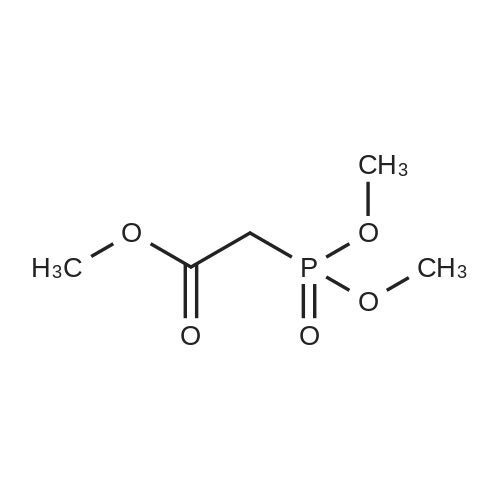
[0103] Dimethyl N-Acetoxymethyl-N-(N-fluorenylmethyloxycarbonyl-L-leucyl)-N-acetoxymethyl-4,5-dehydro-L-homoglutamate(60). A suspension of sodium hydride (60 % in mineral oil, 6 mg, 0.15 mmol) in dry THF (1.5 mL) was cooledto -10 C and a solution of methyl 2-(dimethoxyphosphoryl)acetate (27 mg, 0.15 mmol) in dry THF (1 mL) was addeddropwise. The mixture was stirred for 45 minutes, and then a solution of the aldehyde (41) (54 mg, 0.10 mmol) in dryTHF (1 mL) was slowly added, while the temperature was mantained at -10 C for 35 minutes. The reaction mixture waspoured into water and extracted with diethyl ether. The organic layer was dried, filtered and evaporated as usual, andthe residue was purified by rotatory chromatography (hexane/EtOAc, 80:20), affording the olefin (60) (49 mg, 82%) asa yellowish slurry: [alpha]D = -22.30 (c 0.38, CHCl3); IR (CHCl3) vmax. 3431, 1745, 1721, 1674, 1511 cm-1; 1H NMR (500MHz, CDCl3, 26 C) deltaH 0.94 (3H, d, J = 6.7 Hz, Me), 0.98 (3H, d, J = 6.6 Hz, Me), 1.42-1.56 (2H, m, 3?-H2), 1.70 (1 H,m, 4?-H), 2.10 (3H, s, Ac), 2.92 (2H, dd, J = 7.3, 7.4 Hz, 3-H2), 3.67 (3H, s, OMe), 3.68 (3H, s, OMe), 4.21 (1 H, dd, J =7.2, 7.2 Hz, CHFmoc), 4.34 (1 H, dd, J = 7.0, 10.6 Hz, OCHa-fluorenyl), 4.40 (1 H, dd, J = 7.4, 10.6 Hz, OCHb-fluorenyl),4.55 (1 H, dd, J = 7.0, 8.3 Hz, 2-H), 4.83 (1 H, ddd, J = 4.2, 9.1, 9.4 Hz, 2?-H), 5.34 (1 H, d, J = 9.1 Hz, NH), 5.43 (1 H, d, J = 12.4 Hz, NCHaO), 5.55 (1 H, d, J = 12.4 Hz, NCHbO), 5.85 (1 H, d, J = 15.6 Hz, 5-H), 6.80 (1 H, ddd, J = 7.6, 8.1,15.6 Hz, 4-H), 7.30 (2H, dd, J = 7.5, 7.5 Hz, Ar), 7.39 (2H, dd, J = 7.4, 7.5 Hz, Ar), 7.58 (1 H, d, J = 7.5 Hz, Ar), 7.75 (1H, d, J = 7.6 Hz, Ar); 13C NMR (125.7 MHz, CDCl3, 26 C) deltaC 20.6 (CH3), 21.6 (CH3), 23.2 (CH3), 24.7 (CH), 31.6 (CH2),42.9 (CH2), 47.2 (CH), 49.9 (CH), 51.4 (CH3), 52.6 (CH3), 59.6 (CH), 67.1 (CH2), 71.7 (CH2), 120.0 (2 x CH), 124.0(CH), 125.1 (2 x CH), 127.0 (2 x CH), 127.7 (2 x CH), 141.3 (2 x C), 143.8 (CH), 143.9 (2 x C), 156.5 (C), 166.3 (C),169.9 (C), 170.5 (C), 174.9 (C); MS (EI) m/z (relative intensity) 594 (M+, <1), 339 (M+ - fluorenylmethyl -OCO - HOMe,<1), 308 ([Fmoc-NH-CHCH2CHMe2]+, 3), 279 ([Fmoc-NH-CHCO]+, 3), 200 ([CH2=NH-CH(CO2Me)CH2CH=CHCO2Me+ H]+, 6), 179 ([fluorenyl-CH2]+, 60), 178 ([fluorenyl-CH2 - H]+, 100), 165 ([fluorenyl]+, 25); HRMS calcd for C32H38N2O9,594.2577, found 594.2573; calculated for C16H23N2O6, 339.1556, found 339.1564; calculated for C20H22NO2, 308.1651,found 308.1647; calculated for C14H11, 179.0861, found 179.0857; calculated for C14H10, 178.0783, found 178.0790;calculated for C13H9, 165.0704, found 165.0707. Elemental analysis: Calculated for C32H38N2O9: C, 64.63; H, 6.44; N,4.71; found C, 64.75; H, 6.68; N, 4.88. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping