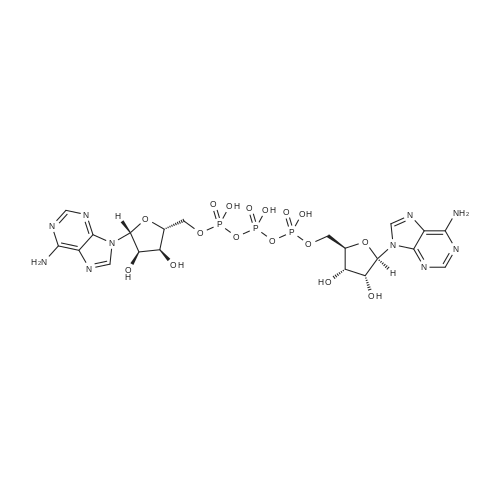
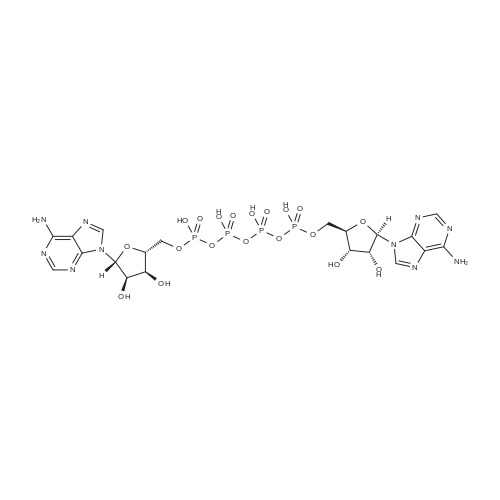
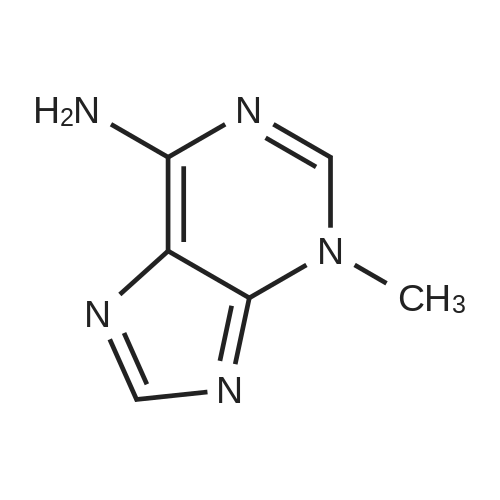
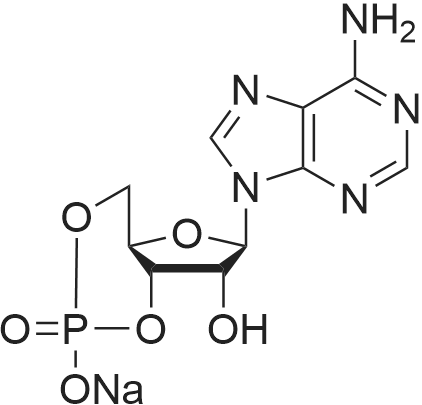
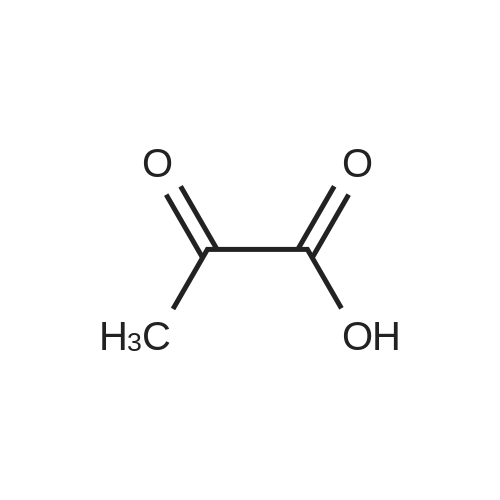
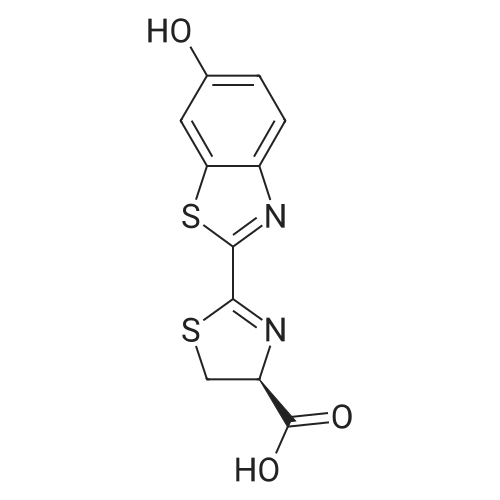
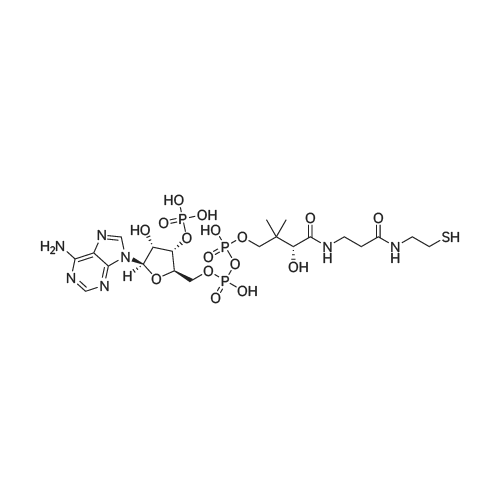
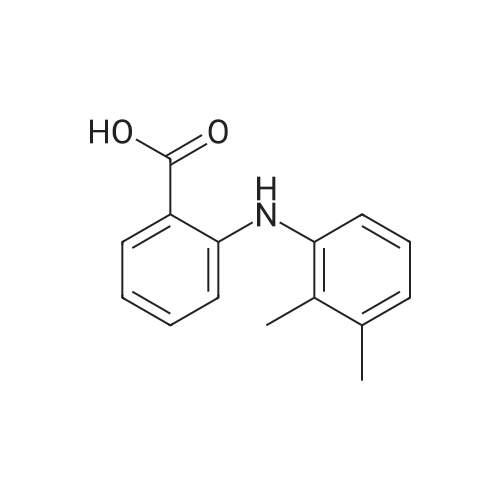
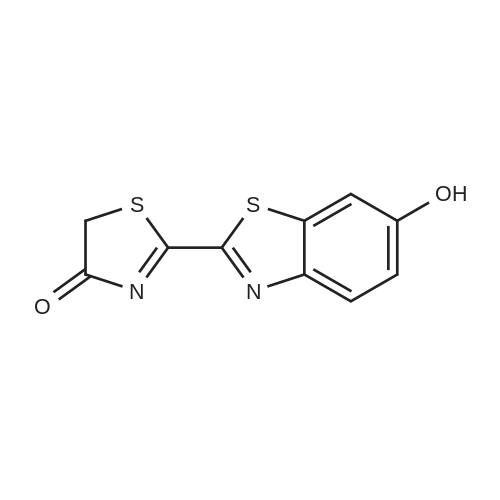
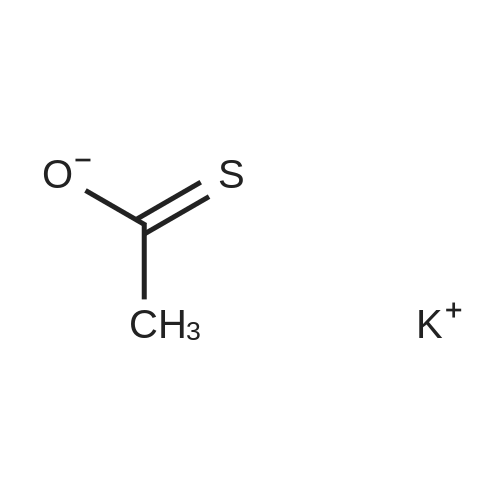
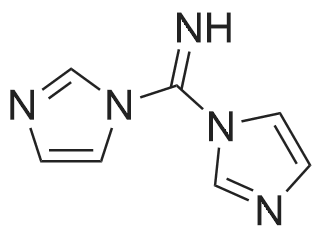
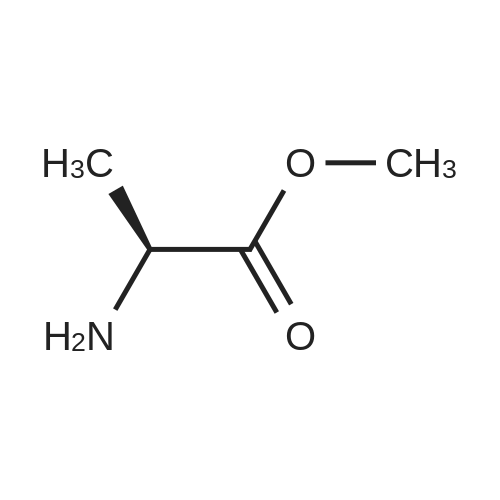
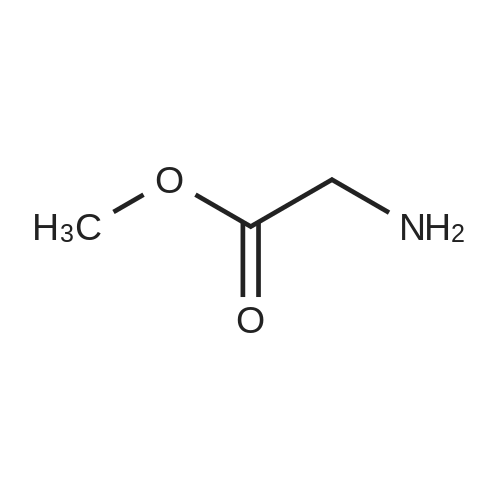
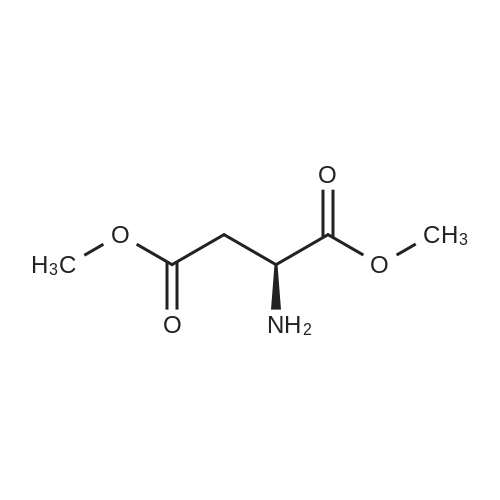
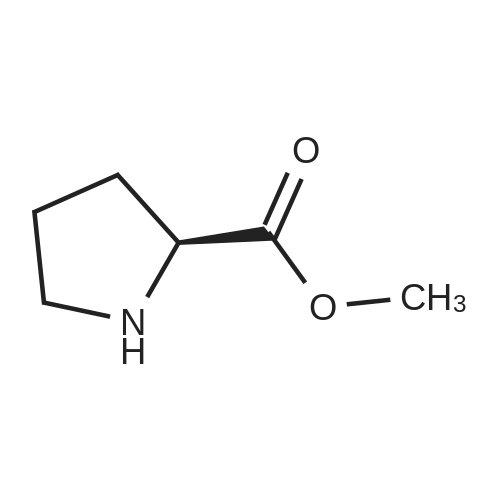
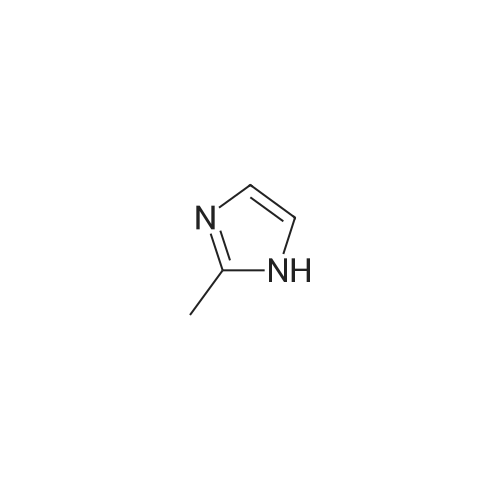
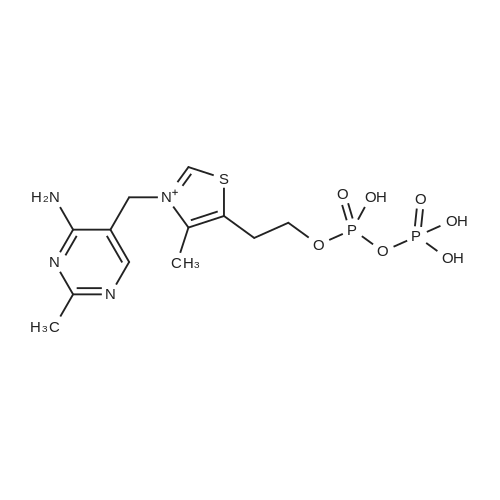
| 45% |
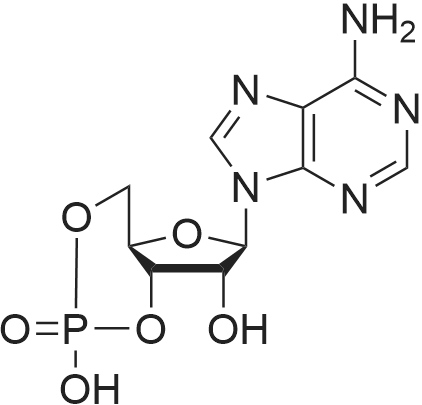
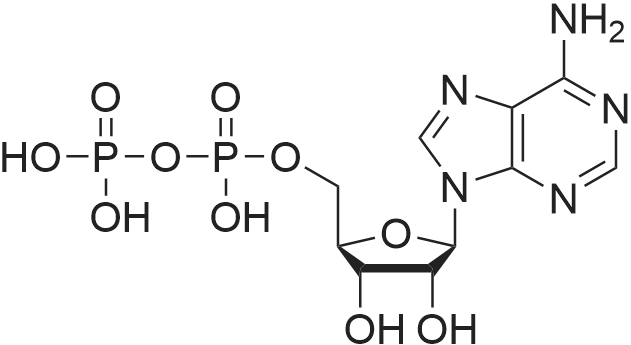
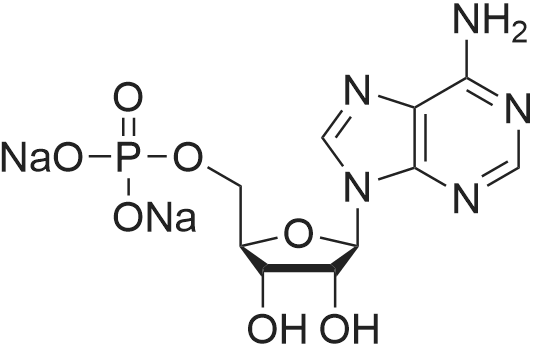
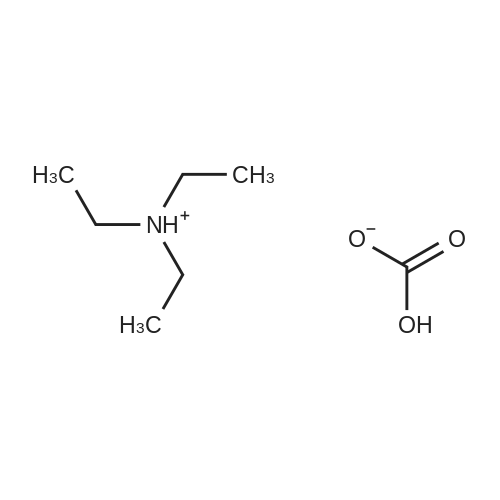
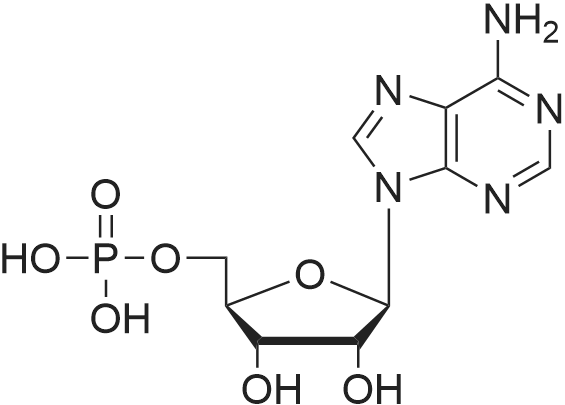
Stage #1: 5'-adenosine monophosphate With triethylamine; 1,1'-carbonyldiimidazole In N,N-dimethyl-formamide at 20℃; for 14h;
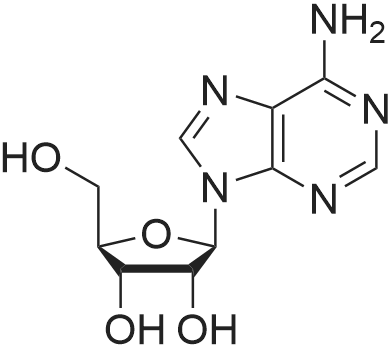
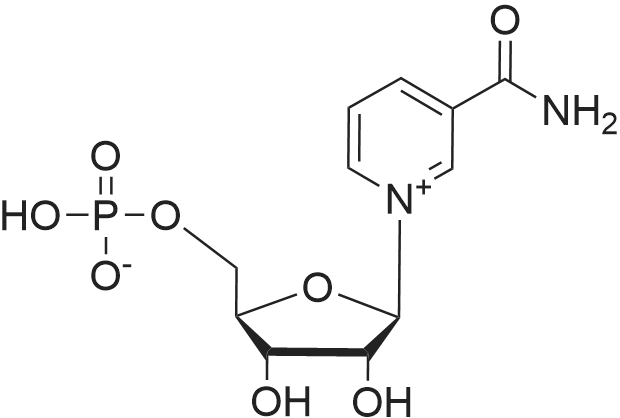
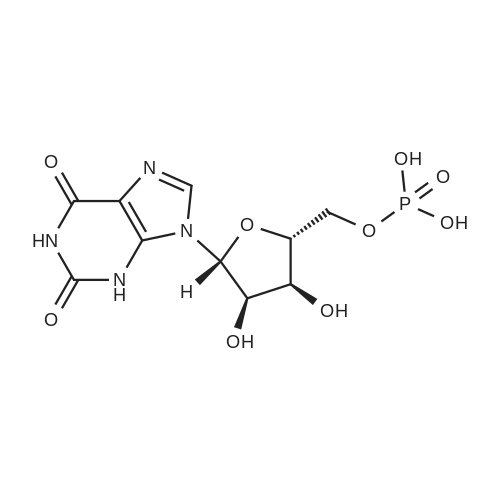
Stage #2: ((2R,3R,4R,5R)-5-(3-carbamoylpyridin-1-ium-1-yl)-3-hydroxy-4-(prop-2-yn-1-yloxy)tetrahydrofuran-2-yl)methyl hydrogen phosphate In N,N-dimethyl-formamide at 20℃; for 96h; |
1
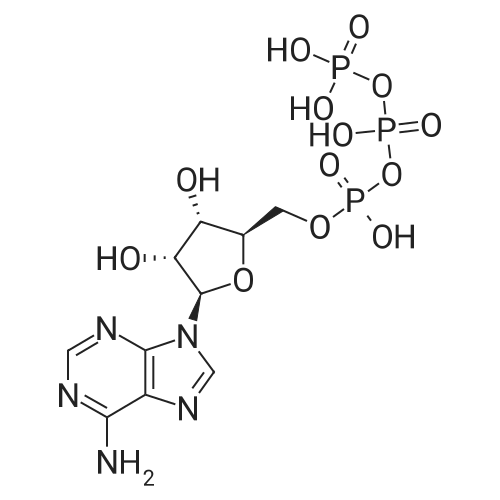
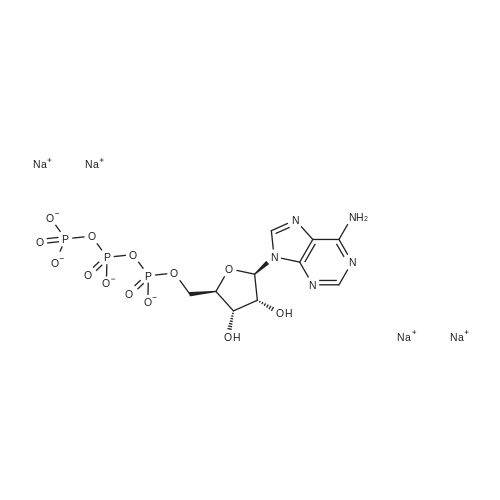
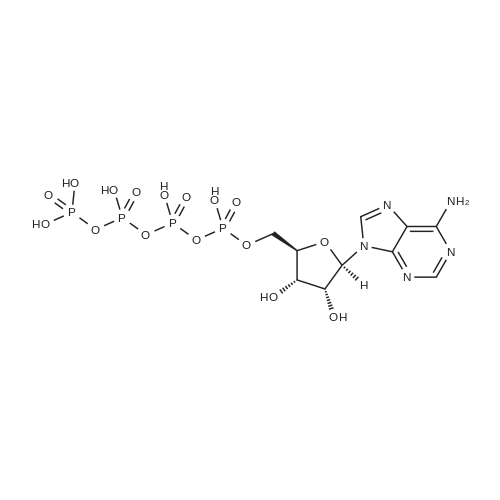
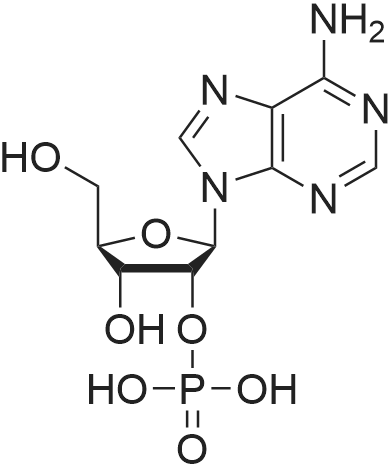
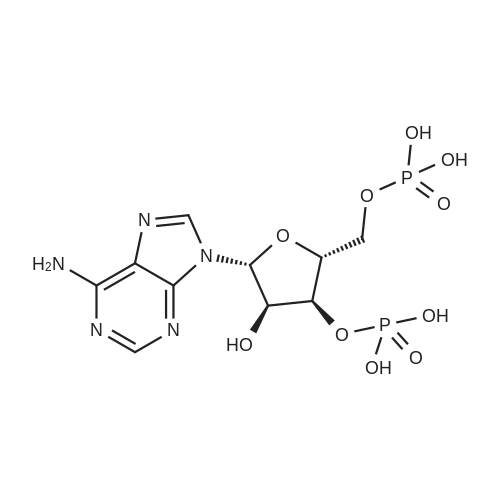
[0319] General procedure for the synthesis of l-((2R,3R,4R,5R)-5-((((((((2R,3S,4R,5R)-5-(6- amino-9H-purin-9-yl)-3,4-dihydr-oxytetrahydrofuran-2-yl)methoxy)(hydroxy)p- hosphoryl)oxy)oxidophosphoryl)o-xy)methyl)-4-hydroxy-3-(prop-2-yn-l-yloxy)tet- rahydrofuran-2-yl)-3-carbamoy-lpyridin-l-ium (10, NAD+ 1): To a stirred solution of Adenosine 5 '-monophosphate (5'-AMP) (52 mg, 0.15 mmol, 1.5 eq) in dried DMF (2 mL) were added 1, 1-carbonyldiimidazole (CDI) (63 mg, 0.50 mmol, 5 eq) and triethylamine (23 μ., 0.16 mmol. 1.6 eq). The reaction mixture was stirred at room temperature for 14 hours, and then quenched with 0.100 ml dried methanol. The solvent was removed under vacuum and the residue was coevaporated 3 times each with 1.00 ml of dried DMF. The activated 5'- AMP was dissolved in dried DMF (1 mL) and compound (9, NM1) (37 mg, 0.10 mmol, 1.0 eq) was added. After stirring at room temperature for 4 days, H20 was added to quench the reaction at 0 °C. The resulting mixture was continued stirring at room temperature for 24 hours. The reaction was then concentrated in vacuo and the crude product was purified via preparative HPLC (C18-A column, 150X4.6 mm, 5 μπι) (mobile phase A: 0.1% formic acid (aq), mobile B: 0.1% formic acid in acetonitrile; flow rate = 1.0 ml/min; 0-16 min: 0-6.7% B, 16-18 min: 6.7-0%) B). Fractions containing the desired product were concentrated and lyophilized to yield NAD+ 1 (32 mg, 45%> yield) as a colorless solid. (0549) [0320] l-((2R,3R,4R,5R)-5-((((((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydr- oxytetrahydrofuran-2-yl)methoxy)(hydroxy)phosphoryl)oxy)oxidophosphoryl)o-xy)methyl)- 4-hydroxy-3-(prop-2-yn-l-yloxy)tetrahydrofuran-2-yl)-3-carbamoy-lpyridin-l-ium (10, NAD+ 1). 1H NMR (400 MHz, D20): δ 2.87 (br, 1H, CH), 4.12-4.14 (m, 1H, CH), 4.24-4.33 (m, 5H, 2CH2+CH), 4.40 (br, 1H, CH), 4.51 (t, 1H, J= 4.0 Hz, CH2), 4.63 (d, 1H, J= 2.8 Hz, CH), 4.71 (t, 1H, J= 5.2 Hz, CH), 4.87-4.89 (m, 2H, 2CH), 6.06 (d, 1H, J= 5.2 Hz, CH), 6.59 (d, 1H, J= 5.6 Hz, CH), 8.09-8.13 (m, 1H, ArH), 8.31 (br, 1H, ArH), 8.61 (br, 1H, ArH), 8.86 (d, 1H, J= 8.0 Hz, ArH), 8.01 (d, 1H, J= 6.0 Hz, ArH), 9.19 (s, 1H, ArH); 13C NMR (100 MHz, D20): δ 59.0, 65.2, 69.3, 70.2, 74.6, 76.7, 78.5, 78.7, 84.1, 87.3, 87.9, 95.6, 126.8, 132.1, 140.9, 143.7, 145.0, 147.6, 148.5, 151.8, 165.2.; HRMS (ESI) Calcd. For C24H28N7Na2Oi4P2+ (M+H)+ requires 746.0965, Found: 746.0955. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping