| 1: 85 % ee
2: 94 % ee |
In 1,2-dichloro-ethane at 0 - 20℃; for 24 - 25h; Molecular sieve; |
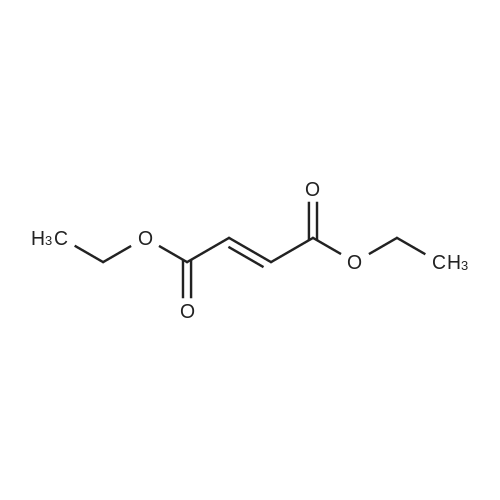
2.1. Cyclopropanation and Purification of Compound 3; An improved Evans cyclopropanation protocol was used for this synthesis using the Cu catalyst prepared from copper (I) triflate and chiral ligand 10. Other ligands and Rh catalysts were tried but all afforded lower diastereoselectivity. The major by-products from the reaction were the cis-isomer, 11 and 12 from the dimerization of ethyl diazoacetate. Solvent plays a significant role in enantioselectivity, diastereoselectivity, and formation of the dimer impurities. As shown in Table 1, a variety of solvents, including coordinating and non-coordinating ones, gave good to excellent conversions (74-98%), except for THF (45%). The diastereoselectivity varied from 80:20 (trans:cis, 1,2-dichloroethane) to 93:7 (trans:cis, MTBE), and ee varied from 85% (1,2-dichloroethane) to 99% (many solvents including MTBE). MTBE gave the best results and was used as the solvent for our first GMP campaign. A significant amount of precipitate was formed when the catalyst was prepared in MTBE. In early studies, this precipitate was removed by filtration prior to the cyclopropanation. However, conversions and ethyl diazoacetate accumulation varied from batch to batch. The situation was greatly improved by generation of the catalyst in situ without filtration. The solid catalyst was completely dissolved after the addition of styrene, giving a clear solution before addition of ethyl diazoacetate. Similar diastereoselectivity and enantioselectivity were obtained. In the prep lab, the cyclopropanation reaction was run in two batches. The first batch used the procedure with the solid catalyst removed and 2.4 kg (assayed, 85% yield after NaBH4 treatment, see below) of 3 was obtained with a trans/cis ratio of 92:8 and 98.8% ee for the trans. The conversion for the reaction was only 95% with 2.0 equiv of ethyl diazoacetate used. The second batch used the procedure with in situ generated catalyst without solid removal. Complete conversion was observed with the use of 1.5 equiv of ethyl diazoacetate. Again, 2.4 kg (assayed, 85% yield after NaBH4 treatment) of 3 was obtained with a trans/cis ratio of 88:12 and 98.9% ee for the trans. |
| 1: 99 % ee
2: 97 % ee |
In tert-butyl methyl ether at 0 - 20℃; for 24 - 25h; Molecular sieve; |
2.1. Cyclopropanation and Purification of Compound 3; An improved Evans cyclopropanation protocol was used for this synthesis using the Cu catalyst prepared from copper (I) triflate and chiral ligand 10. Other ligands and Rh catalysts were tried but all afforded lower diastereoselectivity. The major by-products from the reaction were the cis-isomer, 11 and 12 from the dimerization of ethyl diazoacetate. Solvent plays a significant role in enantioselectivity, diastereoselectivity, and formation of the dimer impurities. As shown in Table 1, a variety of solvents, including coordinating and non-coordinating ones, gave good to excellent conversions (74-98%), except for THF (45%). The diastereoselectivity varied from 80:20 (trans:cis, 1,2-dichloroethane) to 93:7 (trans:cis, MTBE), and ee varied from 85% (1,2-dichloroethane) to 99% (many solvents including MTBE). MTBE gave the best results and was used as the solvent for our first GMP campaign. A significant amount of precipitate was formed when the catalyst was prepared in MTBE. In early studies, this precipitate was removed by filtration prior to the cyclopropanation. However, conversions and ethyl diazoacetate accumulation varied from batch to batch. The situation was greatly improved by generation of the catalyst in situ without filtration. The solid catalyst was completely dissolved after the addition of styrene, giving a clear solution before addition of ethyl diazoacetate. Similar diastereoselectivity and enantioselectivity were obtained. In the prep lab, the cyclopropanation reaction was run in two batches. The first batch used the procedure with the solid catalyst removed and 2.4 kg (assayed, 85% yield after NaBH4 treatment, see below) of 3 was obtained with a trans/cis ratio of 92:8 and 98.8% ee for the trans. The conversion for the reaction was only 95% with 2.0 equiv of ethyl diazoacetate used. The second batch used the procedure with in situ generated catalyst without solid removal. Complete conversion was observed with the use of 1.5 equiv of ethyl diazoacetate. Again, 2.4 kg (assayed, 85% yield after NaBH4 treatment) of 3 was obtained with a trans/cis ratio of 88:12 and 98.9% ee for the trans. |
|
In Isopropyl acetate at 0 - 20℃; for 24 - 25h; Molecular sieve; |
2.1. Cyclopropanation and Purification of Compound 3; An improved Evans cyclopropanation protocol was used for this synthesis using the Cu catalyst prepared from copper (I) triflate and chiral ligand 10. Other ligands and Rh catalysts were tried but all afforded lower diastereoselectivity. The major by-products from the reaction were the cis-isomer, 11 and 12 from the dimerization of ethyl diazoacetate. Solvent plays a significant role in enantioselectivity, diastereoselectivity, and formation of the dimer impurities. As shown in Table 1, a variety of solvents, including coordinating and non-coordinating ones, gave good to excellent conversions (74-98%), except for THF (45%). The diastereoselectivity varied from 80:20 (trans:cis, 1,2-dichloroethane) to 93:7 (trans:cis, MTBE), and ee varied from 85% (1,2-dichloroethane) to 99% (many solvents including MTBE). MTBE gave the best results and was used as the solvent for our first GMP campaign. A significant amount of precipitate was formed when the catalyst was prepared in MTBE. In early studies, this precipitate was removed by filtration prior to the cyclopropanation. However, conversions and ethyl diazoacetate accumulation varied from batch to batch. The situation was greatly improved by generation of the catalyst in situ without filtration. The solid catalyst was completely dissolved after the addition of styrene, giving a clear solution before addition of ethyl diazoacetate. Similar diastereoselectivity and enantioselectivity were obtained. In the prep lab, the cyclopropanation reaction was run in two batches. The first batch used the procedure with the solid catalyst removed and 2.4 kg (assayed, 85% yield after NaBH4 treatment, see below) of 3 was obtained with a trans/cis ratio of 92:8 and 98.8% ee for the trans. The conversion for the reaction was only 95% with 2.0 equiv of ethyl diazoacetate used. The second batch used the procedure with in situ generated catalyst without solid removal. Complete conversion was observed with the use of 1.5 equiv of ethyl diazoacetate. Again, 2.4 kg (assayed, 85% yield after NaBH4 treatment) of 3 was obtained with a trans/cis ratio of 88:12 and 98.9% ee for the trans. |
|
In α,α,α-trifluorotoluene at 0 - 20℃; for 24 - 25h; |
2.1. Cyclopropanation and Purification of Compound 3; An improved Evans cyclopropanation protocol was used for this synthesis using the Cu catalyst prepared from copper (I) triflate and chiral ligand 10. Other ligands and Rh catalysts were tried but all afforded lower diastereoselectivity. The major by-products from the reaction were the cis-isomer, 11 and 12 from the dimerization of ethyl diazoacetate. Solvent plays a significant role in enantioselectivity, diastereoselectivity, and formation of the dimer impurities. As shown in Table 1, a variety of solvents, including coordinating and non-coordinating ones, gave good to excellent conversions (74-98%), except for THF (45%). The diastereoselectivity varied from 80:20 (trans:cis, 1,2-dichloroethane) to 93:7 (trans:cis, MTBE), and ee varied from 85% (1,2-dichloroethane) to 99% (many solvents including MTBE). MTBE gave the best results and was used as the solvent for our first GMP campaign. A significant amount of precipitate was formed when the catalyst was prepared in MTBE. In early studies, this precipitate was removed by filtration prior to the cyclopropanation. However, conversions and ethyl diazoacetate accumulation varied from batch to batch. The situation was greatly improved by generation of the catalyst in situ without filtration. The solid catalyst was completely dissolved after the addition of styrene, giving a clear solution before addition of ethyl diazoacetate. Similar diastereoselectivity and enantioselectivity were obtained. In the prep lab, the cyclopropanation reaction was run in two batches. The first batch used the procedure with the solid catalyst removed and 2.4 kg (assayed, 85% yield after NaBH4 treatment, see below) of 3 was obtained with a trans/cis ratio of 92:8 and 98.8% ee for the trans. The conversion for the reaction was only 95% with 2.0 equiv of ethyl diazoacetate used. The second batch used the procedure with in situ generated catalyst without solid removal. Complete conversion was observed with the use of 1.5 equiv of ethyl diazoacetate. Again, 2.4 kg (assayed, 85% yield after NaBH4 treatment) of 3 was obtained with a trans/cis ratio of 88:12 and 98.9% ee for the trans. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping