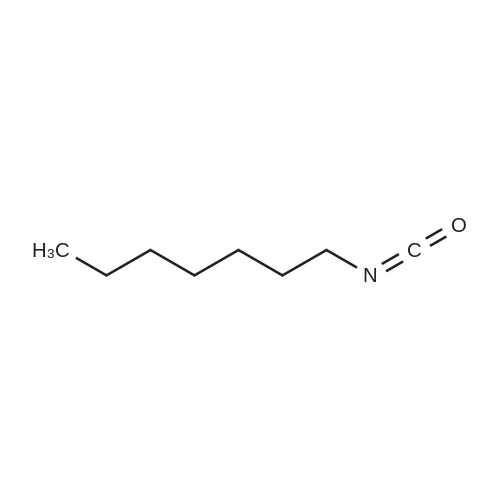
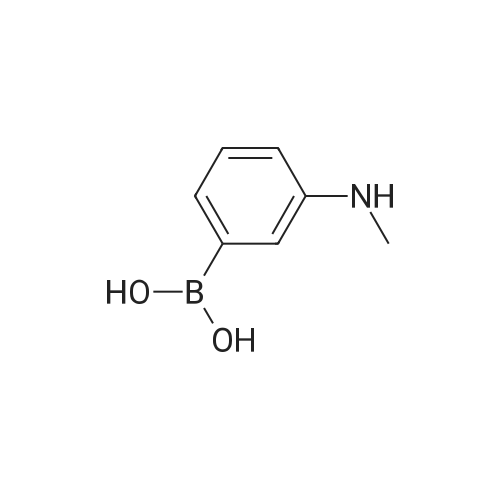
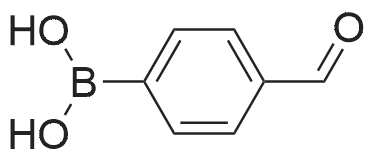
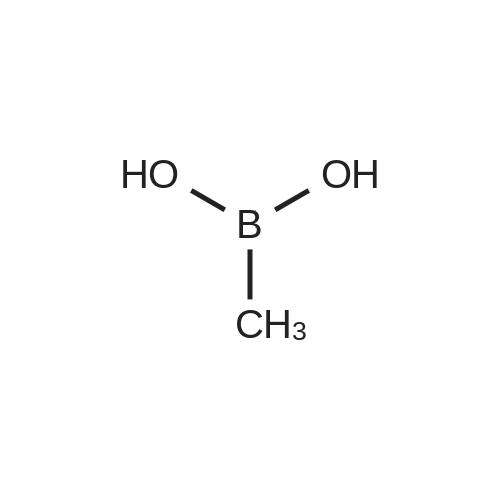
| 99% |
With potassium carbonate; In acetonitrile; at 90℃; |
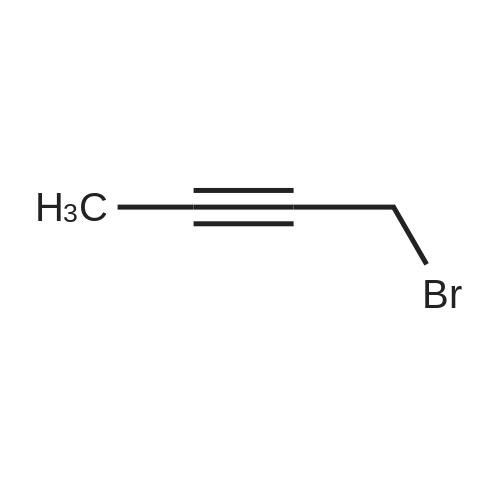
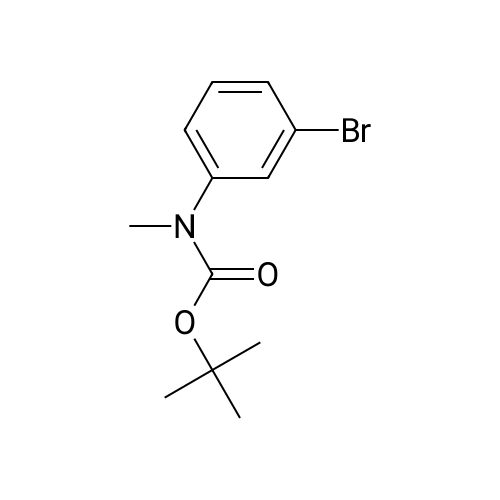
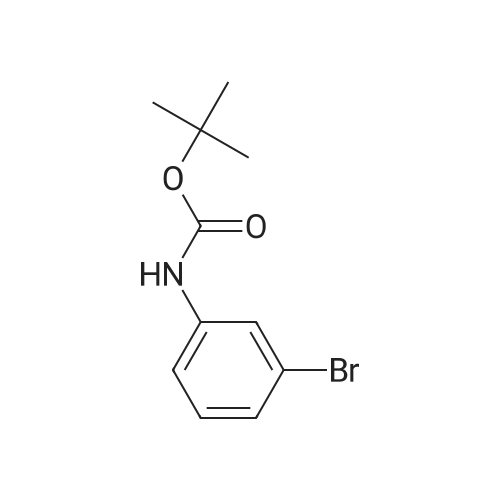
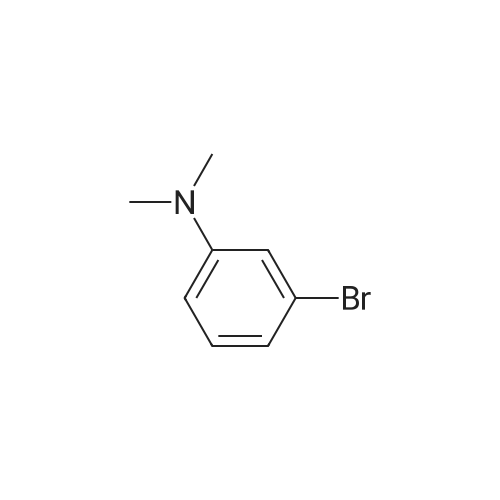
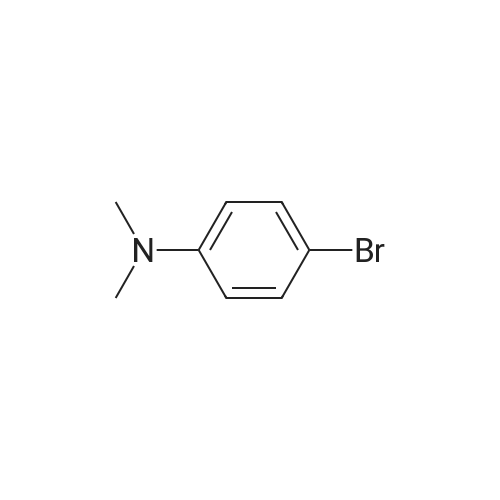
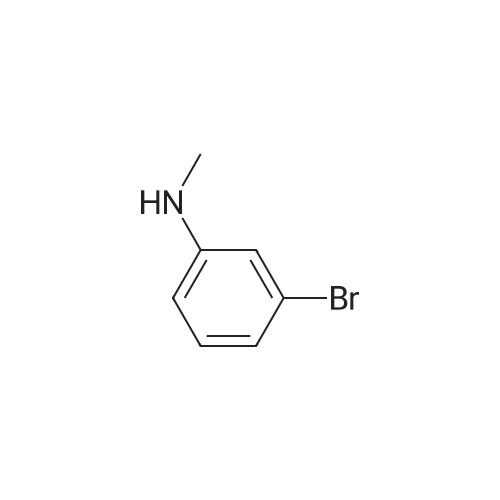
Compound 6 was synthesized in accordance with the following scheme. Potassium carbonate (7.46 g, 54.0 mmol, 2.0 eq.) was suspended in acetonitrile (20 mE), 3-bromo-N-methyl- aniline (4.96 g, 26.7 mmol, 1 eq.) and allyl bromide (3.87 g, 31.3 mmol, 1.2 eq.) were added, and the mixture was heated and refluxed overnight at 90 C. while being stirred. The reaction mixture was filtered through celite, the filtration residue and celite were then washed using ethyl acetate, and the solvent was removed under reduced pressure from a mixture of filtrate and washing solution. The resulting residue was purified by medium-pressure silica gel chromatography (eluent n-hexane/ethyl acetate=100/0 to 98/2) toobtain the objective compound 6 (5.98 g, 99%). 1H NMR (400 MHz, CDCl3): δ 2.95 (s, 3H),3.91-3.93 (m, 2H), 5.15-5.21 (m, 2H), 5.79-5.89 (m, 1H),6.63-6.65 (m, 1H), 6.82-6.86 (m, 2H), 7.06-7.10 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 38.1, 55.0, 110.9, 115.0,116.4, 119.1, 123.5, 130.4, 133.1, 150.7; HRMS (ESIj:Calcd for [M+H], 226.02259, Found, 226.02210 (-0.5 mmu). |
| 73% |
With potassium carbonate; In acetonitrile; at 80℃; |
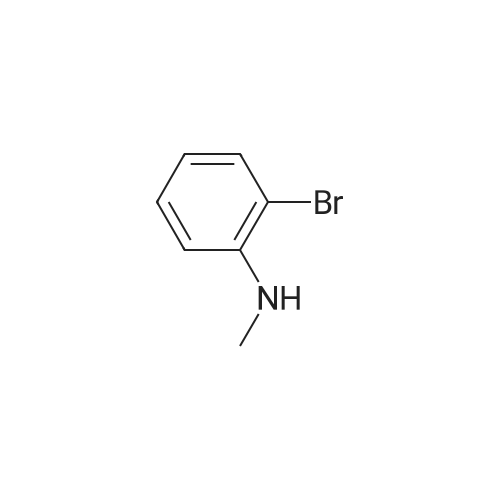
<strong>[66584-32-5]N-methyl-3-bromoaniline</strong> (2.27 g, 12.2 mmol), K2CO3 (4.20 g, 30.4 mol), and allyl bromide (4.00 g, 33.1 mmol) were dissolved in MeCN (50 mL) and stirred overnight at 80 C. After cooling to room temperature and filtering, the solvent of the filtrate was removed under reduced pressure. The remaining oil droplets were purified by silica gel column chromatography (CH2Cl2/n-hexane=), and N-allyl-<strong>[66584-32-5]N-methyl-3-bromoaniline</strong> was obtained (2.02 g, 8.94 mol, yield 73%). (0243) 1H NMR (300 MHz, CDCl3): δ=2.93 (s, 3H), 3.89-3.91 (m, 2H), 5.10-5.18 (m, 2H), 5.74-5.87 (m, 1H), 6.60 (dd, J=8.4 Hz, 2.6 Hz, 1H), 6.78-6.81 (m 2H), 7.05 (m, 1H). 13C NMR (100 MHz, CDCl3): δ=38.1, 55.1, 110.9, 115.0, 116.5, 119.1, 123.5, 130.4, 133.1, 150.7 |
| 72% |
With sodium carbonate; In tetrahydrofuran;Reflux; |
[Synthesis of Compound J5] (0194) Compound J4 (5.00 g, 26.9 mmol, 1 Eq) was dissolved in tetrahydrofuran (50 mL). Next, sodium carbonate (7.85 g, 56.9 mmol, 2.1 Eq) and allyl bromide (3.5 mL, 40.3 mmol, 1.5 Eq) were added and heated and refluxed overnight. More allyl bromide (3.5 mL, 40.3 mmol, 1.5 Eq) was added and heated and refluxed for two days. After air-cooling, the reaction solution was separated by filtration, and the filtrate was concentrated. The residue obtained was separated and purified by silica gel column chromatography (eluent: hexane/ethyl acetate=100/0 to 90/10), and compound J5 (5.53 g, 72%) was obtained as a colorless, transparent liquid. (0195) 1H NMR (CDCl3): δ 7.05 (t, J=8.1 Hz, 1H), 6.83-6.79 (m, 2H), 6.61 (dd, J=8.4 Hz, 2.2 Hz, 1H), 5.81 (ddt, J=17.0 Hz, 10.4 Hz, 4.9 Hz, 1H), 5.16 (dq, J=10.4 Hz, 1.6 Hz, 1H), 5.14 (dq, J=17.0 Hz, 1.6 Hz, 1H), 3.90 (dt, J=4.9 Hz, 1.6 Hz, 2H), 2.93 (s, 3H).; 13C NMR (CDCl3): δ 150.7 (C), 133.1 (CH), 130.4 (CH), 123.5 (C), 119.1 (CH), 116.5 (CH2), 115.1 (CH), 111.0 (CH), 55.1 (CH2), 38.2 (CH2); HRMS-ESI (m/z): [M+H]+ calcd for C10H13NBrN: 226.02259. found: 226.02478 (-2.2 mDa, -9.7 ppm). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping