* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With caesium carbonate In N,N-dimethyl-formamide at 100℃; Inert atmosphere
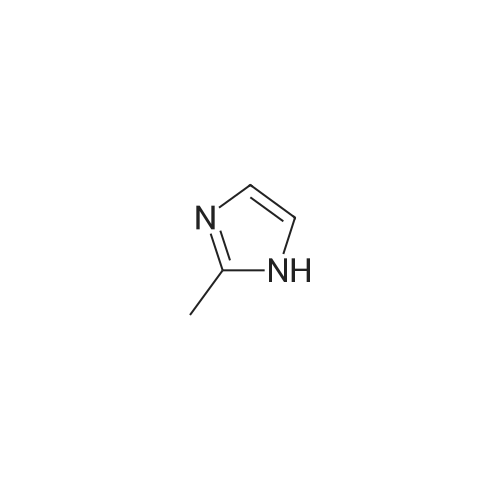
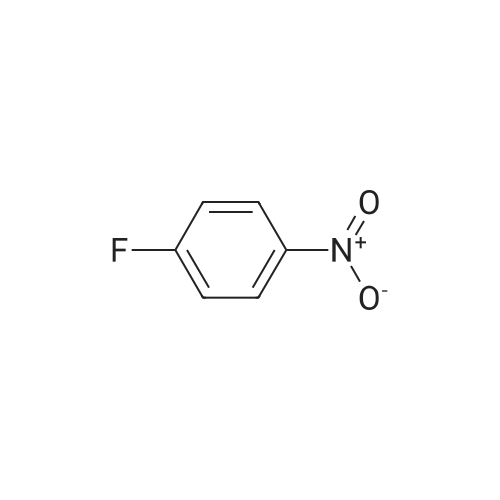
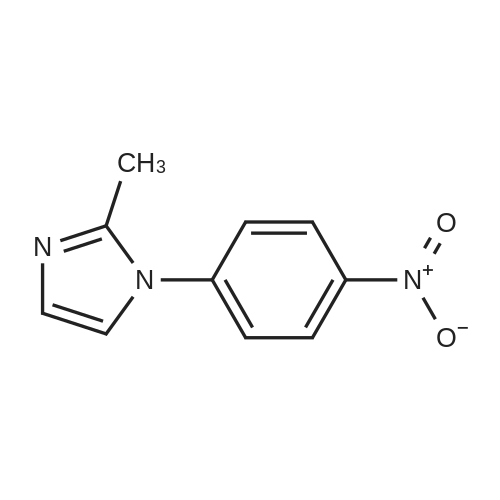
A mixture of l-fluoro-4-nitrobenzene (10 g, 70.9 mmol), 2-methyl-lH- imidazole (5.8 g, 70.9 mmol), and Cs2C03 (34.7 g, 106.4 mmol) in degassed DMF (200 mL) was heated at 100°C under nitrogen overnight. When TLC indicated that l-fluoro-4-nitrobenzene was consumed, the reaction mixture was concentrated in vacuo. The residue was diluted with water (300 mL), and a grey precipitate was formed and was isolated to give 2-methyl-l-(4- nitrophenyl)-lH-imidazole (12.8 g, yield 89percent).
88%
With potassium carbonate In N,N-dimethyl-formamide at 50℃; for 18 h;
5 To 1 -fluoro-4-nitrobenzene (11.0 g, 0.078 mol) in DMF (40 mL) were added 2- methyl-lH-imidazole (6.41 g, 0.078 mol) and potassium carbonate (16.16 g, 0.117 mol) and the reaction stirred at 500C for 18 h. Residual solids were filtered off and the solvent removed from the filtrate. The residues were dissolved in ethyl acetate/water and extracted into ethyl acetate. The solvent was removed and, the residues triturated with 10 diethyl ether to give the title compound (13.93 g, 88percent) as an off-white solid. δη (DMSO- d6) 8.38 (2H, d, J8.8 Hz), 7.78 (2H, d, J9.1 Hz), 7.46 (IH, s), 6.99 (IH, s), 2.38 (3H, s). LCMS (ES+) 204.0 (M+H+).
Reference:
[1] Advanced Synthesis and Catalysis, 2016, vol. 358, # 4, p. 597 - 609
[2] Patent: WO2011/75478, 2011, A1, . Location in patent: Page/Page column 53
[3] Patent: WO2009/71888, 2009, A1, . Location in patent: Page/Page column 83
[4] Patent: US5298520, 1994, A,
[5] Journal of Medicinal Chemistry, 1995, vol. 38, # 10, p. 1799 - 1810
[6] Bioorganic and Medicinal Chemistry Letters, 2004, vol. 14, # 5, p. 1221 - 1227
[7] Journal of Medicinal Chemistry, 2005, vol. 48, # 6, p. 1729 - 1744
[8] Patent: US5128351, 1992, A,
[9] Patent: US5077409, 1991, A,
[10] Patent: EP991638, 2005, B1, . Location in patent: Page/Page column 52
2
[ 693-98-1 ]
[ 586-78-7 ]
[ 73225-15-7 ]
Yield
Reaction Conditions
Operation in experiment
80%
With C40H34CuIN6O6; sodium hydroxide In dimethyl sulfoxide at 100℃; for 4 h; Sealed tube
General procedure: For the catalysis reaction, the catalyst C1 (12 mg,0.01 mmol), imidazole (1.0 mmol), aryl halide(1.0 mmol), NaOH (80 mg, 2.0 mmol), and dimethylsulfoxide (DMSO, 5 mL) were taken in a sealed tube. The reaction mixture was stirred at 100 °C for 4 h and then cooled to room temperature. After adding 5 mL of H2O, the solution was extracted with ethyl acetate. The organic layer was then dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure.The N-arylated product was finally obtained by columnchromatography on silica gel.
Reference:
[1] Organic Letters, 2009, vol. 11, # 15, p. 3294 - 3297
[2] Chemistry - A European Journal, 2009, vol. 15, # 36, p. 8971 - 8974
[3] Indian Journal of Chemistry - Section A Inorganic, Physical, Theoretical and Analytical Chemistry, 2018, vol. 57A, # 2, p. 181 - 185
[4] Patent: US6020357, 2000, A,
[5] Patent: EP946508, 2009, B1, . Location in patent: Page/Page column 64-65
3
[ 636-98-6 ]
[ 693-98-1 ]
[ 73225-15-7 ]
Reference:
[1] New Journal of Chemistry, 2014, vol. 38, # 9, p. 4267 - 4274
[2] Journal of Organic Chemistry, 2009, vol. 74, # 5, p. 1971 - 1976
[3] Organic and Biomolecular Chemistry, 2017, vol. 15, # 26, p. 5503 - 5512
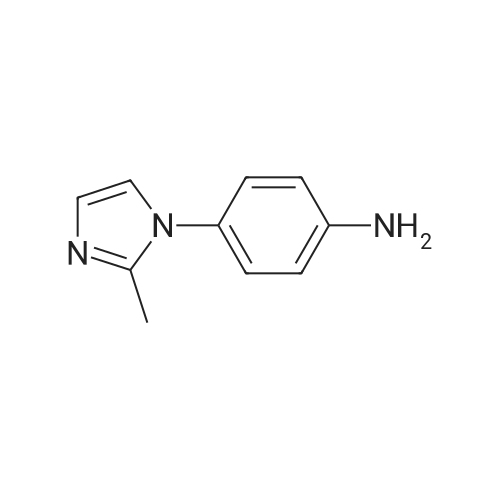
Intermediate 27 (13.9 g, 68 mmol) was dissolved in ethanol to a final concentration of 0.05M. The solution was passed through a palladium on carbon catalyst cartridge (using an η-cube reactor from ThalesNano) at 2 mL/min, 25°C and full 20 hydrogen to give the title compound (11.69 g, 99percent) as an off-white solid. 5η (DMSOd6) 7.08 (1η, s), 7.01 (2η, d, J8.5 Hz), 6.82 (IH, s), 6.63 (2H, d, J8.5 Hz), 5.35 (2H, s), 2.51 (3H, s). LCMS (ES+) 174.0 (M+H+).
92%
With hydrogen In methanol at 20℃; for 8 h;
To a solution 2-methyl-l-(4-nitrophenyl)-lH-imidazole (12.8 g, 63 mmol) in MeOH (150 mL) was added Pd/C (3 g). The resulting mixture was stirred at roomtemperature for 8 hours under hydrogen. When TLC indicated the starting material was consumed, the mixture was filtered and concentrated in vacuo to give 4-(2-methyl-lH-imidazol- l-yl)aniline (10 g, yield 92percent).
Reference:
[1] Patent: WO2009/71888, 2009, A1, . Location in patent: Page/Page column 83
[2] Advanced Synthesis and Catalysis, 2016, vol. 358, # 4, p. 597 - 609
[3] Patent: WO2011/75478, 2011, A1, . Location in patent: Page/Page column 53
[4] Bioorganic and Medicinal Chemistry Letters, 2004, vol. 14, # 5, p. 1221 - 1227
[5] Journal of Medicinal Chemistry, 2005, vol. 48, # 6, p. 1729 - 1744
[6] Patent: US6020357, 2000, A,
[7] Patent: US5077409, 1991, A,
[8] Patent: US5128351, 1992, A,
[9] Patent: US4301169, 1981, A,
[10] Patent: EP946508, 2009, B1, . Location in patent: Page/Page column 65
[11] Patent: EP991638, 2005, B1, . Location in patent: Page/Page column 52-53
With potassium carbonate In N,N-dimethyl-formamide at 140℃; for 5h; Inert atmosphere;
89%
With caesium carbonate In N,N-dimethyl-formamide at 100℃; Inert atmosphere;
15; 1
A mixture of l-fluoro-4-nitrobenzene (10 g, 70.9 mmol), 2-methyl-lH- imidazole (5.8 g, 70.9 mmol), and Cs2C03 (34.7 g, 106.4 mmol) in degassed DMF (200 mL) was heated at 100°C under nitrogen overnight. When TLC indicated that l-fluoro-4-nitrobenzene was consumed, the reaction mixture was concentrated in vacuo. The residue was diluted with water (300 mL), and a grey precipitate was formed and was isolated to give 2-methyl-l-(4- nitrophenyl)-lH-imidazole (12.8 g, yield 89%).
88%
With potassium carbonate In N,N-dimethyl-formamide at 50℃; for 18h;
27
5 To 1 -fluoro-4-nitrobenzene (11.0 g, 0.078 mol) in DMF (40 mL) were added 2- methyl-lH-imidazole (6.41 g, 0.078 mol) and potassium carbonate (16.16 g, 0.117 mol) and the reaction stirred at 500C for 18 h. Residual solids were filtered off and the solvent removed from the filtrate. The residues were dissolved in ethyl acetate/water and extracted into ethyl acetate. The solvent was removed and, the residues triturated with 10 diethyl ether to give the title compound (13.93 g, 88%) as an off-white solid. δη (DMSO- d6) 8.38 (2H, d, J8.8 Hz), 7.78 (2H, d, J9.1 Hz), 7.46 (IH, s), 6.99 (IH, s), 2.38 (3H, s). LCMS (ES+) 204.0 (M+H+).
47%
In water; ethyl acetate; N,N-dimethyl-formamide
16.1 1.
1. 1-(4-Nitrophenyl)-2-methylimidazole Sodium hydride (4.87 g, 122.0 mmol, 60% dispersion in oil) was added to a solution of 2-methylimidazole (10 g, 122.0 mmol) in DMF (100 ml) and stirred at room temperature for 0.25 h. 1-Fluoro-4-nitrobenzene (17.18 g, 122.0 mmol) was added to the reaction mixture and stirred at room temperature for 16 h. Water (150 ml) and ethyl acetate (250 ml) were added, the aqueous separated and extracted with ethyl acetate (3*150 ml). The combined extracts were washed with water (3*150 ml), dried (Na2 SO4) and evaporated to give the desired product (11.5 g, 47%); δ (360 MHz, CDCl3) 2.24 (3H, s, Me); 7.06 (1H, d, J=1.5 Hz, Ar-H); 7.10 (1H, d, J=1.5 Hz, Ar-H); 7.50 (2H, d, J=9.5 Hz, Ar-H); 8.38 (2H, d, J=9.5 Hz, Ar-H).
With sodium hydride In tetrahydrofuran for 16h; Heating;
With potassium carbonate In water; dimethyl sulfoxide; ethyl acetate
42 2-Methyl-1-(p-nitrophenyl)imidazole
EXAMPLE 42 2-Methyl-1-(p-nitrophenyl)imidazole A mixture of 50.0 g of p-fluoronitrobenzene, 85.93 g of 2-methylimidazole and 44.9 g of potassium carbonate in 860 ml of dimethylsulfoxide is heated at 120° C. for 24 hours. The mixture is poured into 2.5 liter of water and stored in a refrigerator for two days. The mixture is filtered and cake washed with copious volumes of water and vacuum dried. The cake is dissolved in 1500 ml of ethyl acetate, filtered through hydrous magnesium silicate and the filtrate reduced to about 500 ml, cooled and the resulting solid, filtered, and air dried to give 81.0 g of the desired product as brown crystals, m.p. 135°-137° C.
With sodium hydride In tetrahydrofuran at 0℃; for 16h; Heating / reflux;
65 1-(3'-Aminobenzisoxazol-5'-yl)-3-trifluoromethyl-5-[4'(2''-methylimidazol-1''-yl)phenyl)aminocarbonyl]pyrazole, TFA salt
Example 65 1-(3'-Aminobenzisoxazol-5'-yl)-3-trifluoromethyl-5-[4'(2''-methylimidazol-1''-yl)phenyl)aminocarbonyl]pyrazole, TFA salt To a suspension of NaH (4.8 g, 120 mmol, prewashed with THF (3 x 5 mL) in THF (100 mL) was added a solution of 1-fluoro-4-nitrobenzene (14.1 g, 100 mmol) and 2-methylimidazole (8.2 g, 100 mmol) in THF (50 mL) at 0 °C. The mixture was refluxed for 16 hours and cooled to room temperature. To it was added EtOAc (200 mL) and water (100 mL). The organic layer was separated, washed with water and brine, dried over MgSO4, and concentrated to give the crude nitro compound.
With copper; caesium carbonate; methyl-alpha-D-glucopyranoside In water; dimethyl sulfoxide at 100℃; Sealed tube; Green chemistry;
General Procedure
General procedure: All the reactions were carried out in DMSO-H 2 O (1:1, 2 mL) in asealed vessel. To a 10 mL hydrothermal synthesis reactor was chargedCu powder (6 mg, 0.1 mmol), MG (39 mg, 0.2 mmol), Cs 2 CO 3 (980 mg,3.0 mmol), nitrogen-containing heterocycle (1.5 mmol), amine (3.0mmol), and aryl halide (0.8 mmol). The reaction mixture was stirredfor a specified time at 100-110 °C. After TLC analysis confirmed thecomplete consumption of aryl halides, the mixture was cooled to r.t.(the pH was adjusted if the product was acidic), diluted with EtOAc(10 mL), filtered through a Celite pad, and washed with EtOAc (20-30mL). The organic layer was dried and concentrated. The residue waspurified by silica gel column chromatography to give the product.
85%
With caesium carbonate; copper(II) sulfate; 1,2-bis(2-pyridyl)ethane-N,N'-dioxide In water at 120℃; for 24h;
84%
With D-galacturonic acid; potassium carbonate; copper(I) bromide In water; dimethyl sulfoxide at 80 - 100℃; Inert atmosphere; Green chemistry;
General Procedure for Catalytic Experiments
General procedure: To a 10 mL vial was charged with aryl halide (0.8 mmol), N-containing heterocycle (1.0 mmol), CuBr (0.04 mmol), GalA (0.08mmol), K2CO3 (2.4 mmol), and 50% aq DMSO. The flask wasevacuated and backfilled with argon three times, and the reaction mixture was stirred at appropriate temperature under oil bath for the indicated time. After the complete consumption of aryl halide monitored by TLC, the mixture was then cooled to ambient temperature (if the product was acidic, the mixture was acidified), diluted with ethyl acetate (5 mL), filtered via aCelite pad, and washed with ethyl acetate (10-20 mL). The organic layer was separated by the separating funnel, which was washed successively with water (2 × 10 mL) and brine (2 ×10 mL). The organic layer was dried over anhydrous MgSO4 and concentrated by reduced pressure in a rotary evaporator, which was then purified by column chromatography on silica gel to provide the desired products.
82%
With C20H14CuN2O8S2(2-)*2Na(1+); tetrabutylammomium bromide; sodium hydroxide In water at 100℃; for 12h;
80%
With C40H34CuIN6O6; sodium hydroxide In dimethyl sulfoxide at 100℃; for 4h; Sealed tube;
Experimental
General procedure: For the catalysis reaction, the catalyst C1 (12 mg,0.01 mmol), imidazole (1.0 mmol), aryl halide(1.0 mmol), NaOH (80 mg, 2.0 mmol), and dimethylsulfoxide (DMSO, 5 mL) were taken in a sealed tube. The reaction mixture was stirred at 100 °C for 4 h and then cooled to room temperature. After adding 5 mL of H2O, the solution was extracted with ethyl acetate. The organic layer was then dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure.The N-arylated product was finally obtained by columnchromatography on silica gel.
With hydrogenchloride In N,N-dimethyl-formamide
A Part A.
Part A. Preparation of N-(4-nitrophenyl)-2-methylimidazole 2-Methylimidazole (1.04 g) was treated with 0.56 g 60% sodium hydride in an oil dispersion in 60 mL DMF with cooling. After 0.33H added 4-bromonitrobenzene in three portions over 0.5 H. Let reaction mixture warm to ambient temperature overnight. Diluted mixture with 100 mL of 1.0M HCl solution and extracted three times with 30 mL portions of ethyl acetate. Combined extracts and dried over magnesium sulfate. Concentrated resulting organics in vacuo. Purified crude material by standard chromatographic techniques to give the purified product as a crystalline solid. LRMS (NH3-CI): 204 (M+H, 100); 1 H-NMR (CDCl3) δ: 8.40 (d, 2H), 7.50 (d, 2H), 7.05 (d, 2H), 2.43 (s, 3H).
Stage #1: 2-methylimidazole With sodium hydride In N,N-dimethyl-formamide; mineral oil for 0.33h; Cooling;
Stage #2: para-nitrophenyl bromide In mineral oin; N,N-dimethyl-formamide at 20℃; Cooling;
135.A; 136.A 1-(3-amidinophenyl)-3-methyl-5-[(4'-(2-methylimidazolyl)phenyl)aminocarbonyl]pyrazole and 1-(3-aminocarbenylphenyl)-3-methyl-5-[(4'-(2-methylimidazolyl)phenyl)aminocarbonyl)pyrazole
Part A. Preparation of N-(4-nitrophenyl)-2-methylimidazole. 2-Methylimidazole (1.04 g) was treated with 0.56 g 60% sodium hydride in an oil dispersion in 60 mL DMF with cooling. After 0.33H added 4-bromonitrobenzene in three portions over 0.5 H. Let reaction mixture warm to ambient temperature overnight. Diluted mixture with 100 mL of 1.0M HCl solution and extracted three times with 30 mL portions of ethyl acetate. Combined extracts and dried over magnesium sulfate. Concentrated resulting organics in vacuo. Purified crude material by standard chromatographic techniques to give the purified product as a crystalline solid. LRMS (NH3-CI): 204 (M+H, 100); 1H-NMR(CDCl3)δ: 8.40 (d, 2H), 7.50 (d, 2H), 7.05 (d, 2H), 2.43 (s, 3H).
0.16 g
With quebrachitol; copper; caesium carbonate In water; dimethyl sulfoxide at 100℃; Inert atmosphere; Green chemistry;






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping