* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Chemical and Pharmaceutical Bulletin, 1998, vol. 46, # 4, p. 623 - 630
2
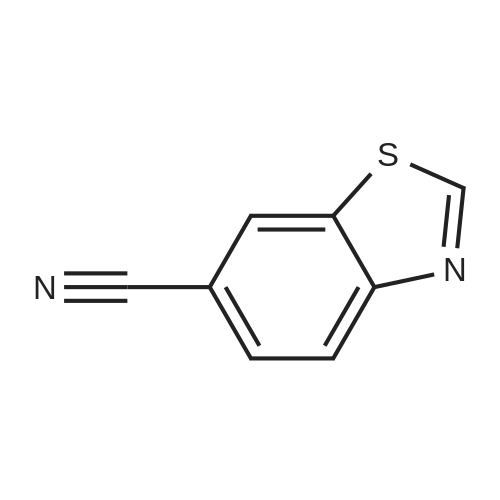
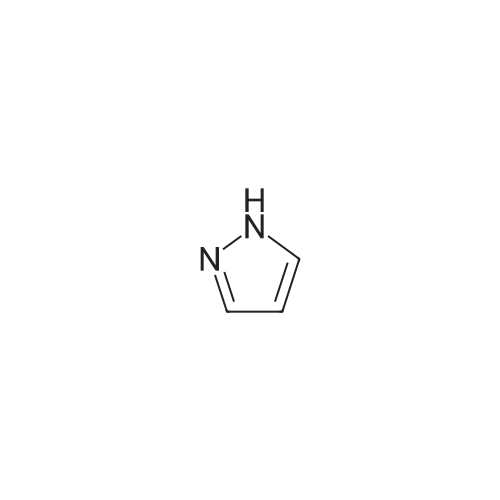
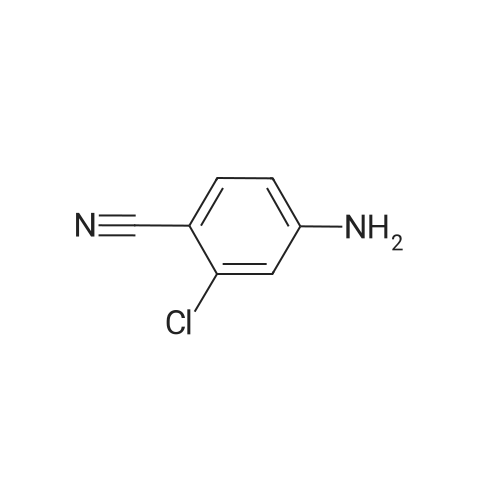
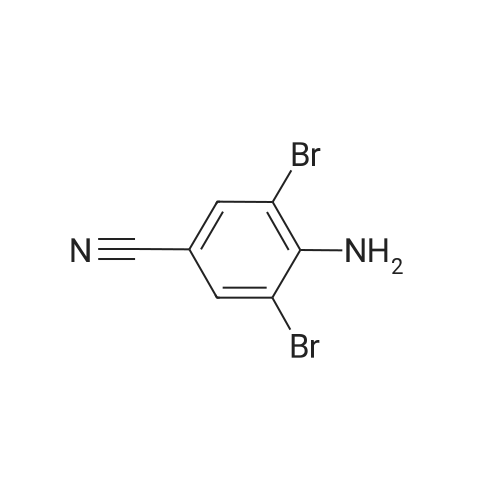
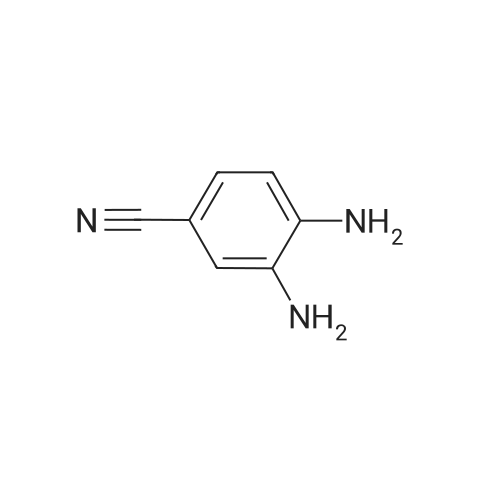
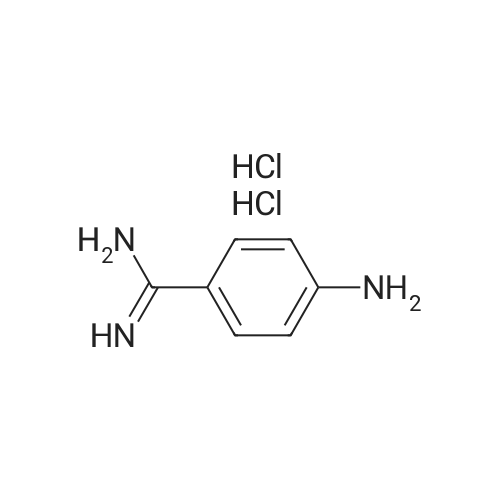
[ 873-74-5 ]
[ 46047-18-1 ]
Yield
Reaction Conditions
Operation in experiment
86%
With sodium azide In water at 100℃; for 5 h; Green chemistry
General procedure: Benzonitrile (1 mmol, 0.103 g), sodium azide (1.1 mmol, 0.0759 g), and 2 mL water were taken in a reaction tube and stirred at room temperature to make homogeneous suspension, and then 20 wtpercent catalyst (ZnO–RGO) was added to the reaction mixture. The reaction mixture was heated to 100 °C for 2 h. The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and centrifuged. The filtrate was treated with 5N HCl (10 mL) and then with ethyl acetate. The organic layer was separated, washed with deionized water, and then dried over anhydrous sodium sulfate and concentrated to give the crude solid crystalline 5-phenyl-1H-tetrazole. It was recrystallized from n-hexane, ethyl acetate, and the yield was about 0.143 g.
81%
With sodium azide; silver nitrate In N,N-dimethyl-formamide at 20 - 120℃; for 5.5 h;
General procedure: Sodiumazide (0.378 g, 0.046 mmol) was added to a solution of AgNO3 (5 mg, 10 mmol)in DMF (5 ml) and reaction mixture was stirred for 5 min, to this stirredsolution benzonitrile 1a (0.4 ml, 0.033 mmol) was added dropwise over theperiod of 1 min at room temperature and stirring continued for 10 min at thesame temperature and then heated at 120 C for 5 h. After consumption of 1a,the reaction mixture was cooled to room temperature and chilled by addingcrushed ice into the reaction mixture followed by addition of 2 N HCl tillreaction mixture reached the pH 2. The reaction mixture was then extractedwith ethyl acetate. The organic layer was dried with anhydrous Na2SO4, andconcentrated to obtain tetrazole 2a in 83percent yield as an off white solid (268 mg).
75%
With sodium azide; acetic acid; urea In water; N,N-dimethyl-formamide at 60 - 110℃; for 9 h;
General procedure: The procedure for the synthesis of the tetrazole 2a is representative. A mixture of sodium azide (0.39 g 0.0060 mol), urea (0.36 g, 0.0060 mol) and water (2.5 mL) was taken in a round–bottom flask and stirred at 60 °C for 1 h. Charged benzonitrile 1a (0.5 g 0.0048 mol), acetic acid(0.5 mL) and DMF (2.5mL) at 60 °C and heat to 110°C stirred for 8 h. After completion of the reaction (as indicated by TLC), the reaction mixture was cooled to room temperature and diluted the reaction mass with water (2.5 mL)and ethyl acetate (5.0 mL) at 25-35 °C. Add 5N aqueous hydrochloric acid (2.5 mL) at 25-35 °C. Stirred for 20- 30 min, the resultant organic layer was separated and the aqueous layer was extracted with ethyl acetate (2.5 mL). The combined organic layer was washed with 40 percent aq.NaCl solution (2.5 mL) and dried over anhydrous Na2SO4 and concentrated to give a crude product, which was isolated using chilled water after 3-4 h maintenance, and eventually filtered off to give 0.67 g (95percent) of an off-white solid.
74%
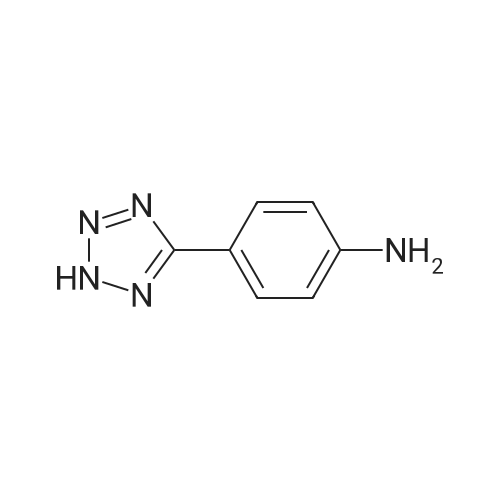
With sodium azide; ammonium chloride In N,N-dimethyl-formamide at 125℃; for 24 h;
Synthesis of 4-(1H-tetrazol-5-yl) aniline (p).A mixture of 4-aminobenzonitrile (100 mg, 0.85 mmol), sodium azide (110.2 mg, 1.69 mmol), DMF (8.5 mL) and ammonium chloride (45.3 mg, 0.85 mmol) was heated in an oil bath for 24 h at 125 °C. When the reaction was deemed complete by TLC the mixture wasacidified with iN HCL and extracted with ethyl acetate (3x). The combined ethyl acetate was washed with brine and dried over Na2SO4, filtered and concentrated to give the crude product. The tetrazole compound was purified by column chromatography on silica gel (90percent CH2C12/MeOH) to yield the desired product (102mg, 74percent).Example 50 - Compound 24 N=N 4-(1 H-tetrazol-5-yl)aniline Chemical Formula: C7H7N5 Molecular Weight: 161.1640 Compound 24 was synthesized according to procedure p, yielding the final product 24 as white powder (74percent). δ (400 MHz, d-MeOD3) 4.34 (brs, 2H, NH2), 6.51 (d, J = 8.9 Hz, 2H, CH), 7.71 (s, J = 8.9 Hz, 2H, CH); d (100 MHz, i -C2D6CO) 1 13.4, 121.6, 126.8, 143.1 , 154.2; LRMS (ES+) Calcd for [C7H7N5 + H] 161.07 found 162.16.
60%
With sodium azide; triethylamine hydrochloride In toluene at 95℃; for 24 h;
[0343] To a solution of 4-aminobenzonitrile (11.8 g, 100 mmol) and triethylamine hydrochloride (17.9 g, 130 mmol) in toluene (550 ml) was added sodium azide (8.45 g, 130 mmol). After stirring for 24 h at 95° C., the reaction mixture was cooled to room temperature and was extraced with water (3.x.60 ml). The combined aqueous phases were acidified with concentrated aqueous HCl to pH 2-3. The product was collected by filtration, washed with water and dried in vacuum. Yield 9.59 g (60percent) pale brown solid. M.p.: 280-281° C., TLC (CH2Cl2/MeOH/AcOH 9:1:0.1): Rf 0.30
54%
With sodium azide; ammonium chloride In N,N-dimethyl-formamideReflux
Example 33. Synthesis of 4-(lH-Tetrazol-5-yl)-phenylamine. The title compound was synthesized following the scheme below and used for synthesis of Compound 285 via Scheme 1.To a solution of 4-amino-benzonitrile (2.36 g, 20 mmol) in dry DMF (20 ml) was added NaN3 (1.6 g, 30 mmol) and NH4C1 (1.6 g, 30 mmol) and the reaction mixture was refluxed overnight. After cooling to room temperature, the resulting mixture was diluted with 40 ml of water and extracted with EtOAc (3x30 ml). The organic layer was washed with brine, dried over Na2S04, filtered and evaporated to give the 4-(lH-Tetrazol-5-yl)-phenylamine (1.5 g, 54percent yield). 1H NMR (400 MHz, DMSO-d6): δ 16.26 (s, 1H), 7.70-7.68 (d, 2H, J = 8.4), 6.70-6.67 (d, 2H, J = 8.4), 5.79-5.76 (s, 2H).
44.4%
With sodium azide; ammonium chloride In N,N-dimethyl-formamide at 120℃; for 12 h;
57.1 4-(1H-Tetrazol-5-yl)-phenylamine To a stirred solution of 4-amino-benzonitrile (300 mg, 2.53 mmol; CAS Reg. No. 873-74-5) in dry DMF (6 ml) was added NH4Cl (547 mg, 10.2 mmol) and NaN3 (660 mg, 10.2 mmol) at room temperature. The reaction mixture was then heated at 120° C. for 12 h. After cooling, TLC shows formation of new spot, filtered the solid material by sintered funnel and washed the solid residue by EtOAc (4*5 ml). Combined organic layers were reduced under pressure at 60° C. and diluted the residue with EtOAc (25 mL). Organic layer was washed with H2O (15 ml), brine (12 ml) and dried over Na2SO4; which was then concentrated under reduced pressure to give the crude material (300 mg). Crude product was then purified by column chromatography [SiO2 (230-400 mesh), MeOH:DCM 5:95] to give the title compound (180 mg, 44.4percent) as light yellow solid.
44%
With sodium azide; triethylamine hydrochloride In toluene for 24 h; Heating / reflux
A mixture of p-amino benzonitrile (2g, 16.9mmol), triethylamine hydrochloride (3.49g, 25.38mmol) and sodium azide (1.65g, 25.38mmol) were taken in anhydrous toluene (20ml) and heated to reflux for 24 hr. The reaction mixture was cooled to room temperature and neutralized with dilute hydrochloric acid. The resultant precipitate was filtered, washed with water then dried to give 4-(lH-tetrazol-5-yl)-ρhenylamine (1.2g, 44percent).
Reference:
[1] RSC Advances, 2015, vol. 5, # 28, p. 21651 - 21658
[2] Synthesis, 1998, # 6, p. 910 - 914
[3] Chemical Communications, 2007, # 48, p. 5182 - 5184
[4] Applied Organometallic Chemistry, 2018, vol. 32, # 8,
[5] RSC Advances, 2016, vol. 6, # 79, p. 75227 - 75233
[6] Synthetic Communications, 2018, vol. 48, # 2, p. 175 - 187
[7] Tetrahedron Letters, 2014, vol. 55, # 11, p. 1879 - 1882
[8] Synlett, 2010, # 3, p. 391 - 394
[9] Journal of Organic Chemistry, 2011, vol. 76, # 21, p. 9090 - 9095
[10] Tetrahedron Letters, 2018, vol. 59, # 5, p. 445 - 449
[11] ACS Medicinal Chemistry Letters, 2013, vol. 4, # 11, p. 1102 - 1107
[12] Patent: WO2013/177534, 2013, A2, . Location in patent: Page/Page column 60; 99; 100
[13] Patent: US2003/232868, 2003, A1, . Location in patent: Page 15
[14] Patent: WO2012/9678, 2012, A1, . Location in patent: Page/Page column 220
[15] Patent: US2010/76027, 2010, A1, . Location in patent: Page/Page column 44
[16] Patent: WO2006/123145, 2006, A1, . Location in patent: Page/Page column 59
[17] Recueil des Travaux Chimiques des Pays-Bas, 1958, vol. 77, p. 1129,1132
[18] Turkish Journal of Chemistry, 2015, vol. 39, # 5, p. 998 - 1011
3
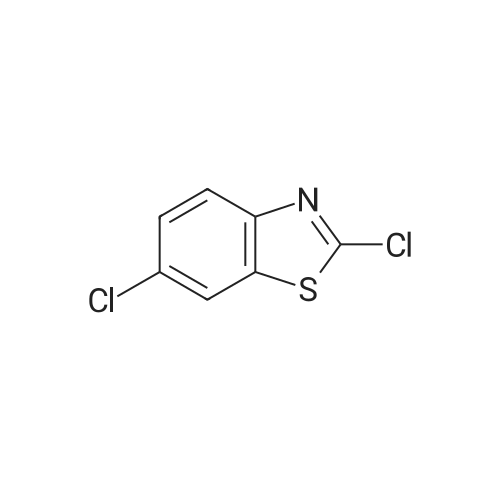
[ 1147550-11-5 ]
[ 873-74-5 ]
[ 19759-66-1 ]
Yield
Reaction Conditions
Operation in experiment
56%
Stage #1: at 13.5℃; for 1 h; Stage #2: With sodium carbonate In water
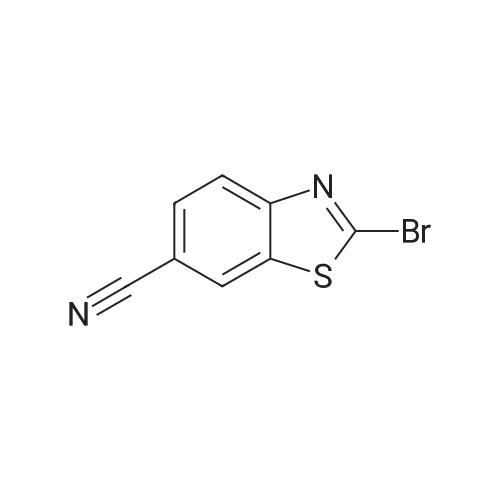
Step A: 2-Chlorobenzothiazole-6-carbonitrile; To a mixture of 4-aminobenzonitrile (23.6 g, 0.2 mol) and ammonium rhodanate (30.4 g, 0.4 mol) was added glacial acetic acid (600 ml.) and the resulting solution was cooled to 13.5 0C on an ice-bath. A mixture of bromine and glacial acetic acid was added drop-wise and slowly. The resulting mixture was stirred for 1 hour at 13.5 0C and filtered. The filter cake was washed with glacial acetic (6 x 100 ml.) and placed in hot water (1000 ml.) with stirring. The mixture was filtered and pH of the filtrate was adjusted to 7 with a saturated sodium carbonate solution. The precipitate was isolated and dried to give 19.5 g (56 percent) of 2-aminobenzo- thiazole-6-carbonitrile. A mixture of concentrated hydrochloric acid (1 13 ml.) and water (52 ml.) was heated at 90 0C while 2-aminobenzothiazole-6-carbonitrile (19 g, 0.108 mol) was added. The mixture was cooled to -5 0C on an ice-bath and a solution of sodium nitrite (7.72 g, 0.1 12 mol) in water (20 ml.) was added drop-wise, keeping the temperature below than 0 0C. When addition was complete the mixture was stirred for 0.5 hour and a solution of CuCI2 (16 g) in water (108 ml.) was added drop-wise. When addition was complete the mixture was stirred for 10 min, and the ice-bath was removed. Stirring was continued for 2 h and the mix- ture was cooled to room temperature. The mixture was filtered and the solid was washed to neutrality with water and dried. This afforded 12.8 g (61 percent) of 2-chlorobenzothiazole-6-carbo- nitrile. 1H-NMR δ 8.14 (s, 1 H), 8.03 (d, 1 H), 7.75 (d, 1 H).
Reference:
[1] Journal of Chemical Research, 2006, # 12, p. 769 - 770
[2] Patent: WO2007/110364, 2007, A1, . Location in patent: Page/Page column 38
[3] European Journal of Medicinal Chemistry, 2016, vol. 115, p. 352 - 360
[4] European Journal of Medicinal Chemistry, 2018, vol. 148, p. 477 - 486
4
[ 333-20-0 ]
[ 873-74-5 ]
[ 19759-66-1 ]
Yield
Reaction Conditions
Operation in experiment
0.70 g
Stage #1: at 10 - 20℃; for 0.5 h; Stage #2: at 20℃; for 16 h;
To a solution of 4-aminobenzonitrile (1.00 g, 8.47 mmol) in acetic acid (12 ml) was added potassium thiocyanate (1.00 g, 16.9 mmol) at 10°C. The reaction mixture was stirred at rt for 30 mm. A solution of bromine (0.5 ml, 10.16 mmol) in acetic acid (3 ml) was added dropwise to the reaction at rt. The reaction mixture was stirred at rt for 16 h. The resulting solid precipitates were collected byfiltration under reduced pressure, washed with acetic acid (10 ml) and dried under vacuum. The obtained precipitates were suspended in ice cold aqueous solution of NH4OH (10 ml) and stirred at rt for 30 mm. The resulting solid precipitates were collected by filtration under reduced pressure, dried under vacuum yielding 2-aminobenzo[djthiazole-6-carbonitrile (0.70 g, 4.00 mmol). This material was directly used in the next step without further purification. LCMS: Method C, 1.62 mi MS: ES+176.13.
Reference:
[1] Heterocycles, 2006, vol. 68, # 11, p. 2285 - 2299
[2] Journal of Medicinal Chemistry, 2009, vol. 52, # 6, p. 1744 - 1756
[3] Journal of Organic Chemistry, 2017, vol. 82, # 18, p. 9312 - 9320
[4] Journal of Medicinal Chemistry, 1999, vol. 42, # 15, p. 2828 - 2843
[5] Organic Letters, 2010, vol. 12, # 15, p. 3567 - 3569
[6] European Journal of Medicinal Chemistry, 2014, vol. 71, p. 24 - 30
[7] Chemical Biology and Drug Design, 2014, vol. 84, # 1, p. 123 - 129
[8] Medicinal Chemistry, 2013, vol. 9, # 4, p. 596 - 607
[9] Research on Chemical Intermediates, 2015, vol. 41, # 8, p. 5599 - 5609
[10] Bioorganic and Medicinal Chemistry Letters, 2015, vol. 25, # 23, p. 5561 - 5565
[11] Patent: WO2017/103614, 2017, A1, . Location in patent: Page/Page column 81
5
[ 873-74-5 ]
[ 19759-66-1 ]
Reference:
[1] Farmaco, 2004, vol. 59, # 4, p. 297 - 305
[2] Patent: US2003/153568, 2003, A1,
[3] Patent: US2003/153568, 2003, A1,
[4] European Journal of Medicinal Chemistry, 2012, vol. 53, p. 41 - 51
[5] Heteroatom Chemistry, 2012, vol. 23, # 4, p. 399 - 410
[6] Medicinal Chemistry Research, 2013, vol. 22, # 1, p. 195 - 210
[7] Journal of Chemical Research, 2014, vol. 38, # 10, p. 611 - 616
[8] Journal of Medicinal Chemistry, 2016, vol. 59, # 21, p. 9814 - 9824
6
[ 540-72-7 ]
[ 873-74-5 ]
[ 19759-66-1 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 1998, vol. 46, # 4, p. 623 - 630
Reference:
[1] Journal of Organic Chemistry, 2018, vol. 83, # 23, p. 14472 - 14488
9
[ 288-13-1 ]
[ 873-74-5 ]
[ 25699-83-6 ]
Reference:
[1] Chemistry - A European Journal, 2014, vol. 20, # 45, p. 14619 - 14623
10
[ 873-74-5 ]
[ 49584-26-1 ]
Reference:
[1] Collection of Czechoslovak Chemical Communications, 2004, vol. 69, # 7, p. 1479 - 1490
[2] Organic Process Research and Development, 2009, vol. 13, # 5, p. 875 - 879
[3] Organic and Biomolecular Chemistry, 2010, vol. 8, # 23, p. 5324 - 5332
11
[ 873-74-5 ]
[ 6393-40-4 ]
Reference:
[1] Journal of Medicinal Chemistry, 1995, vol. 38, # 10, p. 1786 - 1792
[2] Australian Journal of Chemistry, 1994, vol. 47, # 2, p. 247 - 262
[3] Journal of Medicinal Chemistry, 1993, vol. 36, # 12, p. 1746 - 1753
[4] Journal of Medicinal Chemistry, 2014, vol. 57, # 16, p. 6973 - 6988
[5] Journal of the Brazilian Chemical Society, 2018, vol. 29, # 6, p. 1304 - 1317
[6] Patent: US4018790, 1977, A,
12
[ 821-48-7 ]
[ 873-74-5 ]
[ 68104-63-2 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 1998, vol. 8, # 12, p. 1531 - 1536
With N-chloro-succinimide In acetonitrile at 90℃; for 2 h;
Step 1: To a stirred solution of 4-aminobenzonitrile (10.0 g, 84.7 mmol) in MeCN (100 mL) at 90 °C was slowlyadded N-chlorosuccinimide (12.4 g, 93 mmol). After the addition of N-chlorosuccinimide, the reaction mixture was stirredat 90 °C for 2 h. The reaction mixture was then cooled to rt and concentrated under reduced pressure. The residue wasdissolved in 500 mL of CH2Cl2 and washed with 5percent aq NaOH. The organic layer was dried over MgSO4 and concentratedunder reduced pressure to give 4-amino-3-chlorobenzonitrile as a tan solid (12.2 g, 95percent). 1H NMR (300 MHz, CDCl3)δ 7.54 (d, J= 1.7 Hz, 1H), 7.35 (dd, J= 1.8, 8.4 Hz, 1H), 6.77 (d, J = 8.5 Hz, 1H), 4.63 (br s, 2H).
Reference:
[1] Synlett, 1999, # 12, p. 1984 - 1986
[2] Patent: EP2766359, 2016, B1, . Location in patent: Paragraph 0383
[3] Journal of Organic Chemistry, 2017, vol. 82, # 14, p. 7529 - 7537
[4] Organic Letters, 2008, vol. 10, # 1, p. 113 - 116
[5] Synthesis, 1985, # 6/7, p. 669 - 670
15
[ 873-74-5 ]
[ 21803-75-8 ]
[ 20925-27-3 ]
Yield
Reaction Conditions
Operation in experiment
95%
With N-chloro-succinimide In dichloromethane; acetonitrile
3-chloro-4-cyanoaniline. PREPARATION 2 (2-chloro-4-cyanoaniline) To a stirred, 60° C. solution of 4-cyanoaniline (20 g, 0.169 mol) in acetonitrile (200 mL) was added slowly N-chlorosuccinimide (24.8 g, 0.186 mol) to keep the reaction at reflux. After the addition of the N-chlorosuccinimide was complete, the reaction mixture was stirred at 60° C. for two hours. The solvent was then removed under reduced pressure. The residue was then dissolved in dichloromethane, washed with a 5percent sodium hydroxide solution, dried (Na2 SO4) and evaporated under reduced pressure to afford 24.5 g (95percent) of the title compound, 2-chloro-4-cyanoaniline, as a tan solid; m.p. 93°-95° C.
Reference:
[1] Patent: US5034410, 1991, A,
16
[ 873-74-5 ]
[ 17672-27-4 ]
Yield
Reaction Conditions
Operation in experiment
5.2 g
Stage #1: With hydrogenchloride; sodium nitrite In water at -15 - 10℃; for 0.25 h; Stage #2: With hydrogenchloride; tin(ll) chloride In water at -15℃; for 0.5 h;
To a cold solution of 4-cyanoaniline (6.0 g, 0.050 mol) in cone. HCl was added aq. solution of sodium nitrite (3.85 g, 0.055 mmol) at -15°C. The reaction mass was stirred at 0-10°C for 15 minutes and filtered off to remove insolubles. The filtrate was added to stannous chloride in cone. HCl (24.0 g, 0.166 mmol). The reaction mass was stirred at -15°C for 30 minutes. The reaction mass was filtered to afford 5.2 g of desired product.1H NMR (300 MHz, DMSO d6): δ 7.04 (d, 2H), 7.70 (d, 2H), 9.17 (br s, 1H), 10.66 (br s, 2H).
Reference:
[1] Organic Letters, 2015, vol. 17, # 24, p. 6258 - 6261
[2] Journal of the American Chemical Society, 1944, vol. 66, p. 1849
[3] Journal of Organic Chemistry, 2012, vol. 77, # 23, p. 10699 - 10706
[4] Patent: WO2013/186692, 2013, A1, . Location in patent: Page/Page column 73
[5] Molecules, 2016, vol. 21, # 11,
17
[ 873-74-5 ]
[ 17672-27-4 ]
Yield
Reaction Conditions
Operation in experiment
78%
With sodium nitrite In hydrogenchloride; water
1. 4-Cyanophenylhydrazine. Hydrochloride To a cooled (-15° C.) and stirred suspension of 4-aminobenzonitrile (50 g, 423 mmol) in concentrated hydrochloric acid (550 ml) was added dropwise a solution of sodium nitrite (31.5 g, 457 mmol) in water (200 ml) at such a rate as to maintain the temperature below -10° C. After the addition was finished, the reaction mixture was quickly filtered to remove solids and the filtrate was added portionwise to a cooled (-20° C.) and stirred solution of tin (II) chloride dihydrate (477 g, 2.1 mol) in concentrated hydrochloric acid (370 ml) at such a rate as to maintain the temperature below -10° C. After further 15 minutes at -10° to 0° C., the white precipitate was collected by filtration, washed with diethyl ether (4*250ml) and dried to give 56 g (78percent) of the title compound; mp 235°-237° C. (ethanol-water 1:1); 1 H NMR (250MHz, D6 -DMSO) δ10.50 (3H, br s, -N+H3) 9.10 (1H, br s, --NH--), 7.71 (2H, d, J=8.8Hz, Ar--H), 7.03 (2H, d, J=8.8Hz, Ar--H); m/z (CI) 132 (M+ -1).
78%
With sodium nitrite In hydrogenchloride; water
1.4-Cyanophenylhydrazine. Hydrochloride To a cooled (-15° C.) and stirred suspension of 4-aminobenzonitrile (50 g, 423 mmol) in concentrated hydrochloric acid (550 ml) was added dropwise a solution of sodium nitrite (31.5 g, 457 mmol) in water (200 ml) at such a rate as to maintain the temperature below -10° C. After the addition was finished, the reaction mixture was quickly filtered to remove solids and the filtrate was added portionwise to a cooled (-20° C.) and stirred solution of tin (II) chloride dihydrate (477 g, 2.1 mol) in concentrated hydrochloric acid (370 ml) at such a rate as to maintain the temperature below -10° C. After further 15 minutes at -10° to 0° C., the white precipitate was collected by filtration, washed with diethyl ether (4*250 ml) and dried to give 56 g (78percent) of the title compound; mp 235°-237° C. (ethanolwater 1:1); 1 H NMR (250 MHz, DMSO-d6) δ 10.50 (3H, br s, --N+ H3), 9.10 (1H, br s, --NH--), 7.71 (2H, d, J=8.8 Hz, Ar-H), 7.03 (2H, d, J=8.8 Hz, Ar-H); m/z (CI) 132 (M+ - 1).
Reference:
[1] Patent: US5968967, 1999, A,
[2] Patent: US5514682, 1996, A,
[3] Patent: US5298520, 1994, A,
18
[ 873-74-5 ]
[ 3032-92-6 ]
Reference:
[1] Journal of Materials Chemistry, 2010, vol. 20, # 43, p. 9775 - 9786
19
[ 873-74-5 ]
[ 78473-00-4 ]
Reference:
[1] Patent: US5202356, 1993, A,
20
[ 7722-84-1 ]
[ 873-74-5 ]
[ 78473-00-4 ]
Reference:
[1] Patent: US4515800, 1985, A,
[2] Patent: US4517199, 1985, A,
21
[ 873-74-5 ]
[ 58633-04-8 ]
Yield
Reaction Conditions
Operation in experiment
74%
With bromine In 1,4-dioxane; sodium hydroxide
EXAMPLE 25 PREPARATION OF 4-AMINO-3,5-DIBROMOBENZONITRILE STR37 To a stirred solution of 100 mg (0.847 mmoles) of p-aminobenzonitrile in 3.6 mL dioxane chilled in an ice-bath was added sequentially 356 μL (1.78 moles) of 5N sodium hydroxide solution and mg (1.78 mmoles) of bromine. The ice-water bath was removed and the reaction mixture was stirred further for 1.5 hours. After this time, 21.8 μL (0.423 mmoles) of bromine was added to drive the reaction to completion and stirring was continued for 10 minutes. The mixture was partitioned between ethyl acetate and ice-water and the organic phase was separated. It was washed with brine, dried over anhydrous sodium sulfate, filtered, and evaporated. Purification by plate layer chromatography using hexane-ethyl acetate (7:3) as the eluant provided 175 mg (74percent) of the entitled product. NMR(CDCl3) δ5.1 (bs, 2H), 7.66 (s, 2H).
Reference:
[1] RSC Advances, 2016, vol. 6, # 93, p. 90184 - 90187
[2] Journal fuer Praktische Chemie (Leipzig), 1986, vol. 328, # 4, p. 497 - 514
[3] Journal of Organic Chemistry, 1998, vol. 63, # 5, p. 1555 - 1565
[4] Journal of the American Chemical Society, 1960, vol. 82, p. 3454 - 3456
[5] Synthesis (Germany), 2013, vol. 45, # 11, p. 1497 - 1504
[6] Patent: US5455239, 1995, A,
22
[ 873-74-5 ]
[ 58633-04-8 ]
Yield
Reaction Conditions
Operation in experiment
74%
With bromine In 1,4-dioxane; sodium hydroxide
EXAMPLE 1 Preparation of 4-Amino-3,5-dibromobenzonitrile STR15 To a stirred solution of 100 mg (0.847 mmoles) of p-aminobenzonitrile in 3.6 mL dioxane chilled in an ice-bath was added sequentially 356 μL (1.78 mmoles) of 5 N sodium hydroxide solution and 284 mg (1.78 mmoles) of bromine. The ice-water bath was removed and the reaction mixture was stirred further for 1.5 hours. After this time, 21.8 μL (0.423 mmoles) of bromine was added to drive the reaction to completion and stirring was continued for 10 minutes. The mixture was partitioned between ethyl acetate and ice-water and the organic phase was separated. It was washed with brine, dried over anhydrous sodium sulfate, filtered, and evaporated. Purification by plate layer chromatography using hexane-ethyl acetate (7:3) as eluant provided 175 mg (74percent) of the entitled product. NMR(CDCl3) δ: 5.1 (bs, 2H), 7.66 (s, 2H).
Reference:
[1] Patent: US5192758, 1993, A,
23
[ 420-04-2 ]
[ 873-74-5 ]
[ 5637-42-3 ]
Yield
Reaction Conditions
Operation in experiment
33 g
Stage #1: With nitric acid In ethanol; water for 16 h; Reflux Stage #2: With sodium hydroxide In water
A solution of P-aminobenzonitrile (100 gm), ethanol (500 ml), concentrated nitric acid (36 ml) and aqueous cyanamide (50percent, 54 ml) was heated at reflux. The solution was maintained for 16 hours at reflux. The reaction mass was further cooled to 0° C. and then added methyl tert-butyl ether (500 ml) at 0 to 5° C. The reaction mass was maintained for 5 hours at 0 to 5° C. and separated solid obtained was collected by filtration to obtain 59 gm of guanidine nitrate. Guanidine nitrate (59 gm) was dissolved in water (590 ml) and then added sodium hydroxide solution (1M, 325 ml). The separated solid obtained was filtered and dried to obtain 33 gm of 1-(4-cyanophenyl)guanidine.
30 g
With nitric acid In methanol; water at 10 - 65℃; for 8 h;
100 g of 4-aminobenzonitrile was dissolved in 500 mL of methanol and cooled the reaction mixture to 10-15° C. 161 mL of con. Nitric acid was added to the reaction mixture followed by 65.6 ml of 50percent aqueous solution cynamide to the reaction mixture and maintained the reaction at 65° C. for 8 hrs. Cooled the reaction mass to 0° C. and charged 500 ml of methyl-t-butyl ether at 0° C. The solids were filtered, washed with water and acetone and dried to give 30 g of the product as a solid.
Reference:
[1] European Journal of Medicinal Chemistry, 2010, vol. 45, # 1, p. 244 - 255
[2] Patent: WO2012/1695, 2012, A1, . Location in patent: Page/Page column 10
[3] Patent: US2013/96148, 2013, A1, . Location in patent: Paragraph 0085; 0086
[4] Patent: US2015/336900, 2015, A1, . Location in patent: Paragraph 0027; 0028
24
[ 873-74-5 ]
[ 21803-75-8 ]
[ 20925-27-3 ]
Yield
Reaction Conditions
Operation in experiment
95%
With N-chloro-succinimide In dichloromethane; acetonitrile
3-chloro-4-cyanoaniline. PREPARATION 2 (2-chloro-4-cyanoaniline) To a stirred, 60° C. solution of 4-cyanoaniline (20 g, 0.169 mol) in acetonitrile (200 mL) was added slowly N-chlorosuccinimide (24.8 g, 0.186 mol) to keep the reaction at reflux. After the addition of the N-chlorosuccinimide was complete, the reaction mixture was stirred at 60° C. for two hours. The solvent was then removed under reduced pressure. The residue was then dissolved in dichloromethane, washed with a 5percent sodium hydroxide solution, dried (Na2 SO4) and evaporated under reduced pressure to afford 24.5 g (95percent) of the title compound, 2-chloro-4-cyanoaniline, as a tan solid; m.p. 93°-95° C.
Reference:
[1] Patent: US5034410, 1991, A,
25
[ 887144-94-7 ]
[ 873-74-5 ]
[ 327-74-2 ]
Yield
Reaction Conditions
Operation in experiment
67%
With potassium carbonate; nickel(II) hydroxide In dimethyl sulfoxide at 35℃; for 2 h;
The preparation method of the trifluoromethyl aromatic amine of the present embodiment, the aromatic amine is p-cyanoaniline, the reaction time is 12 h, and the other reaction and post-treatment processes are the same as in the embodiment 28. The preparation method of the trifluoromethyl aromatic amine of the present embodiment, the aromatic amine is aniline, and the nickel compound is nickel hydroxide.The base is potassium carbonate, and the reaction process parameters are: 1-trifluoromethyl-1,2-phenyliodo-3(H)-one (0.5 mmol, 1.0 eq).Aromatic amine (1.5 mmol, 3.0 eq), nickel hydroxide 10 molpercent, potassium carbonate (1.5 mmol, 3.0 eq),DMSO (2 mL) was reacted at 35 ° C for 2 h, and the other reactions and workup procedures were the same as in Example 1.
Reference:
[1] Organic Letters, 2018, vol. 20, # 13, p. 3732 - 3735
[2] Patent: CN108503552, 2018, A, . Location in patent: Paragraph 0126-0130
[3] Organic Letters, 2014, vol. 16, # 6, p. 1768 - 1771
26
[ 2314-97-8 ]
[ 873-74-5 ]
[ 327-74-2 ]
Yield
Reaction Conditions
Operation in experiment
61%
With fac-tris(2-phenylpyridinato-N,C2')iridium(III); potassium carbonate In 1,2-dichloro-ethane at 20℃; for 24 h; Inert atmosphere; Schlenk technique; Irradiation
General procedure: A 25 mL of Schlenk tube equipped with a magnetic stir bar were charged with aniline (1.2 mmol, 3.0 equiv) or heterocycles (0.8 mmol, 2.0 equiv), K2CO3 (0.8 mmol, 2.0 equiv) and fac-Ir(ppy)3 (2.6 mg, 0.004 mmol, 1 mol percent), under air. The vessel was evacuated and backfilled with Ar (3 times), CF3I stock solution (0.56 mL, 0.71 mmol/mL in 1,2-chloroethane or 0.36 mL, 1.11 mmol/mL in DMSO, 1.0 equiv), anhydrous 1,2-dichloroethane (3 mL) were then added. The tube was screw capped and stirred at room temperature under irradiation of blue LEDs (12 W) for 24 hours. The reaction S15 mixture was filtered through a pad of Celite and washed with ethyl acetate (3×5 mL). The filtrate was concentrated. The residue was subjected to column chromatography on silica gel to afford the pure product.
Reference:
[1] Journal of Organic Chemistry, 2014, vol. 79, # 19, p. 8984 - 8989
28
[ 79-08-3 ]
[ 873-74-5 ]
[ 42288-26-6 ]
Yield
Reaction Conditions
Operation in experiment
88%
at 100 - 110℃; for 3 h;
3.2.3 Preparation of [(4-cyanophenyl)amino]acetic acid (V) C7H6N2 C2H3DPψ2 C9H8N2O2 MoI. Wt.: 1 18,14 MoI. Wt.: 138,95 MoI. Wt.: 176,17VIngredientsF: 90 g - 0.75 molG: 211.7 g - 1.5 molSodium bicarbonate: 35 g, 0.42 molThe starting substances F and G were mixed in 1250 ml of water and this suspension was inserted into a bath heated up to 100-110 0C. After three hours of heating the reaction container was removed from the bath, cooled in the fridge and the separated substance was sucked off. The substance was dried in a vacuum drier at the temperature of 100 °C. Yield: Crude product: 122 g (92.8percent), HPLC: 97percentThe crude product was purified by conversion to the sodium salt and re-acidification using an aqueous solution of sodium bicarbonate. The acid was released by means of diluted hydrochloric acid (1 :1). After sucking off and washing with water the product was dried in a vacuum drier (105 °C). Yield: Purified product: 115 g (88percent), HPLC: 99.1percent, water content: 0.13percent; sulphate ash: 1.8percent
Reference:
[1] Patent: WO2009/111997, 2009, A1, . Location in patent: Page/Page column 11
[2] Patent: WO2013/111163, 2013, A2, . Location in patent: Page/Page column 18
[3] Patent: US2015/11589, 2015, A1, . Location in patent: Paragraph 0121-0122
[4] European Journal of Medicinal Chemistry, 2016, vol. 120, p. 148 - 159
29
[ 79-11-8 ]
[ 873-74-5 ]
[ 42288-26-6 ]
Yield
Reaction Conditions
Operation in experiment
73%
Reflux
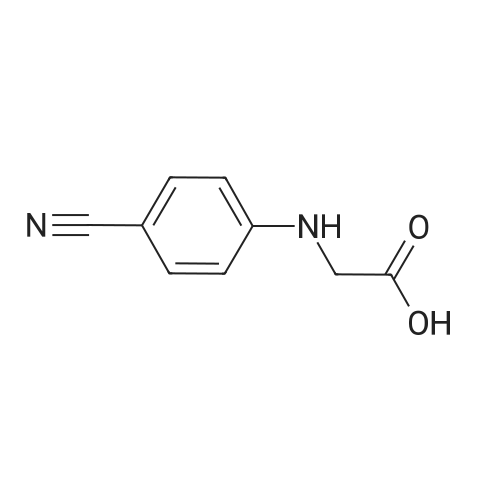
In the 4-cyanato aniline (6.0g, 0 . 05mol) and 1-chloro acetic acid (10g, 0.11mol) adding water in 150 ml, heating to reflux until a large amount of yellow solid is separated out so far, the filter at room temperature, with water, anhydrous ethanol, anhydrous ethyl ether eluviation, that is, to get the yellow solid 4-cyanato anilino-acetic acid (6.4g, yield 73percent). Mass spectrometric (ESI-MS): 177.3(M+H) +, 199.2(M+Na) +; C 9 H 8 N 2 O 2 (176).
58%
Heating / reflux
a) (4-Cyano-phenylamino)-acetic acid A solution of 4-aminobenzonitrile (12 g, 101.6 mmol) and chloroacetic acid (20 g, 211.6 mmol) in water (250 mL) was refluxed until the product began to separate out. After cooled down to the room temperature, the solids were collected by filtration, washed with ether, and dried in vacuo to afford 10.35 g (58percent) of the title compound which was pure enough for the next reaction. 1H NMR (400 MHz, DMSO-d6) δ 12.73 (s, 1H), 7.46 (dd, 2H), 6.93 (t, 1H), 6.65 (dd, 2H), 3.91 (d, 2H).
Reference:
[1] Patent: CN103524559, 2016, B, . Location in patent: Paragraph 0096; 0102-0104
[2] Patent: US2005/107355, 2005, A1, . Location in patent: Page/Page column 30
[3] Journal of Organic Chemistry, 2014, vol. 79, # 16, p. 7772 - 7777
[4] Journal of Organic Chemistry, 1957, vol. 22, p. 78
30
[ 3926-62-3 ]
[ 873-74-5 ]
[ 42288-26-6 ]
Yield
Reaction Conditions
Operation in experiment
73.2%
for 6.5 h; Reflux
e) 2-(4-Cyanophenylamino)acetic acid (compound III) 77.4 g (0.66 mol) of 4-aminobenzonitrile and 150 g (1.31 mol) of sodium chloroacetate were suspended in 1.1 L of water, and the resulting mixture was stirred at reflux temperature for 6.5 hours. After cooling to 0°C, the resulting suspension was stirred at this temperature overnight. The solid was filtered and washed with 200 mL of water. The resulting solid was suspended in 200 mL of ethyl acetate and stirred at room temperature for 1 hour. The solid was filtered, washed with 400 mL of ethyl acetate and dried at 60°C under vacuum for 5 hours to yield 84.5 g of 2-(4-cyanophenylamino)acetic acid as an off-white solid. Yield: 73.2 percent. Purity (HPLC, method 3): 98.4 percent.
73.2%
at 0℃; Reflux
e) 2-(4-Cyanophenylamino)acetic acid (compound III) 77.4 g (0.66 mol) of 4-aminobenzonitrile and 150 g (1.31 mol) of sodium chloroacetate were suspended in 1.1 L of water, and the resulting mixture was stirred at reflux temperature for 6.5 hours. After cooling to 0°C, the resulting suspension was stirred at this temperature overnight. The solid was filtered and washed with 200 mL of water. The resulting solid was suspended in 200 mL of ethyl acetate and stirred at room temperature for 1 hour. The solid was filtered, washed with 400 mL of ethyl acetate and dri e d at 60 ° C under v acuum for 5 hours to yield 84.5 g of 2-(4- cyanophenylamino)acetic acid as an off-white solid. Yield: 73.2 percent. Purity (HPLC, method 3): 98.4 percent.
131 g
With tetrabutylammomium bromide; sodium hydrogencarbonate; potassium iodide In water at 90 - 95℃; for 24 h;
Sodium bicarbonate (21.35 g) was added to a mixture of 4-aminobenzonitrile compound of formula-12 (100 g) and water (1000 ml) followed by sodium 2-chloroacetate (197.42 g). Potassium iodide (5 g) followed by tertiary butyl ammonium bromide (2.5 g) were added to the reaction mixture. The reaction mixture was heated to the 90-95° C. and stirred for 24 hours at the same temperature. After completion of the reaction, the reaction mixture was cooled to 20-25° C. and pHwas adjusted to 7.5 with ammonia. The reaction mixture was stirred for 20 minutes at 20-30° C. Filtered the reaction mixture and ethylacetate was added to the filtrate. The reaction mixture was stirred for 15 minutes. Both the ethylacetate and aqueous layers were separated and the pHof aqueous layer was adjusted to 2.5 using hydrochloric acid. The reaction mixture was stirred for 3 hours at 20-30° C. to precipitate the solid. Filtered the precipitated solid, water followed by hydrochloric acid were added to the obtained solid and stirred for 4 hours at 25-30° C. Filtered the solid, the obtained solid was slurried twice in water for 30-45 minutes and then dried to get the title compound. The same process can be repeated one more time to eliminates the impurities if present. Yield: 131 g
120 g
With tetrabutylammomium bromide; sodium hydrogencarbonate; potassium iodide In water at 85 - 90℃; for 24 h;
Sodium mono chloroacetate (197.42 g), followed by potassium iodide (5 g) and tetrabutyl ammonium bromide (2.5 g) were added to a mixture of 4-aminobenzonitrile (100 g), water (1000 ml) and sodium bicarbonate (42.71 g). The reaction mixture was heated to 85-90° C. and stirred for 24 hours. The reaction mixture was cooled to 25-30° C. and treated with ammonia followed by hydrochloric acid. Filtered the solid, washed with water and then dried to get title compound. Yield: 120 g.
Reference:
[1] Journal of Medicinal Chemistry, 1997, vol. 40, # 18, p. 2843 - 2857
[2] Bioorganic and Medicinal Chemistry Letters, 1996, vol. 6, # 1, p. 81 - 86
32
[ 873-74-5 ]
[ 17626-40-3 ]
Reference:
[1] Journal of Medicinal Chemistry, 1993, vol. 36, # 12, p. 1746 - 1753
33
[ 873-74-5 ]
[ 50397-74-5 ]
Reference:
[1] Synthetic Communications, 2010, vol. 40, # 21, p. 3226 - 3232
[2] Tetrahedron Letters, 2000, vol. 41, # 13, p. 2083 - 2085
[3] Journal of the Chilean Chemical Society, 2011, vol. 56, # 4, p. 863 - 865
[4] Journal of the Iranian Chemical Society, 2012, vol. 9, # 3, p. 321 - 326
[5] Tetrahedron Letters, 2007, vol. 48, # 7, p. 1255 - 1259
[6] Journal of Medicinal Chemistry, 2005, vol. 48, # 18, p. 5823 - 5836
[7] Journal of the Chemical Society, 1956, p. 368,370
[8] Chemistry Letters, 2003, vol. 32, # 2, p. 114 - 115
[9] Advanced Synthesis and Catalysis, 2008, vol. 350, # 13, p. 2052 - 2058
[10] Journal of the American Chemical Society, 2011, vol. 133, # 18, p. 6868 - 6870
[11] Chemistry - A European Journal, 2011, vol. 17, # 49, p. 13665 - 13669
[12] Angewandte Chemie - International Edition, 2012, vol. 51, # 8, p. 1958 - 1961
[13] Synthesis (Germany), 2013, vol. 45, # 11, p. 1497 - 1504
[14] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 23, p. 6363 - 6369
[15] Advanced Synthesis and Catalysis, 2017, vol. 359, # 7, p. 1144 - 1151
[16] Chinese Journal of Chemistry, 2018, vol. 36, # 9, p. 815 - 818
34
[ 873-74-5 ]
[ 2863-98-1 ]
Yield
Reaction Conditions
Operation in experiment
84%
With hydrogenchloride; tin(II) chloride dihdyrate; sodium nitrite In water at -5 - 0℃; for 0.25 h;
To a cooled (−5 to 0°C) stirred suspension of 4-aminobenzonitrile (50g, 423mmol) in concentrated hydrochloric acid (550mL) was added dropwise aqueous sodium nitrite solution (31.5g, 457mmol in 200mL water). To this diazotised solution cooled (0°C) solution of tin (II) chloride dihydrate (477g, 2.1mol) in concentrated hydrochloric acid (370mL) was added while stirring and maintaining the temperature below 0°C. The resulting solution was further stirred for 15min. White precipitates so formed were collected by filtration, washed with diethylether and crystallized from aqueous ethanol. Mp 234–236°C (Lit. mp 235–237°C), yield 60g (84percent).
Reference:
[1] Journal of the American Chemical Society, 2016, vol. 138, # 30, p. 9377 - 9380
[2] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 15, p. 4581 - 4590
[3] Synthetic Communications, 2014, vol. 44, # 24, p. 3563 - 3571
[4] Journal of Medicinal Chemistry, 1994, vol. 37, # 19, p. 3023 - 3032
[5] Angewandte Chemie - International Edition, 2013, vol. 52, # 47, p. 12426 - 12429[6] Angew. Chem., 2013, vol. 125, # 47, p. 12652 - 12656,4
[7] Journal of Agricultural and Food Chemistry, 2014, vol. 62, # 2, p. 381 - 390
[8] Journal of the Chinese Chemical Society, 2018, vol. 65, # 5, p. 538 - 547
35
[ 873-74-5 ]
[ 61033-86-1 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2014, vol. 87, p. 372 - 385
Reference:
[1] Journal of the American Chemical Society, 2012, vol. 134, # 30, p. 12466 - 12469
[2] Journal of the American Chemical Society, 2012, vol. 134, # 43, p. 18147 - 18147,1
38
[ 873-74-5 ]
[ 68827-43-0 ]
Reference:
[1] Journal of Medicinal Chemistry, 1993, vol. 36, # 12, p. 1746 - 1753
39
[ 13675-18-8 ]
[ 873-74-5 ]
[ 126747-14-6 ]
Reference:
[1] Chemistry - A European Journal, 2014, vol. 20, # 22, p. 6608 - 6612
[2] Journal of Organic Chemistry, 2014, vol. 79, # 21, p. 10568 - 10580
40
[ 873-74-5 ]
[ 126747-14-6 ]
Reference:
[1] Chemistry - A European Journal, 2014, vol. 20, # 22, p. 6608 - 6612
41
[ 873-74-5 ]
[ 33348-34-4 ]
Yield
Reaction Conditions
Operation in experiment
88%
With dihydrogen peroxide; iodine In methanol at 20℃; for 48 h;
To a solution of 4-aminobenzonitrile (2.4 g 20 mmol) and -30percent H202 (not titrated before use) in MeOH (30 ml) a solution of l2 (5.05 g, 12 mmol) in MeOH (50 ml) was added at room temperature and the resulting mixture was stirred for 48 h, while a fresh H202 (2 ml) was added every day. The mixture was concentrated under reduced pressure and treated with saturated solution of Na2S203 until most of the colour disappeared. The solid formed was filtered off, diluted to 300 ml with EtOAc, washed with saturated, H20, brine, dried over anhydrous MgS04, filtered and the filtrate evaporated to dryness. The residue was purified by crystallization from EtOH to give the title compound (3 g). The residue was recrystallised from the mixture of CH2CI2/hexane to give more of the title compound (1 .4 g). Total yield 4.4 g (88percent). 1 H NMR (CDCIg) 7.87 (d, 1 H, J = 1 .8 Hz) ; 7.37 (dd, 1 H, J = 1 .8, 8.4), 6.68 (d, 1 H, J = 8.4 Hz), 4.62 (broad s, 2H).
36%
With iodine; silver sulfate In ethanol at 20℃; for 18 h;
INTERMEDIATE 56 - PREPARATION of 4-Amino-3-iodobenzonitrile. Iodine (0.645 g; 2.54 mmol) was added to a stirred mixture of silver sulphate (0.791 g;2.54 mmol) and 4-aminobenzonitrile (0.300 g; 2.54 mmol) in ethanol (10 ml_). The reaction mixture was then stirred at room temperature for 18 hours and filtered over celite. The volatiles were removed under reduced pressure and the residue was partitioned between ethyl acetate and a saturated aqueous solution of sodium thiosulfate. The organic layer was washed with brine, dried and concentrated under reduced pressure. The crude residue was purified by flash chromatography on silica gel (eluent 2 to 40 percent ethyl acetate in heptane) to afford 0.222 g (36percent) of the title compound as a white solid.1 H NMR (DMSO-de) δ 7.96 (d, 1 H), 7.45 (dd, 1 H), 6.76 (d, 1 H), 6.22 (s, 2H).
36%
With iodine; silver sulfate In ethanol at 20℃; for 18 h;
Iodine (0.645 g; 2.54 mmol) was added to a stirred mixture of silver sulphate (0.791 g; 2.54 mmol) and 4-aminobenzonitrile (0.300 g; 2.54 mmol) in ethanol (10 mL). The reaction mixture was then stirred at room temperature for 18 hours and filtered over celite. The volatiles were removed under reduced pressure and the residue was partitioned between ethyl acetate and a saturated aqueous solution of sodium thiosulfate. The organic layer was washed with brine, dried and concentrated under reduced pressure. The crude residue was purified by flash chromatography on silica gel (eluent 2 to 40percent ethyl acetate in heptane) to afford 0.222 g (36percent) of the title compound as a white solid. [0654] 1H NMR (DMSO-d6) δ 7.96 (d, 1H), 7.45 (dd, 1H), 6.76 (d, 1H), 6.22 (s, 2H).
36%
With iodine; silver sulfate In ethanol at 20℃; for 18 h;
INTERMEDIATE 142 - PREPARATION of 4-amino-3-iodobenzonitrile; Iodine (0.645 g; 2.54 mmol) was added to a stirred mixture of silver sulphate (0.791 g; 2.54 mmol) and 4-aminobenzonitrile (0.300 g; 2.54 mmol) in ethanol (10 ml_). The reaction mixture was stirred at room temperature for 18 hours and filtered over celite. The volatiles were removed under reduced pressure and the residue was partitioned between ethyl acetate and a saturated aqueous solution of sodium thiosulphate.. The organic layer was washed with brine, dried and concentrated under reduced pressure. The crude residue was purified by flash chromatography on silica gel (eluent 2 to 40 percent ethyl acetate in heptane) to afford 0.222 g (36percent) of the title compound as a white solid. 1H NMR (DMSO-de) δ 7.96 (d, 1 H), 7.45 (dd, 1 H), 6.76 (d, 1 H), 6.22 (s, 2H).
76%
With Iodine monochloride In water; acetic acid
EXAMPLE 10 Synthesis of 4-Amino-3-iodobenzonitrile (31) To a solution of 5.9 g (0.05 mole) of 4-aminobenzonitrile (30) (Aldrich 14,775-3) in 25 mL of glacial acetic acid was added dropwise a solution of 8.12 g (0.05 mole) of iodine monochloride (Aldrich 20,822-1) in 5 mL of glacial acetic acid. During the addition, the temperature rose to 40°. The solution was stirred at room temperature for 20 minutes. A solid developed in the reaction mixture and the deep brown color of the solution started fading gradually. The mixture was poured into 250 mL of water and stirred for 10 minutes to give a pale brown solid which was filtered and recrystallized from methanol/water containing one gram of activated charcoal (Darco S51) yielding 9.3 g (76percent) of white crystals of 31, mp 110-112°. Infrared (IR) and NMR analysis gave the following results: IR (potassium bromide): 3454 and 3346 (NH2), 2214 (CN), 1621, 1496 cm1. 1H nmr (90 MHz, CDCI3):δ 7.91 (1H; d, 4JH6-H2=1.8 Hz; H-2), 7.41 (1H; dd, 3JH5-H6=8.4, 4JH2-H6=1.8 Hz; H-6), 6.73 (1 H; d, 3JH6-H5=8.4 Hz, H-5), 4.67 (2H, br s, NH2).
10 g
With N-iodo-succinimide In N,N-dimethyl acetamide at 45 - 50℃; for 72 h;
Step 1 : Preparation of 4-amino-3-iodobenzonitrile To a solution of 4-aminobenzonitrile (10 g) in DMA (50 mL) was added N- iodosuccinamide (19.80 g).The reaction mixture was stirred at 45-50°C for 72 h. The reaction mass was quenched in water and obtained solid was filtered to afford 10 g of title product.
10 g
With N-iodo-succinimide In N,N-dimethyl acetamide at 45 - 50℃; for 72 h;
To a solution of 4-aminobenzonitrile (10 g) in DMA (50 mL) was added N-iodosuccinamide (19.80 g).The reaction mixture was stirred at 45-50°C for 72 h. The reaction mass wasquenched in water and obtained solid was filtered to afford 10 g of title product.
Reference:
[1] Journal of the American Chemical Society, 2008, vol. 130, # 43, p. 14339 - 14345
[2] European Journal of Organic Chemistry, 2001, # 24, p. 4607 - 4613
[3] Organic Letters, 2005, vol. 7, # 13, p. 2543 - 2546
[4] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 9, p. 2722 - 2725
[5] Patent: WO2014/63199, 2014, A1, . Location in patent: Page/Page column 56
[6] Angewandte Chemie - International Edition, 2018, vol. 57, # 25, p. 7528 - 7532[7] Angew. Chem., 2018, vol. 130, # 25, p. 7650 - 7654,5
[8] Asian Journal of Chemistry, 2011, vol. 23, # 1, p. 41 - 43
[9] Journal of Medicinal Chemistry, 2002, vol. 45, # 16, p. 3497 - 3508
[10] Bioorganic and Medicinal Chemistry, 2009, vol. 17, # 24, p. 8149 - 8160
[11] Journal of Medicinal Chemistry, 2005, vol. 48, # 18, p. 5823 - 5836
[12] Chemical Communications, 2017, vol. 53, # 24, p. 3481 - 3484
[13] Journal of Medicinal Chemistry, 1994, vol. 37, # 21, p. 3655 - 3662
[14] Patent: WO2012/80221, 2012, A1, . Location in patent: Page/Page column 89
[15] Patent: US2013/274260, 2013, A1, . Location in patent: Paragraph 0653-0654
[16] Patent: WO2010/142801, 2010, A1, . Location in patent: Page/Page column 190
[17] Journal of the Chemical Society - Perkin Transactions 1, 1999, # 4, p. 529 - 534
[18] Bioorganic and Medicinal Chemistry Letters, 2002, vol. 12, # 15, p. 2019 - 2022
[19] Bioorganic and Medicinal Chemistry Letters, 2003, vol. 13, # 24, p. 4385 - 4388
[20] Journal of Organic Chemistry, 2002, vol. 67, # 18, p. 6395 - 6405
[21] Angewandte Chemie - International Edition, 2007, vol. 46, # 12, p. 2074 - 2077
[22] Patent: US6482982, 2002, B1,
[23] Journal of Organic Chemistry, 2015, vol. 80, # 21, p. 10806 - 10816
[24] Synthesis (Germany), 2016, vol. 48, # 6, p. 855 - 864
[25] Patent: WO2016/46782, 2016, A1, . Location in patent: Page/Page column 139; 138
[26] Patent: WO2016/55947, 2016, A1, . Location in patent: Page/Page column 122
With hydrogenchloride In tetrahydrofuran; ethanolReflux
To a solution of S3 (0.13 g, 0.52mmol) in THF:EtOH(1:3, mL) was added S4 (0.07 g, 0.56 mmol) and 1N HCl (1.04 ml, 1.04mmol). The reaction was refluxed overnight and evaporated. The crude product was purified by flash chromatography on silica (50percentEtOAc inhexanes) to afford a white powder (0.07 g, 42percent).1H NMR (CDCl3, 400 MHz) δ 7.99 (d, J= 4.0 Hz, 1H), 7.56-7.76 (m, 5H), 7.00 (s, 2H), 6.53 (brs, 1H), 5.56 (br s, 1H), 2.36 (s, 3H), 2.26 (s, 6H). MS (ESI) m/z 330 [M+H]+. HPLC (214 nm) tr 18.5 min, 100 percent
Reference:
[1] Bioorganic and Medicinal Chemistry, 2014, vol. 22, # 19, p. 5241 - 5248
44
[ 873-74-5 ]
[ 244767-67-7 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 18, p. 5213 - 5216
45
[ 873-74-5 ]
[ 14544-47-9 ]
Reference:
[1] Journal of Materials Chemistry A, 2018, vol. 6, # 34, p. 16655 - 16663
[2] Angewandte Chemie - International Edition, 2018, vol. 57, # 51, p. 16754 - 16759[3] Angew. Chem., 2018, vol. 130, p. 16996 - 17001,6
[4] RSC Advances, 2016, vol. 6, # 33, p. 28047 - 28054
[5] Journal of Materials Chemistry A, 2017, vol. 5, # 44, p. 22933 - 22938
[6] Journal of Materials Chemistry A, 2018, vol. 6, # 2, p. 374 - 382
[7] Chemical Communications, 2018, vol. 54, # 61, p. 8450 - 8453
[8] Patent: WO2004/106311, 2004, A2, . Location in patent: Page 109-110
[9] Chinese Journal of Chemistry, 2015, vol. 33, # 1, p. 90 - 94
[10] Chemical Communications, 2015, vol. 51, # 49, p. 10050 - 10053
[11] Chemical Communications, 2016, vol. 52, # 22, p. 4128 - 4131
[12] ChemSusChem, 2017, vol. 10, # 5, p. 921 - 929
[13] Langmuir, 2018, vol. 34, # 2, p. 685 - 692
[14] Chemical Communications, 2018, vol. 54, # 81, p. 11475 - 11478
[15] Journal of Materials Chemistry A, 2018, vol. 6, # 40, p. 19532 - 19541
46
[ 873-74-5 ]
[ 219763-85-6 ]
Reference:
[1] Patent: US6348474, 2002, B1,
47
[ 873-74-5 ]
[ 219763-87-8 ]
Reference:
[1] Patent: US6348474, 2002, B1,
48
[ 873-74-5 ]
[ 315228-79-6 ]
Reference:
[1] Patent: EP2766359, 2016, B1,
49
[ 38446-95-6 ]
[ 873-74-5 ]
[ 333986-52-0 ]
Yield
Reaction Conditions
Operation in experiment
47%
Stage #1: With sodium tris(acetoxy)borohydride; acetic acid In tetrahydrofuran at 0 - 20℃; for 3.25 h; Stage #2: With sodium hydrogencarbonate In tetrahydrofuran; water; ethyl acetate
Preparation 31 tert-butyl 4-[(4-cyanophenyl)amino]piperidine-1-carboxylate To a mixture of 4-aminobenzonitrile (0.5g, 4.23mmol) and tert-butyl 4-oxocyclohexanecarboxylate (1.26g, 6.35mmol) in THF (5ml) at 0°C was added acetic acid (0.5ml, 8.5mmol) and sodium triacetoxyborohydride (1.35g, 6.35mmol). The mixture was stirred at this temperature for 15min and at room temperature for 3h. Ethyl acetate and 5percent solution of NaHCO3 were added and the organic layer separated, washed with water, brine and dried over magnesium sulphate. The solvent was concentrated and the residue purified by column chromatography with a mixture of hexane/ethyl acetate (from 5/1 to 1/1) to give the desired compound (yield=47percent). LRMS: m/z 302 (M+1)+ Retention time: 6.33 min (Method B) 1H NMR (300 MHz, CHLOROFORM-d) d ppm 1.37 (m, 2 H) 1.47 (s, 9 H) 2.03 (m, 2 H) 2.93 (t, J=11.95 Hz, 2 H) 3.40 - 3.54 (m, 1 H) 4.04 - 4.13 (m, 3 H) 6.56 (d, J=9.06 Hz, 2 H) 7.43 (d, J=8.79 Hz, 2 H)
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 23, p. 6950 - 6954
51
[ 873-74-5 ]
[ 80945-83-1 ]
Reference:
[1] Patent: WO2007/110364, 2007, A1,
52
[ 873-74-5 ]
[ 1000577-94-5 ]
Reference:
[1] Angewandte Chemie - International Edition, 2012, vol. 51, # 8, p. 1958 - 1961
53
[ 10132-07-7 ]
[ 873-74-5 ]
[ 1398507-08-8 ]
Yield
Reaction Conditions
Operation in experiment
73%
at 20 - 102℃; for 21 h;
Example 7: Preparation of 4-(4-amino-6-chloropyrimidin-2-ylamino)benzonitrile ("Compound la")To a solution of 4-aminobenzonitrile (ABN; 7 g; 59.3 mmol) in 2-butanol (190 ml), 2,6- dichloropyrimidin-4-amine ("DCAP"; 10.2 g; 62.2 mmol) was added at room temperature. The resulting suspension was heated to reflux (102 °C) and stirred for 20 hours. The suspension was then cooled to 20-25 °C and stirred for lhour at 20-25 °C. A solid was separated from the suspension by filtration and washed with 2-butanol (20 ml) and dried (2h/ 50 °C/ 10 mbar). Yield: 12.20 g (73.0 percent) of 4-(4-amino-6-chloropyrimidin-2-ylamino)benzonitrile hydrochloride.Purity (HPLC/ MS): 99.0 Area percent
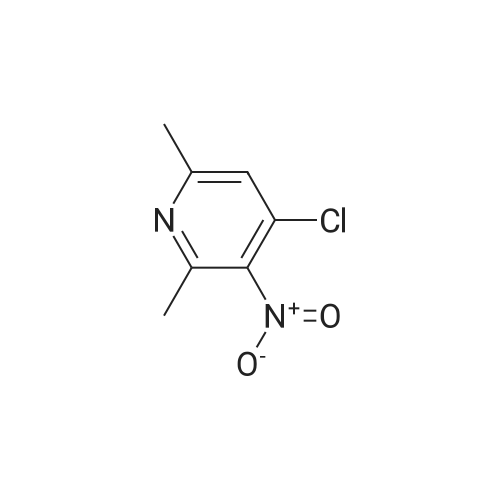
(c) 4-(4-Cyanophenyl)amino-2,6-dimethyl-3-nitropyridine A solution of <strong>[15513-48-1]4-chloro-2,6-dimethyl-3-nitropyridine</strong> (9.80 g, 52.5 mmol) and 4-aminobenzonitrile (6.20 g, 52.5 mmol) in ethanol (160 ml) was stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and the residue was dissolved in dichloromethane (200 ml), and washed with saturated aqueous sodium bicarbonate (100 ml). The organic phase was dried (MgSO4) and concentrated under reduced pressure to give a gum which was crystallized by adding ether (100 ml) and sonicating for 5 minutes.
In ethanol; dichloromethane;
(c) 4-[N-(4-Cyanophenyl)amino]-2,6-dimethyl-3-nitropyridine A solution of <strong>[15513-48-1]4-chloro-2,6-dimethyl-3-nitropyridine</strong> (see part (b)) (9.80 g, 52.5 mmol) and 4-aminobenzonitrile (6.20 g, 52.5 mmol) in ethanol (160 ml) was stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and the residue was dissolved in dichloromethane (200 ml) and washed with saturated aqueous sodium bicarbonate (100 ml). The organic phase was dried (MgSO4) and concentrated under reduced pressure to give a gum which was crystallized by adding ether (100 ml) and sonicating for 5 minutes. The title compound was obtained as a yellow solid which was filtered off and dried in vacuo, (9.80 g, 70percent) m.p. 171°-172° C. 1 H-NMR (300 MHz, CDCl3): delta=2.49 (3H, s), 2.76 (3H, s), 6.96 (1H, s), 7.35 (2H, d, J 8 Hz), 7.74 (2H, d, J 8 Hz), 8.69 (1H, br s) p.p.m.
In ethanol; dichloromethane;
(c) 4-(4-Cyanophenyl)amino-2,6-dimethyl-3-nitropyridine A solution of <strong>[15513-48-1]4-chloro-2,6-dimethyl-3-nitropyridine</strong> (9.80 g, 52.5 mmol) and 4-aminobenzonitrile (6.20 g, 52.5 mmol) in ethanol (160 ml) was stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and the residue was dissolved in dichloromethane (200 ml), and washed with saturated aqueous sodium bicarbonate (100 ml). The organic phase was dried (MgSO4) and concentrated under reduced pressure to give a gum which was crystallized by adding either (100 ml) and sonicating for 5 minutes. The yellow solid (9.80 g, 70percent) was filtered off and dried in vacuo , mp 171-172°C. 1H NMR (300 MHz, CDCl3) 2.49 (3H, s), 2.76 (3H, s), 6.96 (1H, s), 7.35 (2H, d, J 8Hz), 7.74 (2H, d, J 8Hz), (1H, br s).
With N-chloro-succinimide; In acetonitrile; at 90℃; for 2h;
Step 1: To a stirred solution of 4-aminobenzonitrile (10.0 g, 84.7 mmol) in MeCN (100 mL) at 90 C was slowlyadded N-chlorosuccinimide (12.4 g, 93 mmol). After the addition of N-chlorosuccinimide, the reaction mixture was stirredat 90 C for 2 h. The reaction mixture was then cooled to rt and concentrated under reduced pressure. The residue wasdissolved in 500 mL of CH2Cl2 and washed with 5% aq NaOH. The organic layer was dried over MgSO4 and concentratedunder reduced pressure to give 4-amino-3-chlorobenzonitrile as a tan solid (12.2 g, 95%). 1H NMR (300 MHz, CDCl3)delta 7.54 (d, J= 1.7 Hz, 1H), 7.35 (dd, J= 1.8, 8.4 Hz, 1H), 6.77 (d, J = 8.5 Hz, 1H), 4.63 (br s, 2H).
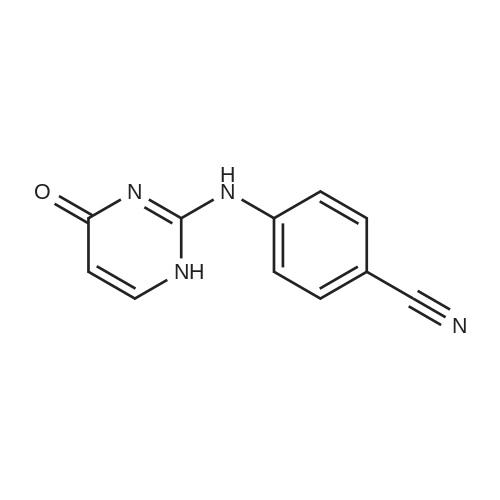
4-((6-oxo-1,6-dihydropyrimidin-2-yl)amino)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
73.6%
at 180℃; for 8h;Inert atmosphere;
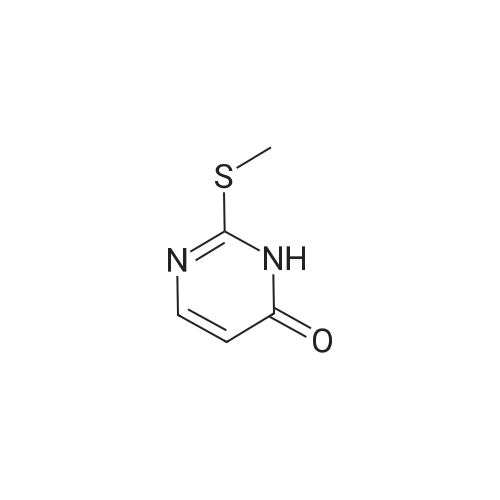
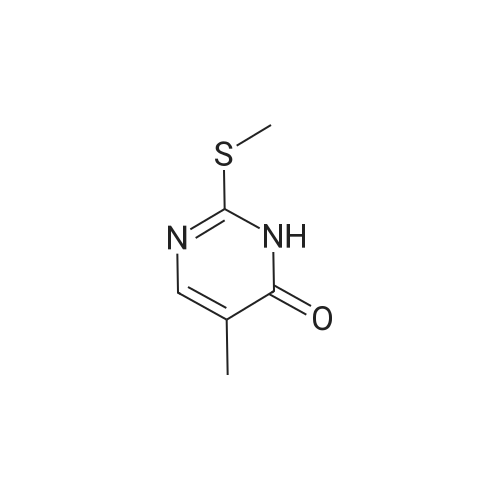
2- (methylthio) pyrimidin-4 (3H) -one (3 g, 21 mmol) was weighed4-aminobenzonitrile (2.99 g, 25 mmol)In a 50 mL round bottom flask, nitrogen protected,Slowly warmed to 180 ° C for 8 h.After cooling the reaction, 20 mL of acetonitrile was added,Filtration, filter cake with acetonitrile washing, TLC detection without 4-aminobenzonitrile residue,Dry the filter cake to obtain a pale yellow solid4 - ((4-oxo-1,6-dihydropyrimidin-2-yl) amino) benzonitrile was obtained in 73.6percent
73.6%
at 180℃; for 8h;Inert atmosphere;
2-(Methylthio)pyrimidin-4(3H)-one (3 g, 21 mmol) and 4-aminobenzonitrile (2.99 g, 25 mmol) were weighed in a 50 mL round bottom flask.Nitrogen protection,Slowly warm up to 180 ° C,Reaction 8h.After the reaction was cooled, it was sonicated by adding 20 mL of acetonitrile, filtered, and the filter cake was washed with acetonitrile, and the residue of 4-aminobenzonitrile was not detected by TLC, and the cake was dried to give a pale yellow solid which was 4-((4-oxo-1,6) - dihydropyrimidin-2-yl)amino)benzonitrile, yield 73.6percent,
With bromine; acetic acid; In water; at 10 - 60℃; for 7h;
4-Aminobenzonitrile (11.8 g, 100 mmol) was dissolved in 100 ml of acetic acid and potassium thiocyanate (29.1 g,300 mmol) was cooled to about 10 C, and liquid bromine (16 g, 100 mmol) was slowly added dropwise. After the addition, the mixture was allowed to warm to room temperature and stirred for 1 hour.The temperature was raised to 60 C and the reaction was continued for 6 hours.Concentrate under reduced pressure, distill off most of the acetic acid, add 100 mL of water, 200 ml of ethyl acetate and stir for 0.5 hour.Filtering, washing,Drying to give the intermediate A 2-amino-6-cyanobenzothiazole(12.4g, yield 71%).
General procedure: A mixture of 0.1mol of appropriate 4-substituted amines and 0.1mol of Potassium thiocyanate (KCNS) in 100mL glacial acetic acid (AcOH) were cooled in an ice bath and stirred for 10-20min, and then 0.1mol bromine in glacial acetic acid was added dropwise at such a rate to keep the temperature below 10C throughout the addition. The reaction mixture was stirred at room temperature for 2-4h, the hydrobromide (HBr) salt thus separated out was filtered, washed with acetic acid, dried, dissolved in hot water and basified to pH 11.0 with ammonia solution (NH4OH) and the resulting precipitate was filtered, washed with water and dried to get the desired product 2a-k. The progress of the reaction was monitored by Thin Layer Chromatography using toluene:acetone (8:2) solvent system.
General procedure: To 100 mL glacial acetic acid, 0.1 mol of appropriate para-substituted amine derivative and equimolecular amount of potassium thiocyanate was added and reaction mixture was kept in an ice bath and cooled. The mixture was allowed to cool at this temperature up to 20 min. Then, in the reaction mixture, bromine in glacial acetic acid (0.1 mol) was added very slowly to maintain the temperature of the reaction mixture below 10 C and was stirred at room temperature for 2 to 4 h to obtain the hydrobromide (HBr) salt. The salt was then filtered, washed with acetic acid, dried on vacuum oven and basified to pH 11.0 with NH3·H2O. The solid precipitate thus formed was filtered, washed with water and dried on vacuum oven to yield the intermediates 3a-k. TLC technique involving toluene : acetone (V/V, 8:2) solvent system was used to monitor the reaction progress [26].
General procedure: The appropriate para-substituted amine derivative (1a-j; 0.1 mol) and an equimolecular amount of potassium thiocyanate were added to 100 mL glacial acetic acid, with cooling of the reaction mixture in an ice bath. The mixture was left at this temperature for up to 20 min then bromine (0.1 mol) in glacial acetic acid was added very slowly, to maintain the temperature of the reaction mixture below 10 C, then the mixture was stirred at room temperature for 2-4 h to furnish the hydrobromide (HBr) salt. The salt was then isolated by filtration, washed with acetic acid, dried in a vacuum oven, then dissolved in sufficient aqueous ammonia solution to ensure the pH was 11.0. The solid precipitate thus formed was filtered, washed with water, and dried in a vacuum oven to yield the intermediates 4a-j. The progress of the reaction was monitored by TLC with toluene-acetone 8:2 as mobile phase.
With bromine; In acetic acid;Cooling with ice;
General procedure: A mixture of 0.1 mol of 4-substituted aniline and 0.1 mol ofPotassium thiocyanate (KCNS) in 100 ml glacial acetic acid (AcOH) was cooled inan ice bath and stirred for 10 to 20 min, and then 0.1 mol bromine in glacialacetic acid was added dropwise at such a rate to keep the temperature below 10C throughout the addition. Theprogress of the reaction was monitored by Thin Layer Chromatography usingtoluene: acetone (8:2) solvent system. The reaction mixture was stirredat room temperature for 2 to 4 hrs, the hydrobromide (HBr) salt thus separatedout was filtered, washed with acetic acid, dried, dissolved in hot water and basified to pH 11.0 with ammonia solution (NH4OH)and the resulting precipitate was filtered, washed with water and dried to getthe desired product 2a-q.
0.70 g
To a solution of 4-aminobenzonitrile (1.00 g, 8.47 mmol) in acetic acid (12 ml) was added potassium thiocyanate (1.00 g, 16.9 mmol) at 10C. The reaction mixture was stirred at rt for 30 mm. A solution of bromine (0.5 ml, 10.16 mmol) in acetic acid (3 ml) was added dropwise to the reaction at rt. The reaction mixture was stirred at rt for 16 h. The resulting solid precipitates were collected byfiltration under reduced pressure, washed with acetic acid (10 ml) and dried under vacuum. The obtained precipitates were suspended in ice cold aqueous solution of NH4OH (10 ml) and stirred at rt for 30 mm. The resulting solid precipitates were collected by filtration under reduced pressure, dried under vacuum yielding 2-aminobenzo[djthiazole-6-carbonitrile (0.70 g, 4.00 mmol). This material was directly used in the next step without further purification. LCMS: Method C, 1.62 mi MS: ES+176.13.
With tris(2,2'-bipyridyl)ruthenium dichloride; In acetonitrile; at 20℃; for 18h;Irradiation;
General procedure: A mixture ofaniline 1 (1 mmol), NH4SCN 2 (1.0 mmol), ruthinium (2 mol%), and CH3CN (3mL) was taken in a flask open to air and stirred at rt for 10-18 h (Table 2). Aftercompletion of the reaction (monitored by TLC), water (5 mL) was added andthe mixture was extracted with ethyl acetate (3 5 mL). The combinedorganic phase was dried over anhydrous Na2SO4, filtered, and evaporatedunder reduced pressure. The resulting crude product was purified by silica gelchromatography using a mixture of hexane/ethyl acetate (4:1) as eluent toafford an analytically pure sample of product 2. All the compounds 2 areknown and were characterized by comparison of their spectral data with thosereported in the literature.
56%
Step A: 2-Chlorobenzothiazole-6-carbonitrile; To a mixture of 4-aminobenzonitrile (23.6 g, 0.2 mol) and ammonium rhodanate (30.4 g, 0.4 mol) was added glacial acetic acid (600 ml.) and the resulting solution was cooled to 13.5 0C on an ice-bath. A mixture of bromine and glacial acetic acid was added drop-wise and slowly. The resulting mixture was stirred for 1 hour at 13.5 0C and filtered. The filter cake was washed with glacial acetic (6 x 100 ml.) and placed in hot water (1000 ml.) with stirring. The mixture was filtered and pH of the filtrate was adjusted to 7 with a saturated sodium carbonate solution. The precipitate was isolated and dried to give 19.5 g (56 %) of 2-aminobenzo- thiazole-6-carbonitrile. A mixture of concentrated hydrochloric acid (1 13 ml.) and water (52 ml.) was heated at 90 0C while 2-aminobenzothiazole-6-carbonitrile (19 g, 0.108 mol) was added. The mixture was cooled to -5 0C on an ice-bath and a solution of sodium nitrite (7.72 g, 0.1 12 mol) in water (20 ml.) was added drop-wise, keeping the temperature below than 0 0C. When addition was complete the mixture was stirred for 0.5 hour and a solution of CuCI2 (16 g) in water (108 ml.) was added drop-wise. When addition was complete the mixture was stirred for 10 min, and the ice-bath was removed. Stirring was continued for 2 h and the mix- ture was cooled to room temperature. The mixture was filtered and the solid was washed to neutrality with water and dried. This afforded 12.8 g (61 %) of 2-chlorobenzothiazole-6-carbo- nitrile. 1H-NMR delta 8.14 (s, 1 H), 8.03 (d, 1 H), 7.75 (d, 1 H).
General procedure: The appropriate 4-substituted aniline (11; 20 mmol), glacial acetic acid (20 mL) and ammonium thiocyanate (40 mmol) were cooled in an ice bath and stirred for 10 min. A solution of bromine (20 mmol) in AcOH (20 mL) was then added over 20 min. The reaction mixture was stirred for 21 h at room temperature. The benzothiazole hydrobromide salt was filtered, washed with acetic acid and dried. It was subsequently dissolved in hot water and neutralized with aqueous ammonia solution (25%), filtered, washed with water anddried under vacuum, recrystallized with aqueous ethanol to obtain 6-substituted 1,3-benzothiazol-2-amine (12) in good yield,
2-Amino-1,3-benzothiazole-6-carbonitrile: 4-Aminobenzonitrile is dissolved in acetic acid (or a weak protic acid) and the solution is cooled to about 16-30 C., preferably 16-18 C. Potassium thiocyanate is added and the flask is then equipped with an addition funnel. The addition funnel is charged with bromine and acetic acid. This dark solution is then added to the benzonitrile solution in a dropwise fashion under good agitation and allowed to stir for about 12-20 hours, preferrably about 16 hours. The slurry is then drowned into water and filtered. The presscake is washed well with water, reslurried in dilute aqueous alkali and filtered. Again the presscake is washed well with water to obtain the title compound.
With bromine; In acetic acid;
Preparation 1 2-Amino-1,3-benzothiazole-6-carbonitrile Two grams of 4-aminobenzonitrile was dissolved in about 40 mL acetic acid and the solution was cooled to about 16 C. About 3.3g of potassium thiocyanate was added and the flask was equipped with an addition funnel. The addition funnel was charged with about 2.7 g bromine and about 5 mL acetic acid. This dark solution was then added to the benzonitrile solution in a dropwise fashion under good agitation and allowed to stir for about 16 hours. The slurry was then drowned into water and filtered. The presscake was washed well with water, reslurried in dilute aqueous alkali and filtered. Again the presscake was washed well with water. After drying in vacuo, about 2 grams of the title compound was isolated. 1H NMR 6.8 (d, 1H, J=8.7 Hz), 6.9 (br s, 2H), 7.6 (dd, 1H, J 2 Hz, J 8.7 Hz), 8.0 (d, 1H, J 2 Hz), LC/MS 2.34 min, 174 (M-H-), RP-HPLC RT 7.7 minutes.
With hydrogenchloride; In diethyl ether; hexane; acetone;
EXAMPLE A5 To a flask under argon was added <strong>[28969-60-0]4-amino-2,5,6-trichloropyrimidine</strong> (0.08564 mol), 4-amino-benzonitrile (0.1071 mol), 1-methyl-2-pyrrolidinone (17 ml) and HCl in diethylether (1M; 85.6 ml). The mixture was placed in an oil bath at 130 C. under a stream of nitrogen until the ether was gone. An additional 10 ml of 1-methyl-2-pyrrolidinone was added. The mixture was heated at 145 C. for 16 hours under argon. 1,4-Dioxane was added. The mixture was refluxed, cooled, then filtered. The filtrate was evaporated. The residue was dissolved in CH2C1-2, washed with 1 N NaOH, then filtered. The solid was dissolved in 2-propanone, evaporated onto silica gel, and chromatographed using 1-3% 2-propanone in hexane as eluent. The pure fractions were collected and the solvent was evaporated, yielding 1.63 g (6.8%) of 4-[(4-amino-5,6-dichloro-2-pyrimidinyl)amino]benzonitrile (interm. 12).
1.63 g (6.8%)
With hydrogenchloride; In diethyl ether; hexane; dichloromethane; acetone;
EXAMPLE A5 To a flask under Argon was added <strong>[28969-60-0]4-amino-2,5,6-trichloropyrimidine</strong> (0.08564 mol), 4-amino-benzonitrile (0.1071 mol), 1-methyl-2-pyrrolidinone (17 ml) and HCl in diethylether (1M; 85.6 ml). The mixture was placed in an oil bath at 130 C. under a stream of nitrogen until the ether was gone. An additional 10 ml of 1-methyl-2-pyrrolidinone was added. The mixture was heated at 145 C. for 16 hours under argon. 1,4-Dioxane was added. The mixture was refluxed, cooled, then filtered. The filtrate was evaporated. The residue was dissolved in CH2Cl2, washed with 1 N NaOH, then filtered. The solid was dissolved in 2-propanone, evaporated onto silica gel, and chromatographed using 1-3% 2-propanone in hexane as eluent. The pure fractions were collected and the solvent was evaporated, yielding 1.63 g (6.8%) of 4-[(4-amino-5,6-dichloro-2-pyrimidinyl)amino]benzonitrile (interm. 12).
With sodium hydroxide; sulfuryl dichloride; In chloroform;
EXAMPLE 15 (+-)-(2E,4E) N-(1,2-Dimethylpropyl)-5-[trans-2-(3,5-dichloro-4-bromophenyl)cyclopropyl]penta-2,4-dienamide (compound 61) P-Aminobenzonitrile (11.8 g) (ex Aldrich) in dry chloroform (250 ml) under nitrogen was treated with sulphuryl chloride (4.05 g) (ex BDH) maintaining reaction temperature below 35. After 2 hours at reflux the mixture was poured onto ice and made alkaline with 2M sodium hydroxide solution. Work up in the usual manner gave 3,5-dichloro-4-aminobenzonitrile (18.2 g) NMR 1 H 7.35(2H,s), 4.70(2H,bs).
With N-chloro-succinimide; In dichloromethane; acetonitrile;
3-chloro-4-cyanoaniline. PREPARATION 2 (2-chloro-4-cyanoaniline) To a stirred, 60 C. solution of 4-cyanoaniline (20 g, 0.169 mol) in acetonitrile (200 mL) was added slowly N-chlorosuccinimide (24.8 g, 0.186 mol) to keep the reaction at reflux. After the addition of the N-chlorosuccinimide was complete, the reaction mixture was stirred at 60 C. for two hours. The solvent was then removed under reduced pressure. The residue was then dissolved in dichloromethane, washed with a 5% sodium hydroxide solution, dried (Na2 SO4) and evaporated under reduced pressure to afford 24.5 g (95%) of the title compound, 2-chloro-4-cyanoaniline, as a tan solid; m.p. 93-95 C.
1. Preparation of 4-cyano-2,6-dichlorobenzamine. Reaction of 4-cyanobenzamine (10 g., 0.085 m, Aldrich Chem. Co.) with 292 ml of 6N HCl and 30% H2 O2 (17.2 mL, 0.17 m) led to the formation of a white crystalline compound with a melting point of 113-115 C. The yield of this compound was 12.3 g.
N-[5-(5,6-dimethoxy-3-methyl-1,4-benzoquinon-2-yl)methyl-2-acetoxybenzoyl]-4-cyanoaniline[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In dichloromethane;
Example 111 N-[5-(5,6-Dimethoxy-3-methyl-1,4-benzoquinon-2-yl)methyl-2-acetoxybenzoyl]-4-cyanoaniline 4-Cyanoaniline (0.189 g, 1.60 mmol), triethylamine (0.162 g, 1.60 mmol) and <strong>[37091-73-9]2-chloro-1,3-dimethylimidazolinium chloride</strong> (0.271 g, 1.60 mmol) were added to a methylene chloride solution (20 ml) of 5-(5,6-dimethoxy-3-methyl-1,4-benzoquinon-2-yl)methyl-2-acetoxybenzoic acid (0.200 g, 0.535 mmol) and the resulting solution was stirred at room temperature for 12 hours. The reaction solution was poured into ice water and then extracted with methylene chloride. The extract was washed with water and then dried, and the solvent was removed by distillation. The obtained residue was purified by preparative thin-layer chromatography (hexane: ethyl acetate = 1:2) to obtain the titled compound (0.141 g, 0.297 mmol, 56percent).
With sodium t-butanolate;palladium diacetate; 2'-dicyclohexylphosphanyl-6-hydroxy-biphenyl-3-sulfonic acid; In tert-butyl alcohol; for 30h;Heating / reflux;Product distribution / selectivity;
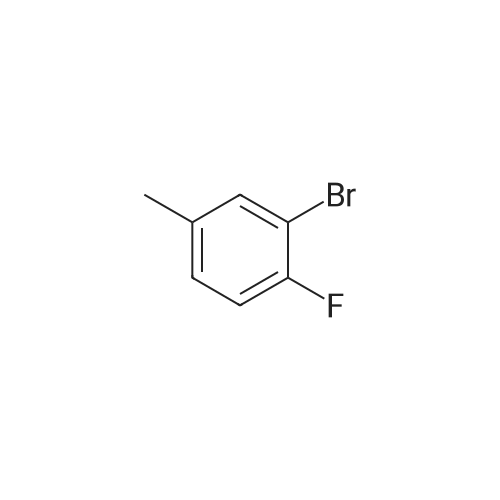
EXAMPLE 5Coupling of <strong>[452-62-0]3-bromo-4-fluorotoluene</strong> with 4-aminobenzonitrile to give 4'-cyano-2-fluoro-5-methyldiphenylamine; 189 mg (1 mmol) of <strong>[452-62-0]3-bromo-4-fluorotoluene</strong>, 118 mg of 4-aminobenzonitrile (1 mmol), 192 mg (2 mmol) of sodium tert-butoxide, 2.2 mg of palladium(II) acetate (1 mol %) and 4.5 mg of the HBPNS ligand (1 mol %) were heated to reflux in 6 ml of degassed tert-butanol for 30 h. After cooling, the reaction mixture was added to 10 ml of water and the mixture was extracted with 10 ml of toluene. To remove tert-butanol residues, the toluene phase was washed with 5 ml of water and concentrated on a rotary evaporator. After drying under reduced pressure, 178 mg (0.79 mmol, 79%) of the product were obtained.
A solution of P-aminobenzonitrile (100 gm), ethanol (500 ml), concentrated nitric acid (36 ml) and aqueous cyanamide (50%, 54 ml) was heated at reflux. The solution was maintained for 16 hours at reflux. The reaction mass was further cooled to 0C and then added methyl tert-butyl ether (500 ml) at 0 to 5C. The reaction mass was maintained for 5 hours at 0 to 5C and separated solid obtained was collected by filtration to obtain 59 gm of guanidine nitrate.Guanidine nitrate (59 gm) was dissolved in water (590 ml) and then added sodium hydroxide solution (1M, 325 ml). The separated solid obtained was filtered and dried to obtain 33 gm of l-(4-cyanophenyl)guanidine.
33 g
A solution of P-aminobenzonitrile (100 gm), ethanol (500 ml), concentrated nitric acid (36 ml) and aqueous cyanamide (50%, 54 ml) was heated at reflux. The solution was maintained for 16 hours at reflux. The reaction mass was further cooled to 0 C. and then added methyl tert-butyl ether (500 ml) at 0 to 5 C. The reaction mass was maintained for 5 hours at 0 to 5 C. and separated solid obtained was collected by filtration to obtain 59 gm of guanidine nitrate. Guanidine nitrate (59 gm) was dissolved in water (590 ml) and then added sodium hydroxide solution (1M, 325 ml). The separated solid obtained was filtered and dried to obtain 33 gm of 1-(4-cyanophenyl)guanidine.
30 g
With nitric acid; In methanol; water; at 10 - 65℃; for 8h;
100 g of 4-aminobenzonitrile was dissolved in 500 mL of methanol and cooled the reaction mixture to 10-15 C. 161 mL of con. Nitric acid was added to the reaction mixture followed by 65.6 ml of 50% aqueous solution cynamide to the reaction mixture and maintained the reaction at 65 C. for 8 hrs. Cooled the reaction mass to 0 C. and charged 500 ml of methyl-t-butyl ether at 0 C. The solids were filtered, washed with water and acetone and dried to give 30 g of the product as a solid.
General procedure: The mixture of 2-(methylthio)-pyrimidin-4(1H)-ones 12a-c (100 mmol) and 4-cyanoaniline (35.4 g, 300 mmol) was slowly heated to 180-190 C and maintained at this temperature for 8 h. After cooling, the hard mixture was crushed by ultrasound treatment in CH3CN (150 mL). Then the solid was filtered off and washed with CH3CN until there was no residue 4-cyanoaniline detected by TLC.
In neat (no solvent); at 180 - 200℃;Inert atmosphere;
General procedure: The 2-thiouracil derivatives (0.1 mol) were added into the sodium hydroxide solution (1.05 mol/L, 100 mL) at room temperature. After dissolution, the solution was slowly added with iodomethane (17.74 g, 0.125 mol) as methylating agent. After the reaction mixture was stirred for 24 h at rt, the precipitate was filtered off, washed with H2O, and dried to give 2-(methylthio)pyrimidin-4(1H)-ones 11a-c, which were used without further purification. Compound 11a-c (80 mmol) and 4-cyanoaniline (27.74 g, 240 mmol) were pounded into a fine powder and mixed thoroughly. Then the mixture was slowly heated to 180-200 C, and reacted in solvent-free condition at this temperature for about 8 h. After cooled to room temperature, the mixture was smashed in acetonitrile with ultrasonic machine. The precipitate was filtered off, washed with acetonitrile (3×10 mL) and then dichloromethane (2×10 mL). The product 12a-c were dried and used without further purification. The common blocks 12a-c (4 mmol) together with t-BuOK (0.45 g, 4 mmol) were then dissolved in DMSO (10 mL) and stirred for 15 min at room temperature. After that, the corresponding 13 (4 mmol) dissolved in DMSO (3 mL) was added slowly into the mixture. Stirring at room temperature for 2-6 h, the reaction was stopped when 13 was not detected by TLC. The mixture was poured into 100 ml H2O and neutralized with 3N HCl. The resulting target compounds 3a-w were collected by filtration and recrystallized from methanol and dichloromethane.
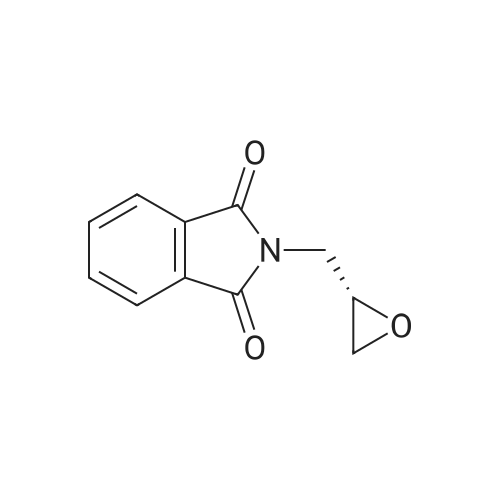
A mixture of 4-aminobenzonitrile (52.0 g, 0.440 mol) and (S)-glycidyl phthalimide (89.4 g, 0.440 mol) was stirred at 1050C for 13 hours. The reaction mixture was cooled to 600C, and the resulting solid with amorphous lump shape was pulverized. Then, 3.0 L of an isopropanol/methylene chloride solution (1:2) was added thereto, and stirred for 2 hours while refluxing (at the time, the solid with amorphous lump shape are dissolved and a light microcrystalline solid is formed). The suspension was distilled to remove 2L of methylene chloride while further adding 1.0 L of isopropanol thereto. Then, the suspension was kept at room temperature for 15 hours, and stirred at 00C for 1 hour, and filtered under a reduced pressure. The solid thus obtained was washed with 300 mL of isopropanol cooled to 00C and dried to obtain the compound (II) (122 g, 84%) as a light yellow solid.TLC : Rf = 0.3 (50% EtOAc/Hex)1H NMR (600 MHz, DMSO-d6) delta 7.90-7.82 (m, 4H), 7.45 (d, J = 7 Hz, 2H), 6.70 (d, J = 7 Hz, 2H), 4.07-4.14 (m, IH), 3.64-3.58 (m, 2H), 3.22-3.18 (m, IH), 3.13-3.17 (m, IH)
78%
With magnesium(II) perchlorate; In 1,4-dioxane; for 10h;Reflux;
A mixture of 4-aminobenzonitrile (9.7g, 82.1mmol), (S)-(+)-glycidyl phthalimide (20.0g, 98.5mmol) and Mg(ClO4)2 (0.2g, 0.82mmol) in 1,4-dioxane (150mL) was refluxed for 10h. After the completion of reaction, the solvent was removed and the residue was treated with ethyl acetate (100mL). The organic layer was washed with water (2×30mL) and evaporated to afford 8 (20.5g, 78% yield) as a yellow solid; MS (ESI) m/z (%): 322.1 [M+H]+.
With tris-(dibenzylideneacetone)dipalladium(0); sodium t-butanolate; DavePhos; In toluene; at 85℃;Inert atmosphere;
General procedure: Compound 8 (7.5 mmol), 9a-c (10.5 mmol), Pd2dba3 (0.19 mmol), 2'-(dicyclohexyl phosphino)-N,N-dimethylbiphenyl-2-amine (0.75 mmol) and NaOtBu (10.5 mmol) were weighed in a 50 mL RB flask and dry toluene (20 mL) was added and flushed with argon for 15 min. Subsequently the reaction mixture was stirred at 85 C overnight, cooled to room temperature, diluted with EtOAc and filtered through a pad of celite and concentrated to obtain a dark brown solid which was flash chromatographed on silica gel eluting with 30% EtOAc in hexanes.
With potassium tert-butylate; In N,N-dimethyl-formamide; at 20℃; for 1.0h;Cooling with ice;
General procedure: EXAMPLE 1 Preparation of 5-(4"-cyanovinyl-2",6"-dimethylphenoxy)-N1-(4'-cyanophenyl)-4-trifluoromethyl-2-nitroaniline (Compound 1) 4-cyano aniline (130 mg, 1.1 mmol) and <strong>[400-70-4]2,4-dichloro-5-nitrobenzotrifluoride</strong> (260 mg, 1 mmol) were used as starting materials. A general synthetic method of intermediate I was employed to get 5-chloro-N-(4-cyanophenyl)-2-nitro-4-trifluoromethylaniline (I-1) (281 mg) with a yield of 82%, as a yellow solid, and with a melting point of 180-182 C.; MS m/z (%) 359.2 (M+NH4, 100). I-1 and 4"-formyl-2",6"-dimethylphenol were coupled by Method A in Preparation Example 2. The product was dissolved in 15 mL of THF, and was reacted with (EtO)2P(O)CH2CN (1.5 mmol) and an equivalent mole of potassium t-butoxide in THF by Method C in Preparation Example 2 under stirring for about 3 h. The product obtained by conventional post-treatment was isolated and purified through medium pressure column (dichloromethane/methanol) to get Compound 1, with a melting point of 248-250 C. and a yield of 87%. 1H NMR delta ppm 2.15 (6H, s, 2*CH3), 5.85 (1H, d, J=16.4 Hz, =CH), 6.27 (1H, s, ArH-6), 7.10 (2H, d, J=8.8 Hz, ArH-2',6'), 7.21 (2H, s, ArH-3",5"), 7.32 (1H, d, J=16.4 Hz, CH=), 7.52 (2H, d, J=8.8 Hz, ArH-3',5'), 8.67 (1H, s, ArH-3), 9.91 (1H, s, NH); MS m/z (%) 501.1 (M+Na, 100).; Method A. 2, 4-dichloro-5-substituent nitrobenzene (1 equiv) and 4-cyanoaniline (1.1 equiv) were dissolved in DMF (3 ml), and under the condition of ice bath, potassium t-butoxide was added in batches, and the resultant solution was stirred for about 1 h at room temperature. The reaction solution was poured into ice-water, the pH was adjusted to about 6 with 5% aqueous hydrochloric acid solution, and yellow precipitates formed. The mixture was filtered, washed with water until it was neutral, dried, and isolated and purified through medium pressure column, to get pure intermediate I.
With toluene-4-sulfonic acid; In 1,4-dioxane;Inert atmosphere; Reflux;
A mixture of compound 2 (10 g, 69.2 mmol), 2,6-p-bromoaniline (8.18 g, 69.2 mmol) and p-toluenesulfonic acid(21.45 g, 124.56 mmol) was dissolved in 100 mL dioxane under reflux with nitrogen and refluxed overnight. The reaction system was cooled to room temperature, saturated sodium bicarbonate solution was added and extracted with ethyl acetate three times. The organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate,The residue was purified by column chromatography (petroleum ether: ethyl acetate = 6: 1) to give 9.8 g of compound 3 in a yield of 62%.
With toluene-4-sulfonic acid; at 100 - 110℃; for 14h;
Example 1:Preparation of 4-(4-methoxypyrimidin-2-ylamino)benzonitrile1,4-Dioxane (100 ml) was added to a mixture of <strong>[22536-63-6]<strong>[22536-63-6]2-chloro-4-methoxypyrimidin</strong>e</strong> (20 gm) as obtained in preparative example 2, 4-aminobenzonitrile (16.33 gm) and p- toluene sulfonic acid (42.12 gm). The mixture was then heated to 100 to 110C and stirred for 14 hours. The solution was then cooled to room temperature and basified with saturated sodium bicarbonate solution. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with water and then concentrated to obtain a residual solid. The residual solid obtained was slurried in isopropyl alcohol at room temperature for 30 minutes and filtered. The solid obtained was then dried to give 17.3 gm of 4-(4-methoxypyrimidin-2-ylamino)benzonitrile.
17.3 g
With toluene-4-sulfonic acid; In 1,4-dioxane; at 100 - 110℃; for 14h;
1 ,4-Dioxane (100 ml) was added to a mixture of <strong>[22536-63-6]<strong>[22536-63-6]2-chloro-4-methoxypyrimidin</strong>e</strong> (20 gm), 4-aminobenzonitrile (16.33 gm) and p-toluene sulfonic acid (42.12 gm). The mixture was then heated to 100 to 1 10C and stirred for 14 hours. The solution was then cooled to room temperature and basified with saturated sodium bicarbonate solution. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with water and then concentrated to obtain a residual solid. The residual solid obtained was slurried in isopropyl alcohol at room temperature for 30 minutes and filtered. The solid obtained was then dried to give 17.3 gm of 4-(4-methoxypyrimidin-2-ylamino)benzonitrile.
17.3 g
With toluene-4-sulfonic acid; In 1,4-dioxane; at 100 - 110℃; for 14h;
Example 1 Preparation of 4-(4-methoxypyrimidin-2-ylamino)benzonitrile [0080] 1,4-Dioxane (100 ml) was added to a mixture of <strong>[22536-63-6]<strong>[22536-63-6]2-chloro-4-methoxypyrimidin</strong>e</strong> (20 gm) as obtained in preparative example 2, 4-aminobenzonitrile (16.33 gm) and p-toluene sulfonic acid (42.12 gm). The mixture was then heated to 100 to 110 C. and stirred for 14 hours. The solution was then cooled to room temperature and basified with saturated sodium bicarbonate solution. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with water and then concentrated to obtain a residual solid. The residual solid obtained was slurried in isopropyl alcohol at room temperature for 30 minutes and filtered. The solid obtained was then dried to give 17.3 gm of 4-(4-methoxypyrimidin-2-ylamino)benzonitrile.
17.3 g
With toluene-4-sulfonic acid; In 1,4-dioxane; at 100 - 110℃; for 14h;
Step-III: Preparation of 4-(4-methoxypyrimidin-2-ylamino)benzonitrile 1,4-Dioxane (100 ml) was added to a mixture of <strong>[22536-63-6]<strong>[22536-63-6]2-chloro-4-methoxypyrimidin</strong>e</strong> (20 gm), 4-aminobenzonitrile (16.33 gm) and p-toluene sulfonic acid (42.12 gm). The mixture was then heated to 100 to 110 C. and stirred for 14 hours. The solution was then cooled to room temperature and basified with saturated sodium bicarbonate solution. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with water and then concentrated to obtain a residual solid. The residual solid obtained was slurried in isopropyl alcohol at room temperature for 30 minutes and filtered. The solid obtained was then dried to give 17.3 gm of 4-(4-methoxypyrimidin-2-ylamino)benzonitrile.
Example 7: Preparation of 4-(4-amino-6-chloropyrimidin-2-ylamino)benzonitrile ("Compound la")To a solution of 4-aminobenzonitrile (ABN; 7 g; 59.3 mmol) in 2-butanol (190 ml), 2,6- dichloropyrimidin-4-amine ("DCAP"; 10.2 g; 62.2 mmol) was added at room temperature. The resulting suspension was heated to reflux (102 C) and stirred for 20 hours. The suspension was then cooled to 20-25 C and stirred for lhour at 20-25 C. A solid was separated from the suspension by filtration and washed with 2-butanol (20 ml) and dried (2h/ 50 C/ 10 mbar). Yield: 12.20 g (73.0 %) of 4-(4-amino-6-chloropyrimidin-2-ylamino)benzonitrile hydrochloride.Purity (HPLC/ MS): 99.0 Area %
With toluene-4-sulfonic acid; In isopropyl alcohol; at 20 - 85℃; for 4h;
Example 5:Preparation of rilpivirineTo a mixture of (E)-3-( 4-(2-chloropyrimidin-4-ylamino )-3,5-dimethylphenyl)acrylonitrile ( 40 gm), 4-aminobenzonitrile (17 gm), isopropanol ( 400 ml)5 and p-toluenesulfonic acid (20 ml) were added at room temperature. The reaction mixturewas then heated to 80 to 85C and stirred for 4 hours. The reaction mass was then cooledto 0C and pH was adjusted to 10 to 11 with aqueous ammonia solution. The separatedsolid was filtered and then dried to provide 40 gm of rilpivirine.Chromatographic purity: 96.0%;10 Content of Z-isomer: 3.5%.
With palladium diacetate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; sodium t-butanolate; In 1,4-dioxane; at 110℃; for 16.0h;Inert atmosphere;
General procedure: Stock solution of 5 mol% Pd catalyst in dioxane was prepared as follows. A flask was charged with Pd(OAc)2 (0.025 mmol, 5.6 mg) and Xantphos (0.03 mmol, 17 mg); dry dioxane (5 mL) was added and the resulting solution was stirred for 15 min under an argon atmosphere. Next, a 50 mL round bottomed flask was charged with 2,5-dichloro-5-fluoropyridine (0.5 mmol, 83 mg), an aniline 2 (0.6 mmol) and t- BuONa (0.7 mmol, 67 mg). Freshly prepared stock solution of Pd catalyst was added and the resulting solution was stirred for 2 min under an argon atmosphere. The round bottomed flask was kept in 110 C preheated oil bath and reaction mixture was refluxed for 16 h, then allowed to cool down to room temperature and filtered through Celite. Celite cake was washed with 100 mL dichloromethane, combined organic phases were evaporated to dryness under reduced pressure and the residue separated with an automated chromatography system using Silica Flash Cartridges applying a heptane-ethyl acetate gradient (from 100% heptane to 100% ethylacetate in 35 minutes, 35 mL/min).
With hydrogenchloride; In 1,4-dioxane; 1-methyl-pyrrolidin-2-one; diethyl ether; at 130 - 145℃; for 16.0h;Inert atmosphere;
Example A5 To a flask under argon was added <strong>[28969-60-0]4-amino-2,5,6-trichloropyrimidine</strong> (0.08564 mol), 4-amino-benzonitrile (0.1071 mol), 1-methyl-2-pyrrolidinone (17 ml) and HCl in diethylether (1M; 85.6 ml). The mixture was placed in an oil bath at 130 C under a stream of nitrogen until the ether was gone. An additional 10 ml of 1-methyl-2-pyrrolidinone was added. The mixture was heated at 145 C for 16 hours under argon. 1,4-Dioxane was added. The mixture was refluxed, cooled, then filtered. The filtrate was evaporated. The residue was dissolved in CH2Cl2, washed with 1 N NaOH, then filtered. The solid was dissolved in 2-propanone, evaporated onto silica gel, and chromatographed using 1-3% 2-propanone in hexane as eluent. The pure fractions were collected and the solvent was evaporated, yielding 1.63 g (6.8%) of 4-[(4-amino-5,6-dichloro-2-pyrimidinyl)amino]benzonitrile (interm. 12).
(E)-4-(3-(4-cyanophenyl)triaz-1-enyl)benzimidamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
26%
General procedure: The synthesis of DMZ analogues followed previously reported procedure [19a], see Scheme 2. <strong>[2498-50-2]4-Aminobenzamidine dihydrochloride</strong> (212mg, 1.0mmol) was added to a stirred solution of 12N HCl (0.27mL) and water (1.5mL) in a 10mL flask at 0C and stirring was continued for 15min. To the mixture was added (dropwise) cold (?0C) NaNO2 solution (76mg in 0.27mL water, 1.1 eq.) and stirring was continued for 15min before cold (?0C) NaOAc solution (328mg in 1.5mL water, 4.0 eq.) was added dropwise over 15min to adjust the pH to 6.0. Cold (?0C) aromatic amine solution (1.0mmol in 1.0mL methanol) was added dropwise to the above solution and stirring was continued for another 1-12hat 0C. After the reaction was completed, the solvent was removed under reduced pressure. Water (100mL) was added to the residue and the aqueous mixture was washed with dichloromethane (2×15mL). The aqueous layer was then basified with 2.5% NaOH solution to make the pH>10.0. The desired compound was then extracted from the aqueous layer with ethyl acetate (2×100mL). The organic layer was washed with brine and dried with sodium sulfate. Finally, the solvent was removed under reduced pressure and the final product was obtained with purity >95%.
1 kg of compound of formula II and 692 g of p-aminophenyl cyanide were added to 8 L of pyridine, heated to 110 ° C, reacted for 8 h, cooled to 0 to 5 ° C, stirred for 10 h, filtered and washed with methanol to give Light yellow solid 1206 g, yield, 97percent
4-((5-bromo-2-nitropyridin-3-yl)amino)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78.6%
With palladium diacetate; caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; for 12h;Inert atmosphere;
Palladium acetate (0.1541g, 0.69mmol) and Xantphos (0.3977g, 0.69mmol) were dissolved in 15mL dioxane and stirred at ambient temperature for 15min. 4-Aminobenzonitrile (3.3g, 14.4mmol), intermediate 3 (3.0g, 13.8mmol) and Cs2CO3 (6.7g, 20.6mmol) were added. Then the reaction flask was evacuated and backfilled with nitrogen for three times, the resulting mixture was stirred at 100C for 12h. The mixture was filtrated, and the filtrate was collected and concentrated. The crude residue was purified by silica gel column chromatography to provide the title product 4 as a yellow solid. Yield: 78.6%, mp: 241-244C. 1H NMR (400MHz, DMSO-d6, ppm) delta: 9.43 (s, 1H, NH), 8.27 (d, J=1.9Hz, pyridine-H), 8.19 (d, J=2.0Hz, pyridine-H), 7.80 (d, J=8.7Hz, Ph-H), 7.41 (d, J=8.7Hz, Ph-H). 13C NMR (100MHz, DMSO-d6, ppm) delta: 145.33, 144.95, 140.96, 135.15, 134.27 (2×C), 132.30, 126.21, 120.77 (2×C), 119.54, 105.37. ESI--MS: m/z 317.2 (M-1), 319.2 (M+1). C12H7BrN4O2 (317.98).
2-methyl-propane-2-sulfinic acid [3-(4-cyano-phenylamino)-oxetan-3-yl]-amide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
67%
In methanol; at 60℃; for 6h;Inert atmosphere;
General procedure: To a stirred solution of substituted aniline (1 eq) in methanol (3 mL), <strong>[1158098-73-7]oxetan-3-tert-butylsulfinimine</strong> (1.5 eq) was added under argon atmosphere. Reaction mixture was heated at 60 C for appropriate time (Table 2). After the completion of reaction (TLC), reaction mixture was then concentrated. To the resulting residue water was added and then extracted with ethyl acetate (2 X 3 mL). Organic layer was then dried over anhydrous sodium sulphate and filtered. Solvent was removed under reduced pressure. The resulting products were then purified by chromatography using n-hexane-ethyl acetate (7:3) to afford pure 2-methyl-N-[(3-phenylamino)oxetan-3-yl]-2-propanesulfinamide derivatives. All synthesized compounds were characterized by IR, 1H-NMR, HRMS and 13C-NMR spectroscopic techniques.
With palladium diacetate; triphenylphosphine; sodium t-butanolate; In toluene; at 80℃; for 8h;Inert atmosphere;
5.91 g (50 mmol) of p-aminobenzonitrile, 19.87 g (50 mmol) of 2-bromo-9,9'-diphenylphosphonium, 9.60 g (100 mmol) of sodium tert-butoxide were dissolved in anargon atmosphere.In 500 ml of dehydrated toluene, 0.23 g(1 mmol) of palladium acetate and 0.20 g (1 mmol) of triphenylphosphinewere added under stirring, and the mixture wasreacted at 80 C for 8 hours.After cooling, it was filtered through a Celite/silica gel, and the filtrate was evaporated to remove organic solvent. The residue was crystallized from toluene and dried to give19.34 g (44.50 mmol) of Compound N1.
4-(1H-naphtho[1,8-de][1,3,2]diazaborinin-2(3H)-yl)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
51%
With tert.-butylnitrite; sodium acetate; tetra-(n-butyl)ammonium iodide; dibenzoyl peroxide; In acetonitrile; at 80℃;Schlenk technique; Inert atmosphere;
General procedure: In air, Bpin-B(dan) (0.1 mmol, 1.0 eq.), aryl amide (0.2 mmol, 2.0 eq.), TBAI (0.01 eq.),NaOAc (0.15 eq.), and BPO (0.01 eq.) were sequentially weighed and added to a screw-cappedSchenk tube containing a magnetic stir bar. The vessel was evacuated and refilled with nitrogen forthree times. t-BuONO (0.2 eq.) and MeCN (0.6 mL) were added in turn under N2 atmosphere usingsyringes through a septum which was temporarily used to replace the screw cap. The reaction mixturewas then vigorously stirred at 80 C for the indicated time. The resulting mixture was filtered through a pad of Celite, and the filter cake was washed with ethyl acetate (3 mL x 2). The combined filtratewas evaporated under vacuum to dryness and the residue was purified by column chromatography toyield the desired product.
N-(4-cyanophenyl)-4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)benzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
37%
Compound 7 (0.37 g, 1.2 mmol) was taken in a round bottom flask, dissolved in 5 ml of anhydrous DMF, and the solution was stirred at 0 C, and HOBt (0.21 g, 1.5 mmol) and EDCi (0.38 g) were added to the system. , 2 mmol), DIPEA (0.3 ml, 3 mmol) and a catalytic amount of DMAP. After 1 h of activation, the compound 4-aminobenzonitrile (0.12 g, 1 mmol) was added thereto and allowed to react at room temperature for 12 h. After completion of the reaction, washed with water and extracted 3 times with ethyl acetate, the combined organic phases were washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and concentrated to give crude product pressurization,Column chromatography to give a pale yellow solid compound C1 (0.15g, 37% yield).
4-chloro-(6-methoxy(1,3,5-triazin-2-yl)amino)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
87%
In acetone; at 20℃; for 24h;Alkaline conditions;
A solution of 4-aminobenzonitrile (10 mmole) in 50mL acetone was added dropwise, as described previously [32], to an ice-cold solution of <strong>[3638-04-8]2,4-dichloro-6-methoxy-1,3,5-triazine</strong> 5 (10mmol) and NaHCO3 (10mmol) in 50mL acetone over 10min. The reaction mixture was stirred at rt for 24h and then acetone was concentrated under vacuum, and excess ice water was added. The solid product obtained was filtered, washed with water, and dried to afford the product. White solid; Yield 87%; m.p. 226-227C; 1H NMR (CDCl3) delta: 4.06 (s, 3H, OCH3), 7.49 (br.s., 1H, NH), 7.64(d, 2H, J=8.8Hz, Ar), 7.72 (d, 2H, J=8.8Hz, Ar) ppm; 13C NMR (CDCl3) delta: 52.1 (OMe), 102.2 (CN), 120.3, 133.4, 163.5, 172.3ppm. Anal.Calc for C11H8ClN5O (261.67): C, 50.49; H, 3.08; N, 26.76. Found: C, 50.66; H, 3.19; N, 26.92.
4-((2-methoxypyrimidin-5-yl)amino)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
With tris-(dibenzylideneacetone)dipalladium(0); sodium t-butanolate; XPhos; In 1,4-dioxane; at 120℃; for 1h;Inert atmosphere; Microwave irradiation;
General procedure: A G10 microwave vial equipped with a stir-bar was charged with compound 5-bromo-2-substituted pyrimidine (0.3 mmol), amino compound (0.33 mmol), Pd2dba3 (2 mol%), XPhos (8 mol%), NaOtBu (0.42 mmol) and anhydrous 1,4-dioxane (2 ml). The reaction vial was purged with argon, and heated by microwave irradiation until its temperature was 120 C. Then, the irradiation was automatically controlled to keep the mixture at this temperature for 1h. The reaction mixture was concentrated under reduced pressure and the crude residue was purified by flash chromatography on silica gel. The purified material was dried in vacuo to afford the corresponding products.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping