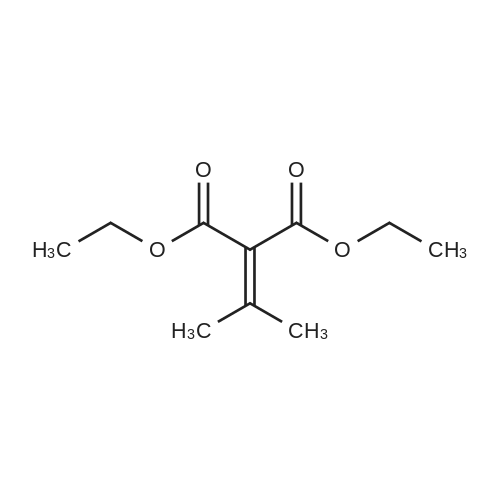
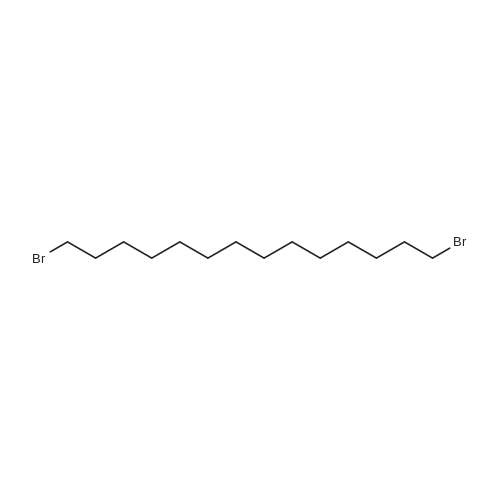
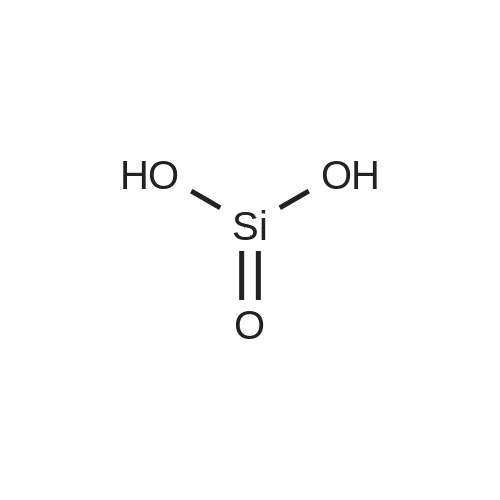
- All Products
- Chemistry
- Life Science
- Materials Science
- Custom Synthesis
- Recommended Products
- About Us | Contact Us
Heterocyclic Building Blocks
Aliphatic Heterocycles (105)
(1R,5S)-3-Azabicyclo[3.1.0]Hexane (11)1,2-Dithiolane (7)1,4-Diazepan-2-One (5)1,4-Diazepan-5-One (9)1,4-Dioxane (17)1,4-Oxazepane (8)10,11-Dihydro-5H-Dibenzo[B,F]Azepine (9)11,12-Didehydro-5,6-Dihydrodibenz[B,F]Azocine (14)3-((((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Glycyl)-2,2-Dimethyloxazolidine-4-Carboxylic Acid (9)3H-Diazirine (11)4-Benzyl-1,4-Oxazepane (5)Tetrahydro-2H-Thiopyran 1,1-Dioxide (8)
Azetidines (2879)
Alcohols (18)Amides (102)Amines (17)Aryls (44)Bromides (7)Carboxylic Acids (17)Chlorides (10)Esters (11)Ethers (18)Fluorinated Building Blocks (20)Ketones (16)Nitriles (5)Sulfamides (5)1-(Azetidin-1-Yl)-2-Bromoethan-1-One (3)1-(Azetidin-3-Yl)-1H-Pyrazole (7)1-(Phenylsulfonyl)Azetidine (7)1-Benzhydrylazetidine (13)1-Benzylazetidine (6)1-Methylazetidine (6)1-Phenylazetidine (8)2,7-Diazaspiro[3.5]Nonane (7)2-Azaspiro[3.3]Heptane (11)2-Imino-N-((6R,7R)-8-Oxo-5-Thia-1-Azabicyclo[4.2.0]Oct-2-En-7-Yl)-2-(Thiazol-4-Yl)Acetamide (3)3-Bromoazetidine (3)3-Methylazetidin-3-Ol (3)3-Methyleneazetidine (8)4-(Azetidin-1-Yl)Piperidine (6)Azetidin-2-One (11)Azetidin-2-Ylmethanol (7)Azetidin-3-Amine (4)Azetidin-3-Ol (11)Azetidin-3-Yl Acetate (3)Azetidin-3-Ylmethanamine (3)Azetidine-2-Carboxylic Acid (6)Azetidine-3-Carboxylic Acid (5)Benzyl Azetidine-1-Carboxylate (9)Methyl Azetidine-2-Carboxylate (3)N-((6R,7R)-8-Oxo-5-Thia-1-Azabicyclo[4.2.0]Oct-2-En-7-Yl)-2-Phenylacetamide (5)Tert-Butyl Azetidine-1-Carboxylate (64)Oxetanes (9)Piperidines (16)Pyridines (4)Pyrimidines (4)Pyrrolidines (12)Spiroes (85)Tetrahydropyrans (6)
Benzimidazoles (4397)
Alcohols (11)Aldehydes (5)Amides (26)Amines (19)Aryls (40)Bromides (8)Carboxylic Acids (6)Chlorides (15)Esters (12)Ethers (17)Fluorinated Building Blocks (14)Sulfides (7)Benzimidazole-Carboxylic Acid (43)(1H-Benzo[D]Imidazol-2-Yl)Methanamine (7)(1H-Benzo[D]Imidazol-2-Yl)Methanol (14)1,2-Diphenyl-1H-Benzo[D]Imidazole (7)1-Benzyl-1H-Benzo[D]Imidazole (6)1-Ethyl-1H-Benzo[D]Imidazole (4)1-Methyl-1H-Benzo[D]Imidazole (47)1-Phenyl-1H-Benzo[D]Imidazole (4)1H-Benzo[D]Imidazol-2-Amine (7)1H-Benzo[D]Imidazol-5-Amine (4)1H-Benzo[D]Imidazol-6-Amine (6)1H-Benzo[D]Imidazole-2-Carbaldehyde (5)1H-Benzo[D]Imidazole-2-Carboxylic Acid (7)1H-Benzo[D]Imidazole-2-Thiol (10)1H-Benzo[D]Imidazole-4-Carboxylic Acid (6)1H-Benzo[D]Imidazole-5-Carboxylic Acid (4)2-((Pyridin-2-Ylmethyl)Thio)-1H-Benzo[D]Imidazole (7)2-(1H-Benzo[D]Imidazol-2-Yl)Ethan-1-Amine (6)2-(Chloromethyl)-1H-Benzo[D]Imidazole (9)2-Benzyl-1H-Benzo[D]Imidazole (7)2-Bromo-1H-Benzo[D]Imidazole (4)2-Chloro-1H-Benzo[D]Imidazole (17)2-Methyl-1H-Benzo[D]Imidazole (20)4-Bromo-1H-Benzo[D]Imidazole (14)4-Methyl-1H-Benzo[D]Imidazole (7)5,6-Dichloro-1H-Benzo[D]Imidazole (4)5-Bromo-1H-Benzo[D]Imidazole (13)5-Chloro-1H-Benzo[D]Imidazole (14)5-Methoxy-1H-Benzo[D]Imidazole (2)5-Methyl-1H-Benzo[D]Imidazole (10)6-Chloro-1H-Benzo[D]Imidazole (25)Methyl (1H-Benzo[D]Imidazol-2-Yl)Carbamate (5)Methyl 1H-Benzo[D]Imidazole-2-Carboxylate (3)Tert-Butyl 1H-Benzo[D]Imidazole-1-Carboxylate (3)Piperidines (18)Pyridines (6)
Benzofurans (3699)
Alcohols (18)Aldehydes (4)Amides (7)Amines (13)Anhydrides (13)Aryls (12)Bromides (18)Carboxylic Acids (9)Chlorides (15)Esters (34)Ethers (2)Fluorinated Building Blocks (11)Iodides (4)Ketones (10)Nitroes (5)Benzofuran-Carboxylic Acid (67)Dimethoxyisobenzofuran (4)Epoxyisobenzofuran (1)Fluorobenzofuran (19)Fluoroisobenzofuran (5)Methoxyisobenzofuran (4)Methylbenzofuran (25)1-(Benzofuran-2-Yl)Ethan-1-One (3)2,2-Dimethyl-2,3-Dihydrobenzofuran (7)2,3-Dihydrobenzofuran-3-Amine (11)2,3-Dihydrobenzofuran-5-Amine (3)2,3-Dihydrobenzofuran-7-Amine (5)2-(Benzofuran-3-Yl)Acetic Acid (3)2-Bromodibenzo[B,D]Furan (6)2-Ethylbenzofuran (3)2-Methylbenzofuran (5)3-Methylbenzofuran (4)4-Bromobenzofuran (3)4-Phenyldibenzo[B,D]Furan (8)5,7,8-Trimethylchromane (3)5-Bromo-2,3-Dihydrobenzofuran (6)5-Chloro-2,3-Dihydrobenzofuran (5)5-Fluoro-2,3-Dihydrobenzofuran (5)5-Methoxyisobenzofuran-1(3H)-One (5)6-Bromo-2,3-Dihydrobenzofuran (7)6-Fluoro-2,3-Dihydrobenzofuran (6)6-Methoxybenzofuran (4)7-Bromo-2,3-Dihydrobenzofuran (6)7-Bromobenzofuran (6)7-Fluoro-2,3-Dihydrobenzofuran (6)Benzofuran-2(3H)-One (7)Benzofuran-2-Carbaldehyde (5)Benzofuran-2-Carboxylic Acid (21)Benzofuran-2-Ylboronic Acid (8)Benzofuran-3(2H)-One (15)Benzofuran-3-Carboxylic Acid (6)Benzofuran-3-Yl(Phenyl)Methanone (11)Methyl Benzofuran-2-Carboxylate (6)Piperidines (5)Spiroes (6)
Benzothiazoles (2441)
Alcohols (6)Aldehydes (5)Amides (23)Amines (30)Aryls (10)Bromides (11)Carboxylic Acids (5)Chlorides (6)Esters (5)Ethers (15)Fluorinated Building Blocks (7)Nitroes (5)Sulfamides (6)Thioesters (12)Benzothiazole-Carboxylic Acid (27)Bromobenzothiazole (7)Dichlorobenzothiazole (10)Methoxybenzothiazole (4)Methylbenzothiazole (7)2-(Methylthio)Benzo[D]Thiazole (6)2-Bromobenzo[D]Thiazole (28)2-Chlorobenzo[D]Thiazole (21)2-Methylbenzo[D]Thiazole (26)4-Bromobenzo[D]Thiazole (9)4-Chlorobenzo[D]Thiazole (7)4-Fluorobenzo[D]Thiazole (6)4-Methylbenzo[D]Thiazole (10)5-Chlorobenzo[D]Thiazole (5)5-Nitrobenzo[D]Thiazole (3)6-Bromobenzo[D]Thiazole (7)6-Chlorobenzo[D]Thiazole (6)6-Fluorobenzo[D]Thiazole (5)6-Methoxybenzo[D]Thiazole (6)6-Methylbenzo[D]Thiazole (6)6-Nitrobenzo[D]Thiazole (3)7-Bromobenzo[D]Thiazole (8)7-Chlorobenzo[D]Thiazole (6)7-Fluorobenzo[D]Thiazole (9)Benzo[D]Thiazol-2-Amine (30)Benzo[D]Thiazol-5-Amine (3)Benzo[D]Thiazol-6-Amine (3)Benzo[D]Thiazol-6-Ol (2)Benzo[D]Thiazole-2-Carbonitrile (11)Benzo[D]Thiazole-2-Thiol (11)Benzo[D]Thiazole-6-Carboxylic Acid (5)Methyl Benzo[D]Thiazole-6-Carboxylate (5)Morpholines (4)
Benzothiophenes (1120)
Amines (6)Aryls (4)Bromides (4)Esters (8)Fluorinated Building Blocks (5)Benzothiophene-Carboxylic Acid (58)2-(Benzo[B]Thiophen-2-Yl)-4,5-Dihydrooxazole (10)2-Methylbenzo[B]Thiophene (4)3-Bromobenzo[B]Thiophene (5)3-Chlorobenzo[B]Thiophene (6)3-Methylbenzo[B]Thiophene (4)4,5,6,7-Tetrahydrobenzo[B]Thiophene (21)5-Bromobenzo[B]Thiophene (3)5-Chlorobenzo[B]Thiophene (4)6-Bromobenzo[B]Thiophene (4)7-Bromobenzo[B]Thiophene (5)Benzo[B]Thiophen-2-Amine (5)Benzo[B]Thiophen-2-Ylboronic Acid (5)Benzo[B]Thiophene-2-Carbaldehyde (7)Benzo[B]Thiophene-2-Carbonitrile (3)Benzo[B]Thiophene-2-Carboxylic Acid (32)Benzo[B]Thiophene-3-Carbaldehyde (6)Ethyl Benzo[B]Thiophene-2-Carboxylate (9)Methyl Benzo[B]Thiophene-2-Carboxylate (27)
Benzoxazoles (2808)
Amides (33)Amines (7)Bromides (10)Carboxylic Acids (4)Chlorides (11)Esters (4)Ethers (6)Fluorinated Building Blocks (5)Nitroes (5)Benzoxazole-Carboxylic Acid (25)2-Chlorobenzo[D]Oxazole (11)2-Methylbenzo[D]Oxazole (26)2-Phenylbenzo[D]Oxazole (22)3-Phenylbenzo[D]Oxazol-3-Ium (8)5-Bromobenzo[D]Oxazole (3)5-Chlorobenzo[D]Oxazole (5)5-Methylbenzo[D]Oxazole (3)6-Chlorobenzo[D]Oxazole (4)Benzo[D]Oxazol-2-Amine (16)Benzo[D]Oxazole-2-Thiol (6)Methyl Benzo[D]Oxazole-2-Carboxylate (3)
Carbazole Series (479)
Carbazolyl-Phenyl-Boronic Acid/Ester (14)1-Methyl-9H-Carbazole (7)2,3,4,9-Tetrahydro-1H-Carbazol-1-One (7)2-Bromo-9H-Carbazole (4)3,9-Diphenyl-9H-Carbazole (7)3-(Tert-Butyl)-9H-Carbazole (3)3-Bromo-9-Phenyl-9H-Carbazole (16)3-Bromo-9H-Carbazole (4)3-Chloro-9H-Carbazole (3)3-Methyl-9H-Carbazole (3)7H-Benzo[C]Carbazole (5)9-([1,1'-Biphenyl]-4-Yl)-9H-Carbazole (9)9-(P-Tolyl)-9H-Carbazole (5)9-Benzyl-9H-Carbazole (7)9-Ethyl-9H-Carbazole (10)
Dioxolanes (1576)
Alcohols (4)Amines (4)Aryls (16)Bromides (6)Carboxylic Acids (4)Esters (7)Ketones (5)Sulfonates (4)(3As,6As)-Tetrahydrofuro[3,4-D][1,3]Dioxole (6)1,3-Dioxolan-2-One (11)2,2-Difluorobenzo[D][1,3]Dioxole (23)2,2-Dimethyl-1,3-Dioxolane (29)2,2-Dimethylbenzo[D][1,3]Dioxole (7)2-Phenyl-1,3-Dioxolane (18)4-Bromobenzo[D][1,3]Dioxole (6)4-Methyl-1,3-Dioxolane (5)5,6-Dihydro-[1,3]Dioxolo[4,5-G]Isoquinolino[3,2-A]Isoquinolin-7-Ium (4)5-Bromobenzo[D][1,3]Dioxole (12)5-Chlorobenzo[D][1,3]Dioxole (5)5-Nitrobenzo[D][1,3]Dioxole (4)9-((3As,4R,6Ar)-Tetrahydrofuro[3,4-D][1,3]Dioxol-4-Yl)-9H-Purine (7)Benzo[D][1,3]Dioxol-5-Amine (3)Spiroes (23)
Furans (6925)
Alcohols (8)Aldehydes (15)Alkenyls (9)Amides (30)Amines (22)Aryls (39)Bromides (13)Carboxylic Acids (35)Chlorides (9)Esters (23)Ethers (10)Fluorinated Building Blocks (4)Ketones (10)Nitriles (7)Nitroes (8)Sulfides (6)Aminodihydrofuran (7)Bromofuran (32)Chlorofuran (2)Chlorofuro (11)Cyanofuran (53)Dibromofuran (5)Dihydrofuran (31)Dihydrofuro (15)Dihydroxydihydrofuran (8)Dimethylfuran (10)Formylfuran (10)Hexahydrofuro (6)Hydroxydihydrofuran (31)Methyldihydrofuran (11)Methylfuran (58)Nitrofuran (10)2,3-Dihydrofuran-3-One (9)2-(Bromomethyl)Furan (3)2-(Chloromethyl)Furan (3)2-(Furan-2-Yl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (6)2-Bromofuran (21)2-Chlorofuran (3)2-Methylfuran (38)2-Nitrofuran (9)3-Bromofuran (13)4-Methylisobenzofuran-1(3H)-One (3)5-Nitro-2,3-Dihydrobenzofuran (3)5-Phenylfuran-2-Carbaldehyde (9)5-Phenylfuran-2-Carboxylic Acid (6)6-Chloro-2,3-Dihydrobenzofuran (3)6-Fluoroisobenzofuran-1(3H)-One (3)7-Chloro-2,3-Dihydrobenzofuran (3)Ethyl Furan-2-Carboxylate (6)Furan-2,5-Dione (8)Furan-2-Carbaldehyde (22)Furan-2-Carbonitrile (8)Furan-2-Carboxylic Acid (32)Furan-2-Ylmethanol (8)Furan-3-Carboxylic Acid (11)Furo[3,2-B]Pyridine (7)Furo[3,2-C]Pyridine (7)Methyl Furan-2-Carboxylate (20)N-(Furan-2-Ylmethyl)Aniline (5)Piperazines (4)Quinolines (3)
Imidazoles (1074)
Alcohols (27)Aldehydes (21)Alkenyls (7)Amides (26)Amines (38)Aryls (50)Bromides (16)Carboxylic Acids (42)Chlorides (15)Esters (34)Ethers (6)Fluorinated Building Blocks (25)Iodides (12)Ketones (7)Nitroes (13)Sulfamides (7)Trifluoromethyls (12)Biimidazole (5)Bromoimidazo (102)Dibromoimidazo (8)Dichloroimidazo (9)Dihydroimidazo (25)Imidazol-Amine (1)Methoxyimidazo (13)Methylimidazo (121)Tetrahydroimidazo (40)Pyridines (4)Pyrrolidines (2)1,1'-Diphenyl-4,4',5,5'-Tetrahydro-1H,1'H-2,2'-Biimidazole (11)1,2-Dimethyl-1H-Imidazole (6)1,3-Diphenyl-1H-Imidazol-3-Ium (14)1,3-Diphenyl-4,5-Dihydro-1H-Imidazol-3-Ium (5)1-(Imidazo[1,2-A]Pyridin-3-Yl)Ethan-1-One (3)1-(Phenylsulfonyl)-1H-Imidazole (5)1-Benzyl-1H-Imidazole (20)1-Decyl-3-Methyl-1H-Imidazol-3-Ium (14)1-Ethyl-1H-Imidazole (3)1-Isopropyl-1H-Imidazole (5)1-Phenyl-1H-Imidazole-4-Carboxylic Acid (5)1-Trityl-1H-Imidazole (15)1H-Imidazo[4,5-B]Pyridine (14)1H-Imidazo[4,5-C]Pyridine (12)1H-Imidazol-2-Amine (6)1H-Imidazole-2-Carboxylic Acid (7)1H-Imidazole-4-Carbonitrile (2)1H-Imidazole-4-Carboxylic Acid (4)2-(Pyridin-2-Yl)-1H-Benzo[D]Imidazole (5)2-Bromo-1H-Imidazole (13)2-Bromoimidazo[1,2-A]Pyridine (5)2-Ethyl-1H-Imidazole (3)2-Methyl-1-Phenyl-1H-Imidazole (11)2-Methylimidazo[1,2-A]Pyridine (8)2-Phenyl-1H-Benzo[D]Imidazole (26)2-Phenyl-1H-Imidazole (26)2-Phenylimidazo[1,2-A]Pyridine (11)2-Propyl-1H-Imidazole (5)3-Bromoimidazo[1,2-A]Pyrazine (6)3-Bromoimidazo[1,2-A]Pyridine (26)3-Bromoimidazo[1,2-A]Pyrimidine (6)3-Iodoimidazo[1,2-A]Pyridine (10)3-Methylimidazo[1,2-A]Pyridine (3)4,5,6,7-Tetrahydro-3H-Imidazo[4,5-C]Pyridine (5)4-(1H-Imidazol-1-Yl)Benzaldehyde (7)4-Bromo-1-Phenyl-1H-Imidazole (9)4-Methyl-1-Phenyl-1H-Imidazole (7)4-Methyl-1H-Imidazole (6)4-Phenyl-1H-Imidazole (8)5,6,7,8-Tetrahydroimidazo[1,2-A]Pyrazine (15)5,6,7,8-Tetrahydroimidazo[1,2-A]Pyridine (12)5,6,7,8-Tetrahydroimidazo[1,5-A]Pyrazine (11)5-Benzyl-1H-Imidazole (1)5-Bromoimidazo[1,2-A]Pyridine (6)5-Chloro-1H-Imidazole (2)5-Fluoro-1H-Benzo[D]Imidazole (12)5-Iodo-1H-Imidazole (4)5-Methylimidazo[1,2-A]Pyridine (4)5-Phenyl-1H-Imidazole (6)6,7-Dihydro-5H-Pyrrolo[1,2-A]Imidazole (9)6-Bromoimidazo[1,2-A]Pyrazine (11)6-Bromoimidazo[1,2-A]Pyridine (14)6-Bromoimidazo[1,2-A]Pyrimidine (5)6-Chloroimidazo[1,2-A]Pyridine (4)6-Chloroimidazo[1,2-B]Pyridazine (30)7-Methylimidazo[1,2-A]Pyridine (3)8-Bromoimidazo[1,2-A]Pyrazine (7)8-Bromoimidazo[1,2-A]Pyridine (15)8-Chloroimidazo[1,2-A]Pyrazine (8)8-Chloroimidazo[1,2-A]Pyridine (7)8-Fluoroimidazo[1,2-A]Pyridine (3)8-Methylimidazo[1,2-A]Pyridine (9)Ethyl 1H-Imidazole-2-Carboxylate (6)Ethyl 2-(Imidazo[1,2-A]Pyridin-3-Yl)Acetate (3)Ethyl Imidazo[1,2-A]Pyridine-2-Carboxylate (25)Ethyl Imidazo[1,2-A]Pyridine-3-Carboxylate (13)Imidazo[1,2-A]Pyridin-3-Amine (7)Imidazo[1,2-A]Pyridin-8-Amine (7)Imidazo[1,2-A]Pyridin-8-Ol (3)Imidazo[1,2-A]Pyridine-2-Carbaldehyde (8)Imidazo[1,2-A]Pyridine-2-Carboxylic Acid (21)Imidazo[1,2-A]Pyridine-3-Carbaldehyde (9)Imidazo[1,2-A]Pyridine-3-Carboxylic Acid (14)Imidazo[1,2-A]Pyridine-7-Carboxylic Acid (5)Imidazo[1,2-A]Pyridine-8-Carboxylic Acid (6)Imidazo[1,2-A]Pyrimidine-3-Carboxylic Acid (5)Imidazo[1,2-C]Pyrimidine (6)Imidazo[1,5-A]Pyrazine (9)Imidazo[2,1-B]Thiazole (14)Methyl 1H-Imidazole-2-Carboxylate (4)Methyl 1H-Imidazole-5-Carboxylate (8)Methyl Imidazo[1,2-A]Pyridine-8-Carboxylate (3)Tert-Butyl 1H-Imidazole-1-Carboxylate (6)
Imidazolidines (1119)
Amides (19)Aryls (10)Carboxylic Acids (3)Methylimidazolidine (3)Oxoimidazolidine (8)Thioxoimidazolidin (9)Spiroes (4)(3Ar,6As)-Tetrahydro-1H-Thieno[3,4-D]Imidazol-2(3H)-One (18)1-Phenylimidazolidin-2-One (8)3-Phenyl-2-Thioxoimidazolidin-4-One (6)3-Phenylimidazolidine-2,4-Dione (4)Imidazolidin-2-One (16)Imidazolidine-2,4-Dione (17)
Indazoles (3061)
Alcohols (17)Aldehydes (9)Amides (6)Amines (18)Aryls (8)Bromides (25)Carboxylic Acids (19)Chlorides (10)Esters (15)Ethers (10)Fluorinated Building Blocks (13)Iodides (12)Nitriles (8)Nitroes (7)Indazol-Amide (3)(1H-Indazol-4-Yl)Boronic Acid (3)(1H-Indazol-6-Yl)Boronic Acid (4)1-(1H-Indazol-1-Yl)Ethan-1-One (7)1-(Tetrahydro-2H-Pyran-2-Yl)-1H-Indazole (17)1-Benzyl-1H-Indazole (5)1-Ethyl-1H-Indazole (6)1H-Indazol-3-Amine (23)1H-Indazol-3-Ol (10)1H-Indazol-4-Amine (12)1H-Indazol-4-Ol (4)1H-Indazol-5-Amine (3)1H-Indazol-6-Amine (5)1H-Indazol-6-Ol (6)1H-Indazol-7-Amine (6)1H-Indazole-3-Carbaldehyde (19)1H-Indazole-3-Carbonitrile (9)1H-Indazole-3-Carboxylic Acid (23)1H-Indazole-4-Carboxylic Acid (9)1H-Indazole-5-Carboxylic Acid (4)1H-Indazole-7-Carboxylic Acid (6)2,3-Dimethyl-2H-Indazole (4)2-Methyl-2H-Indazole (34)3-(Piperidin-4-Yl)-1H-Indazole (5)3-Chloro-1H-Indazole (13)4,5,6,7-Tetrahydro-1H-Indazole (12)4-Chloro-1H-Indazole (18)4-Fluoro-1H-Indazole (22)4-Methoxy-1H-Indazole (7)4-Methyl-1H-Indazole (12)4-Nitro-1H-Indazole (10)5-Bromo-1H-Indazole (14)5-Chloro-1H-Indazole (9)5-Methoxy-1H-Indazole (5)5-Nitro-1H-Indazole (5)6-(Trifluoromethyl)-1H-Indazole (6)6-Bromo-1H-Indazole (23)6-Chloro-1H-Indazole (14)6-Fluoro-1H-Indazole (15)6-Methoxy-1H-Indazole (14)6-Methyl-1H-Indazole (9)6-Nitro-1H-Indazole (9)7-Bromo-1H-Indazole (18)7-Chloro-1H-Indazole (7)7-Fluoro-1H-Indazole (12)7-Methoxy-1H-Indazole (5)7-Nitro-1H-Indazole (11)Methyl 1H-Indazole-3-Carboxylate (23)Methyl 1H-Indazole-4-Carboxylate (10)Methyl 1H-Indazole-5-Carboxylate (3)Methyl 1H-Indazole-6-Carboxylate (9)Methyl 1H-Indazole-7-Carboxylate (8)Methyl 2-Methyl-2H-Indazole-3-Carboxylate (3)Tert-Butyl 1H-Indazole-1-Carboxylate (29)
Indoles (7653)
Alcohols (26)Aldehydes (21)Alkenyls (4)Amides (42)Amines (33)Aryls (51)Bromides (36)Carboxylic Acids (52)Chlorides (27)Esters (61)Ethers (54)Fluorinated Building Blocks (39)Iodides (11)Ketones (20)Nitriles (31)Nitroes (10)Sulfamides (6)Trifluoromethyls (7)Azaindole (44)Benzyloxyindole (1)Biindolinylidene (1)Bromo-Indole-Carboxylic Acid (25)Cyanoindole (5)Dihydroindolo (14)Fluoroindole (5)Formylindole (44)Hydroxyindole (7)Hydroxyisoindoline (2)Isoindole (26)Methoxyindole (9)Nitroindole (6)(1-(Tert-Butoxycarbonyl)-1H-Indol-2-Yl)Boronic Acid (17)(1H-Indol-2-Yl)Boronic Acid (3)(1H-Indol-2-Yl)Methanol (8)(3As,7As)-Octahydro-1H-Indole (5)(9H-Fluoren-9-Yl)Methyl (2-(1H-Indol-3-Yl)Ethyl)Carbamate (12)1,2,3,4-Tetrahydrocyclopenta[B]Indole (5)1,2-Dimethyl-1H-Indole (3)1-(1H-Indol-1-Yl)Ethan-1-One (9)1-(1H-Indol-3-Yl)Ethan-1-One (6)1-(Phenylsulfonyl)-1H-Indole (26)1-(Triisopropylsilyl)-1H-Pyrrolo[2,3-B]Pyridine (8)1-Benzyl-1H-Indole (9)1-Ethyl-1H-Indole (7)1-Methyl-1H-Indole (82)1-Methyl-1H-Indole-2-Carboxylic Acid (8)1-Methyl-1H-Indole-3-Carbaldehyde (9)1-Methyl-1H-Pyrrolo[2,3-B]Pyridine (14)1-Phenyl-1H-Indole (5)1H-Indol-3-Ol (5)1H-Indol-3-Yl Acetate (4)1H-Indol-4-Amine (10)1H-Indol-4-Ol (7)1H-Indol-5-Ol (4)1H-Indol-6-Amine (21)1H-Indol-6-Ol (6)1H-Indol-7-Amine (10)1H-Indole-2-Carbaldehyde (20)1H-Indole-2-Carbonitrile (9)1H-Indole-3-Carbonitrile (17)1H-Indole-3-Carboxylic Acid (28)1H-Indole-4-Carboxylic Acid (10)1H-Indole-5-Carbonitrile (5)1H-Indole-5-Carboxylic Acid (4)1H-Indole-6-Carbonitrile (4)1H-Indole-6-Carboxylic Acid (5)1H-Indole-7-Carbonitrile (6)1H-Indole-7-Carboxylic Acid (7)1H-Pyrrolo[2,3-B]Pyridine-3-Carbaldehyde (14)1H-Pyrrolo[2,3-B]Pyridine-5-Carbaldehyde (5)2,3,4,5-Tetrahydro-1H-Pyrido[4,3-B]Indole (12)2,3,4,9-Tetrahydro-1H-Pyrido[3,4-B]Indole (13)2,3-Dimethyl-1H-Indole (9)2-(1,3,2-Dioxaborolan-2-Yl)-1-Methyl-1H-Indole (6)2-(1H-Indol-1-Yl)Acetic Acid (5)2-(1H-Indol-3-Yl)Acetic Acid (28)2-(1H-Indol-3-Yl)Ethan-1-Amine (4)2-(1H-Indol-3-Yl)Ethan-1-Ol (10)2-(2-Methyl-1H-Indol-3-Yl)Acetic Acid (5)2-(4-Fluorophenyl)-1H-Indole (4)2-Methyl-1H-Indole-3-Carbaldehyde (3)2-Methyl-1H-Pyrrolo[2,3-B]Pyridine (12)3-(1-Oxoisoindolin-2-Yl)Piperidine-2,6-Dione (21)3-(1H-Indol-3-Yl)Propanoic Acid (4)3-(Piperidin-4-Yl)-1H-Indole (8)3-Iodo-1H-Pyrrolo[2,3-B]Pyridine (11)3-Methyl-1H-Indole (41)3H-Indole (8)4-Bromo-1H-Indole (25)4-Chloro-1H-Indole (21)4-Fluoro-1H-Indole (24)4-Fluoro-1H-Pyrrolo[2,3-B]Pyridine (6)4-Fluoroindoline-2,3-Dione (3)4-Methoxy-1H-Indole (11)4-Methyl-1H-Indole (23)4-Nitro-1H-Indole (6)5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)-1H-Indole (5)5-(Benzyloxy)-1H-Indole (8)5-Bromo-1-Methyl-1H-Indole (3)5-Chloro-1H-Indole (64)5-Chloro-1H-Indole-2-Carboxylic Acid (6)5-Chloro-1H-Pyrrolo[2,3-B]Pyridine (13)5-Fluoro-1H-Indole (50)5-Fluoro-1H-Pyrrolo[2,3-B]Pyridine (9)5-Fluoroindolin-2-One (4)5-Methoxy-1H-Indole (11)5-Methoxy-2-Methyl-1H-Indole (5)5-Methyl-1H-Indole (33)5-Nitro-1H-Indole (6)6-(Trifluoromethyl)-1H-Indole (5)6-Bromo-1-Methyl-1H-Indole (3)6-Bromo-1H-Indole (15)6-Bromo-1H-Pyrrolo[2,3-B]Pyridine (6)6-Chloro-1H-Indole (19)6-Chloro-1H-Pyrrolo[2,3-B]Pyridine (13)6-Fluoro-1H-Indole (17)6-Methoxy-1H-Indole (12)6-Methyl-1H-Indole (29)6-Methyl-1H-Pyrrolo[2,3-B]Pyridine (8)6-Nitro-1H-Indole (8)7-Bromo-1H-Indole (28)7-Bromoindoline-2,3-Dione (3)7-Chloro-1H-Indole (29)7-Fluoro-1H-Indole (24)7-Fluoro-1H-Indole-2-Carboxylic Acid (5)7-Methoxy-1H-Indole (8)7-Methyl-1H-Indole (34)7-Nitro-1H-Indole (11)9H-Pyrido[3,4-B]Indole (5)Benzo[Cd]Indol-2(1H)-One (5)Ethyl 1H-Indole-3-Carboxylate (6)Ethyl 5-Chloro-1H-Indole-2-Carboxylate (5)Ethyl 5-Fluoro-1H-Indole-2-Carboxylate (5)Ethyl 6-Chloro-1H-Indole-2-Carboxylate (5)Ethyl 7-Chloro-1H-Indole-2-Carboxylate (5)Methyl 1H-Indole-3-Carboxylate (21)Methyl 1H-Indole-4-Carboxylate (9)Methyl 1H-Indole-5-Carboxylate (7)Methyl 1H-Indole-6-Carboxylate (9)Methyl 1H-Indole-7-Carboxylate (7)Methyl 2-(1H-Indol-3-Yl)Acetate (3)N-(2-(1H-Indol-3-Yl)Ethyl)Acetamide (5)Spiro[Indoline-3,4'-Piperidin]-2-One (18)Tert-Butyl 1H-Indole-1-Carboxylate (18)Tert-Butyl 1H-Pyrrolo[2,3-B]Pyridine-1-Carboxylate (9)Tert-Butyl 2-(1,3,2-Dioxaborolan-2-Yl)-1H-Indole-1-Carboxylate (6)Tert-Butyl 3-(Hydroxymethyl)-1H-Indole-1-Carboxylate (10)Tert-Butyl 3-Bromo-1H-Indole-1-Carboxylate (9)Tert-Butyl 3-Iodo-1H-Indole-1-Carboxylate (7)Tert-Butyl 5-Methoxy-1H-Indole-1-Carboxylate (6)Tryptophan (22)Piperazines (5)Piperidines (4)Pyridines (5)Pyrimidines (7)
Indolines (4494)
Alcohols (13)Alkenyls (3)Amides (73)Amines (9)Aryls (20)Bromides (16)Carboxylic Acids (16)Chlorides (14)Esters (9)Ethers (5)Fluorinated Building Blocks (11)Ketones (11)Nitriles (5)Nitroes (9)Aminoindoline (10)Bromoindoline (17)Bromoisoindolin (5)Bromoisoindoline (6)Chloroindolin (4)Chloroisoindoline (3)Dibromoindoline (12)Dichloroindoline (16)Difluoroindoline (1)Dimethylindoline (21)Dioxoindoline (8)Dioxoisoindolin (72)Fluoroindolin (9)Fluoroindoline (9)Fluoroisoindoline (10)Hydroxyindolin (5)Isoindoline (20)Methoxyindoline (12)Methoxyisoindoline (9)Methylindoline (46)Methylisoindoline (18)Nitroindolin (6)Nitroindoline (11)Nitroisoindoline (8)Oxoindoline (32)Oxoisoindoline (16)Spiro-Indoline-Piperidine (34)(E)-[3,3'-Biindolinylidene]-2,2'-Dione (5)(Z)-3-((1H-Pyrrol-2-Yl)Methylene)Indolin-2-One (8)(Z)-3-(Phenyl(Phenylamino)Methylene)Indolin-2-One (5)(Z)-3-Benzylideneindolin-2-One (7)1-(Indolin-1-Yl)Ethan-1-One (9)1-Benzylindoline-2,3-Dione (5)1-Methylindolin-2-One (14)1-Methylindoline (3)1-Methylindoline-2,3-Dione (8)2-(1,3-Dioxoisoindolin-2-Yl)Propanoic Acid (3)2-Methylindoline (7)2-Methylisoindolin-1-One (7)3,3-Dimethylindolin-2-One (5)3,3-Dimethylindoline (7)4-Bromoindoline (4)4-Chloroindoline-2,3-Dione (6)4-Methylindoline (3)5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Indoline (5)5-Bromoindolin-2-One (5)5-Bromoindoline (8)5-Bromoisoindoline-1,3-Dione (3)5-Chloroindolin-2-One (3)5-Fluoroindoline (3)5-Methoxyindoline (4)5-Methylindolin-2-One (3)5-Methylindoline (3)6-Bromoindolin-2-One (8)6-Bromoindoline-2,3-Dione (4)6-Chloroindolin-2-One (5)6-Chloroindoline (3)6-Chloroindoline-2,3-Dione (5)6-Fluoroindolin-2-One (5)6-Fluoroindoline-2,3-Dione (3)7-Bromoindoline (3)7-Fluoroindolin-2-One (3)7-Fluoroindoline-2,3-Dione (6)7-Methylindoline-2,3-Dione (7)Indoline-2-Carboxylic Acid (3)Methyl Indoline-6-Carboxylate (3)Spiro[Cyclopropane-1,3'-Indolin]-2'-One (6)Tert-Butyl Indoline-1-Carboxylate (24)Spiroes (5)
Isoquinolines (2732)
Alcohols (4)Amides (10)Amines (12)Bromides (11)Carboxylic Acids (4)Chlorides (14)Esters (7)Ethers (8)Fluorinated Building Blocks (7)Nitriles (4)Bromoisoquinoline (69)Chloroisoquinoline (101)Dibromoisoquinoline (9)Dichloroisoquinoline (14)Dihydroisoquinoline (107)Fluoroisoquinoline (78)Methoxyisoquinoline (59)Methylisoquinoline (59)Nitroisoquinoline (23)1,3-Dichloroisoquinoline (14)1-Bromoisoquinoline (20)1-Chloroisoquinoline (20)1-Methylisoquinoline (9)1H-Benzo[De]Isoquinoline-1,3(2H)-Dione (14)2-Methyl-3,4-Dihydroisoquinolin-1(2H)-One (6)3-Bromoisoquinoline (10)3-Chloroisoquinoline (20)3-Methylisoquinoline (8)4-Bromoisoquinolin-1(2H)-One (3)4-Bromoisoquinoline (33)4-Chloroisoquinoline (11)4-Fluoroisoquinoline (6)5,6-Dihydro-[1,3]Dioxolo[4,5-G]Isoquinolino[3,2-A]Isoquinolin-7-Ium (4)5-Bromoisoquinoline (17)5-Chloroisoquinoline (6)5-Methoxyisoquinoline (6)5-Nitroisoquinoline (7)6-Bromo-3,4-Dihydroisoquinolin-1(2H)-One (6)6-Bromoisoquinoline (9)6-Methoxy-3,4-Dihydroisoquinolin-1(2H)-One (3)7-Bromo-3,4-Dihydroisoquinolin-1(2H)-One (6)7-Bromoisoquinoline (6)7-Chloroisoquinoline (6)7-Fluoroisoquinoline (3)7-Methoxyisoquinoline (5)8-Bromoisoquinoline (17)8-Chloroisoquinoline (7)8-Fluoroisoquinolin-1(2H)-One (3)8-Fluoroisoquinoline (10)8-Methoxyisoquinoline (7)Isoquinolin-1-Amine (18)Isoquinolin-1-Ol (9)Isoquinolin-3-Amine (13)Isoquinolin-3-Ol (6)Isoquinolin-4-Amine (16)Isoquinolin-4-Ol (9)Isoquinolin-4-Ylboronic Acid (3)Isoquinolin-5-Amine (9)Isoquinolin-5-Ol (7)Isoquinolin-8-Amine (6)Isoquinoline-1-Carbonitrile (7)Isoquinoline-1-Carboxylic Acid (10)Isoquinoline-3-Carboxylic Acid (7)Isoquinoline-4-Carboxylic Acid (12)Isoquinoline-5-Carboxylic Acid (5)Isoquinoline-5-Sulfonyl Chloride (3)Isoquinoline-6-Carboxylic Acid (3)Methyl Isoquinoline-3-Carboxylate (3)Methyl Isoquinoline-4-Carboxylate (6)Tert-Butyl 3,4-Dihydroisoquinoline-2(1H)-Carboxylate (19)
Isoxazoles (3406)
Alcohols (8)Aldehydes (9)Amides (16)Amines (25)Aryls (54)Bromides (11)Carboxylic Acids (18)Chlorides (19)Esters (11)Ethers (12)Fluorinated Building Blocks (12)Aminoisoxazole (7)Bromoisoxazole (5)Dihydroisoxazol (2)Dimethylisoxazole (17)Phenylisoxazole (28)(3-Phenylisoxazol-5-Yl)Methanol (7)3-Bromoisoxazole (7)3-Methyl-5-Phenylisoxazole (6)3-Methylisoxazole (30)3-Phenylisoxazol-5-Amine (8)5-(Thiophen-2-Yl)Isoxazole (5)5-Cyclopropylisoxazole (5)5-Methyl-3-Phenylisoxazole (4)5-Methylisoxazole (16)5-Phenylisoxazole-3-Carboxylic Acid (15)Ethyl Isoxazole-3-Carboxylate (8)Ethyl Isoxazole-4-Carboxylate (5)Isoxazol-3-Amine (7)Isoxazol-3-Ylmethanol (3)Isoxazol-5-Amine (15)Isoxazol-5-Ylmethanol (5)Isoxazole-3-Carboxylic Acid (8)Isoxazole-5-Carboxylic Acid (7)Methyl 5-Phenylisoxazole-3-Carboxylate (6)N-((5R,6R)-7-Oxo-4-Thia-1-Azabicyclo[3.2.0]Heptan-6-Yl)-3-Phenylisoxazole-4-Carboxamide (1)
Morpholines (4812)
Alcohols (26)Aldehydes (9)Amides (99)Amines (54)Aryls (137)Bromides (30)Carboxylic Acids (34)Chlorides (24)Esters (32)Ethers (17)Fluorinated Building Blocks (17)Ketones (8)Nitriles (5)Nitroes (12)Phenols (3)Sulfamides (11)Trifluoromethyls (5)Ureas (4)Benzylmorpholine (19)Cyanomorpholine (6)Dimorpholine (3)Ethylmorpholine (9)Isopropylmorpholine (9)Methylmorpholine (68)Morpholinobenzaldehyde (11)Morpholinobenzo (13)Morpholinobenzoic (8)Morpholinoethanone (5)Morpholinoethoxy (1)Morpholinoethyl (5)Morpholinomethyl (16)Morpholinophenyl (14)Morpholinopropoxy (6)Morpholinopyrimidin (6)Morpholinosulfonyl (7)Oxomorpholine (6)(R)-3-Phenylmorpholine (7)(S)-3-Phenylmorpholine (10)2,2-Dimethylmorpholine (5)2,6-Dimethylmorpholine (5)2-(Methoxymethyl)Morpholine (4)2-(Morpholin-2-Yl)Acetic Acid (5)2-Ethylmorpholine (3)2-Isopropylmorpholine (4)2-Methylmorpholine (7)3-(4-Morpholinophenyl)Oxazolidin-2-One (5)3-(Methoxymethyl)Morpholine (4)3-Ethylmorpholine (4)3-Isopropylmorpholine (5)3-Methylmorpholine (13)3-Phenylmorpholine (8)4-(2-Fluorophenyl)Morpholine (8)4-(3-Phenoxypropyl)Morpholine (7)4-(O-Tolyl)Morpholine (6)4-(Pyrazin-2-Yl)Morpholine (5)4-(Pyridin-4-Yl)Morpholine (5)4-(Pyrimidin-2-Yl)Morpholine (9)4-Benzylmorpholin-3-One (6)4-Morpholinoaniline (4)4-Morpholinobenzaldehyde (8)4-Morpholinobenzoic Acid (5)4-Phenylmorpholin-3-One (6)6,6-Dimethylmorpholine-3-Carboxylic Acid (5)Benzyl Morpholine-4-Carboxylate (5)Ethyl Morpholine-2-Carboxylate (4)Methyl 2-(Morpholin-3-Yl)Acetate (3)Methyl Morpholine-2-Carboxylate (4)Methyl Morpholine-3-Carboxylate (10)Morpholin-2-Ylmethanamine (8)Morpholin-2-Ylmethanol (12)Morpholin-3-Ylmethanol (14)Morpholine-2-Carbonitrile (4)Morpholine-2-Carboxylic Acid (12)Morpholine-3-Carboxylic Acid (10)Morpholino(Phenyl)Methanone (11)Tert-Butyl (Morpholin-2-Ylmethyl)Carbamate (3)Tert-Butyl 2-(Bromomethyl)Morpholine-4-Carboxylate (3)Tert-Butyl Morpholine-4-Carboxylate (23)Benzothiazoles (4)Piperidines (30)Pyridazines (4)Pyridines (24)Pyrimidines (4)Spiroes (33)Thiazoles (5)
Naphthyridines (942)
Bromides (4)Carboxylic Acids (3)Fluorinated Building Blocks (4)Ketones (4)1,5-Naphthyridine-3-Carboxylic Acid (6)1,6-Naphthyridine (22)1,7-Naphthyridine (23)2,7-Naphthyridine (5)3-Bromo-1,5-Naphthyridine (8)4-Bromo-1,5-Naphthyridine (5)4-Chloro-1,5-Naphthyridine (6)5,6,7,8-Tetrahydro-1,6-Naphthyridine (22)5,6,7,8-Tetrahydro-1,7-Naphthyridine (6)
Oxazines (235)
2,2-Dimethyl-2H-Benzo[B][1,4]Oxazin-3(4H)-One (3)2-Methyl-2H-Benzo[B][1,4]Oxazin-3(4H)-One (3)2H-Pyrido[3,2-B][1,4]Oxazin-3(4H)-One (8)3,4-Dihydro-2H-Pyrido[3,2-B][1,4]Oxazine (5)6-Amino-2H-Benzo[B][1,4]Oxazin-3(4H)-One (3)6-Bromo-2H-Benzo[B][1,4]Oxazin-3(4H)-One (7)6-Bromo-3,4-Dihydro-2H-Benzo[B][1,4]Oxazine (5)6-Chloro-2H-Benzo[B][1,4]Oxazin-3(4H)-One (8)6-Chloro-3,4-Dihydro-2H-Benzo[B][1,4]Oxazine (5)7-Bromo-3,4-Dihydro-2H-Benzo[B][1,4]Oxazine (7)Spiro[Benzo[D][1,3]Oxazine-4,4'-Piperidin]-2(1H)-One (5)
Oxazoles (2023)
Alcohols (9)Amides (10)Amines (16)Aryls (48)Bromides (12)Carboxylic Acids (16)Chlorides (14)Esters (18)Ethers (4)Fluorinated Building Blocks (5)Nitriles (6)Nitroes (4)Bioxazole (12)2-Bromooxazole (7)2-Chlorooxazole (7)2-Methyloxazole (19)2-Phenyloxazole (16)3,4-Diphenylisoxazole (8)4,5-Diphenyloxazole (7)4-(Oxazol-5-Yl)Aniline (3)4-Methyloxazole (9)5-(4-Nitrophenyl)Oxazole (3)5-Methyloxazole (5)6-Bromobenzo[D]Oxazole (3)Ethyl Oxazole-4-Carboxylate (8)Ethyl Oxazole-5-Carboxylate (3)Methyl Oxazole-4-Carboxylate (9)Oxazol-2-Amine (8)Oxazole-4-Carboxylic Acid (4)Oxazolo[4,5-B]Pyridine (7)Oxazolo[5,4-B]Pyridine (5)
Oxazolidines (1192)
Alcohols (6)Amides (50)Amines (4)Anhydrides (5)Aryls (25)Esters (7)Fluorinated Building Blocks (2)Dimethyloxazolidin (3)Dioxooxazolidin (2)Oxazolidinone (2)Phenyloxazolidin (7)Propionyloxazolidin (4)(S)-4-Benzyloxazolidin-2-One (1)3-(4-Morpholinophenyl)Oxazolidin-2-One (5)3-Phenyloxazolidin-2-One (4)4-Isopropyloxazolidine (6)Oxazolidine (21)Oxazolidine-2,5-Dione (9)Spiroes (6)
Oxazolines (1578)
Amides (4)Aryls (8)Esters (6)(3Ar,8As)-2-(Pyridin-2-Yl)-3A,8A-Dihydro-8H-Indeno[1,2-D]Oxazole (6)(3As,8Ar)-2-(Pyridin-2-Yl)-3A,8A-Dihydro-8H-Indeno[1,2-D]Oxazole (7)(4R,5S)-4,5-Diphenyl-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (5)(R)-4-Benzyl-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (7)(R)-4-Phenyl-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (7)(S)-4-Benzyl-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (8)(S)-4-Phenyl-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (6)1,3-Bis(4,5-Dihydrooxazol-2-Yl)Benzene (8)2,2'-(1,3-Diphenylpropane-2,2-Diyl)Bis(4,5-Dihydrooxazole) (5)2,2'-(2-Phenylethane-1,1-Diyl)Bis(4,5-Dihydrooxazole) (5)2,3-Dihydropyrazolo[5,1-B]Oxazole (7)2,6-Bis(4,5-Dihydrooxazol-2-Yl)Pyridine (12)2-(6-Benzhydrylpyridin-2-Yl)-4,5-Dihydrooxazole (10)2-(6-Cyclopropylpyridin-2-Yl)-4,5-Dihydrooxazole (10)2-(Benzo[B]Thiophen-2-Yl)-4,5-Dihydrooxazole (10)2-(Pyridin-2-Ylmethyl)-4,5-Dihydrooxazole (14)2-(Quinolin-2-Yl)-4,5-Dihydrooxazole (7)2-Phenyl-4,5-Dihydrooxazole (18)4,4',5,5'-Tetrahydro-2,2'-Bioxazole (10)4-(Tert-Butyl)-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (16)4-Isopropyl-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (16)Bis((3Ar,8As)-3A,8A-Dihydro-8H-Indeno[1,2-D]Oxazol-2-Yl)Methane (5)Bis((3As,8Ar)-3A,8A-Dihydro-8H-Indeno[1,2-D]Oxazol-2-Yl)Methane (5)Bis(4,5-Dihydrooxazol-2-Yl)Methane (22)
Piperazines (7243)
Alcohols (26)Alkenyls (10)Amides (273)Amines (36)Aryls (139)Bromides (10)Carboxylic Acids (31)Chlorides (22)Esters (38)Ethers (31)Fluorinated Building Blocks (28)Nitriles (17)Nitroes (14)Phenols (7)Sulfamides (12)Sulfones (4)Trifluoromethyls (7)Ureas (6)Isobutylpiperazine (6)Propylpiperazine (10)(9H-Fluoren-9-Yl)Methyl Piperazine-1-Carboxylate (7)1,4-Diphenylpiperazine (7)1-(2-Chlorophenyl)Piperazine (4)1-(2-Fluorophenyl)Piperazine (8)1-(3-Methoxyphenyl)Piperazine (4)1-(4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Phenyl)Piperazine (10)1-(5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridin-2-Yl)Piperazine (6)1-(Methylsulfonyl)Piperazine (5)1-(Phenylsulfonyl)Piperazine (17)1-(Piperidin-4-Yl)Piperazine (6)1-(Pyridin-3-Yl)Piperazine (13)1-(Pyridin-3-Ylmethyl)Piperazine (6)1-Benzhydryl-4-Benzylpiperazine (1)1-Benzhydrylpiperazine (9)1-Benzyl 4-(Tert-Butyl) Piperazine-1,4-Dicarboxylate (10)1-Benzyl-4-Methylpiperazine (10)1-Ethyl-4-Phenylpiperazine (4)1-Ethylpiperazine (6)1-Isopropylpiperazine (4)1-Methyl-4-(Pyridin-2-Yl)Piperazine (5)1-Methyl-4-Phenylpiperazine (22)1-Methylpiperazin-2-One (6)1-Methylpiperazine (21)10-(3-(Piperazin-1-Yl)Propyl)-10H-Phenothiazine (2)2-(4-(Benzyloxy)Phenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (10)2-(Piperazin-1-Yl)Benzonitrile (6)2-(Piperazin-1-Yl)Ethan-1-Amine (8)2-(Piperazin-1-Yl)Ethan-1-Ol (10)2-(Piperazin-1-Yl)Pyrazine (9)2-(Piperazin-1-Yl)Pyrimidine (11)2-Phenylpiperazine (3)3-(Piperazin-1-Yl)Aniline (3)3-(Piperazin-1-Yl)Benzoic Acid (3)3-(Piperazin-1-Yl)Pyridazine (7)3-Methylpiperazin-2-One (7)4-(Piperazin-1-Yl)Benzaldehyde (5)4-(Piperazin-1-Yl)Pyrimidine (13)Benzyl 2-Methylpiperazine-1-Carboxylate (7)Benzyl 3-(Hydroxymethyl)Piperazine-1-Carboxylate (6)Benzyl 3-Methylpiperazine-1-Carboxylate (5)Phenyl(Piperazin-1-Yl)Methanone (20)Piperazine-2,5-Dione (9)Tert-Butyl 3-Oxopiperazine-1-Carboxylate (7)Tert-Butyl 4-(Pyridin-2-Yl)Piperazine-1-Carboxylate (12)Tert-Butyl 4-Benzylpiperazine-1-Carboxylate (13)Tert-Butyl 4-Phenylpiperazine-1-Carboxylate (23)Tert-Butyl Piperazine-1-Carboxylate (30)Furans (4)Indoles (5)Piperidines (5)Pyridines (16)Pyrimidines (25)Spiroes (14)
Piperidines (21679)
Alcohols (132)Aldehydes (17)Alkenyls (18)Amides (436)Amines (177)Aryls (311)Bromides (31)Carboxylic Acids (91)Chlorides (68)Esters (69)Ethers (82)Fluorinated Building Blocks (95)Iodides (4)Ketones (66)Nitriles (54)Nitroes (13)Oximes (5)Phenols (5)Sulfamides (25)Sulfonates (10)Sulfones (6)Sulfonyl Chlorides (4)Trifluoromethyls (8)Bipiperidine (12)Chloropiperidine (6)Dimethylpiperidine (46)Ethynylpiperidine (5)Fluoropiperidine (33)Formylpiperidine (9)Isopropylpiperidine (11)Piperidine-Carboxylate (3)Piperidineacetic (5)Piperidinecarboxylic (6)Piperidinyl-Phenyl-Boronic Acid/Ester (32)Propylpiperidine (8)Azetidines (16)Benzimidazoles (18)Benzisoxazoles (4)Benzofurans (5)Indoles (4)Morpholines (30)Oxadiazoles (4)Oxetanes (8)Piperazines (5)Pyrazines (8)Pyridines (15)Pyrimidines (4)Pyrrolidines (36)Spiroes (139)Tetrahydrofurans (9)Tetrahydropyrans (5)Thiophenes (9)Other Aromatic Heterocycles (14)(9H-Fluoren-9-Yl)Methyl Piperidine-1-Carboxylate (16)(E)-N-(3-Oxo-1-Phenyl-3-(Piperidin-1-Yl)Prop-1-En-2-Yl)Benzamide (6)(R)-2-Phenylpiperidine (10)(R)-3-Aminopiperidin-2-One (3)(R)-N-(Piperidin-3-Yl)-7H-Pyrrolo[2,3-D]Pyrimidin-4-Amine (5)(S)-2-Phenylpiperidine (11)(S)-N-Phenylpiperidine-2-Carboxamide (1)1,3-Dihydrospiro[Indene-2,4'-Piperidine] (5)1,4'-Bipiperidine (8)1-((Benzyloxy)Carbonyl)Piperidine-2-Carboxylic Acid (5)1-((Benzyloxy)Carbonyl)Piperidine-3-Carboxylic Acid (3)1-(Methylsulfonyl)Piperidine (6)1-(Phenylsulfonyl)Piperidine (23)1-(Piperidin-1-Yl)Ethan-1-One (13)1-(Piperidin-4-Yl)Piperazine (6)1-Benzyl 3-Methyl Piperidine-1,3-Dicarboxylate (3)1-Benzyl-N-Methylpiperidin-3-Amine (6)1-Benzylpiperidin-3-One (5)1-Benzylpiperidin-4-Amine (8)1-Benzylpiperidin-4-One (7)1-Isopropylpiperidine (5)1-Methyl-4-Phenylpiperidine (4)1-Methylpiperidin-2-One (3)1-Oxa-8-Azaspiro[4.5]Decane (5)1-Phenylpiperidin-2-One (8)2,2-Dimethylpiperidin-4-One (6)2,2-Dimethylpiperidine (21)2,7-Diazaspiro[3.5]Nonane (7)2,8-Diazaspiro[4.5]Decan-3-One (6)2,8-Diazaspiro[4.5]Decane (5)2-(Piperidin-1-Yl)Ethan-1-Amine (8)2-(Piperidin-1-Yl)Ethan-1-Ol (10)2-(Piperidin-1-Yl)Pyridine (11)2-(Piperidin-1-Yl)Pyrimidine (16)2-(Piperidin-2-Yl)Ethan-1-Ol (8)2-(Piperidin-4-Yl)Ethan-1-Ol (5)2-(Piperidin-4-Yl)Pyridine (5)2-Methylpiperidin-4-One (6)2-Methylpiperidine (14)2-Phenylpiperidine (14)3,3-Difluoropiperidin-4-One (3)3,3-Difluoropiperidine (29)3,3-Dimethylpiperidin-4-One (3)3,3-Dimethylpiperidine (5)3-(1-Oxoisoindolin-2-Yl)Piperidine-2,6-Dione (21)3-(Piperidin-1-Yl)Pyridine (5)3-(Piperidin-4-Yl)-1H-Indazole (5)3-(Piperidin-4-Yl)-1H-Indole (8)3-(Piperidin-4-Yl)Pyridine (5)3-Fluoropiperidin-4-One (3)3-Fluoropiperidine (26)3-Methoxypiperidine (7)3-Methylpiperidin-4-One (3)3-Methylpiperidine (10)4,4-Difluoropiperidine (5)4-(1H-Pyrazol-1-Yl)Piperidine (11)4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)-1,2,3,6-Tetrahydropyridine (11)4-(Azetidin-1-Yl)Piperidine (6)4-(Piperidin-1-Yl)Pyrimidine (9)4-(Pyrrolidin-1-Yl)Piperidine (6)4-Benzylpiperidine (18)4-Hydrazineylpiperidine (5)4-Hydroxypiperidin-2-One (3)4-Methyl-1-Phenylpiperidine (6)4-Methylenepiperidine (14)4-Oxopiperidine-2-Carboxylic Acid (3)4-Phenylpiperidin-4-Ol (10)5-Hydroxypiperidin-2-One (3)6-Azaspiro[2.5]Octane (10)6-Oxopiperidine-2-Carboxylic Acid (3)7-Azaspiro[3.5]Nonane (7)Azepane (25)Benzyl 3-Hydroxypiperidine-1-Carboxylate (7)Benzyl 4-Oxopiperidine-1-Carboxylate (12)Benzyl Piperidin-4-Ylcarbamate (7)Benzyl Piperidine-1-Carboxylate (50)Benzyl-3-Aminopiperidine-1-Carboxylate (6)Ethyl Piperidine-1-Carboxylate (5)Ethyl Piperidine-2-Carboxylate (9)Ethyl Piperidine-3-Carboxylate (14)Ethyl Piperidine-4-Carboxylate (10)Methyl 4-Oxopiperidine-2-Carboxylate (3)Methyl Piperidine-2-Carboxylate (12)Methyl Piperidine-3-Carboxylate (19)N-Benzylpiperidin-4-Amine (3)N-Methylpiperidin-3-Amine (5)N-Phenylpiperidine-2-Carboxamide (2)Phenyl(Piperidin-1-Yl)Methanone (21)Phenyl(Piperidin-4-Yl)Methanone (21)Piperidin-2-Ylmethanamine (6)Piperidin-2-Ylmethanol (11)Piperidin-3-Amine (17)Piperidin-3-Ol (27)Piperidin-3-One (13)Piperidin-3-Ylmethanamine (5)Piperidin-3-Ylmethanol (15)Piperidin-4-Amine (14)Piperidin-4-Ol (44)Piperidin-4-Ylmethanamine (7)Piperidin-4-Ylmethanol (11)Piperidine-2,4-Dione (5)Piperidine-2,6-Dione (13)Piperidine-2-Carboxylic Acid (26)Piperidine-3-Carboxamide (5)Piperidine-3-Carboxylic Acid (14)Piperidine-4-Carboxylic Acid (12)Quinuclidine (5)Spiro[Benzo[D][1,3]Oxazine-4,4'-Piperidin]-2(1H)-One (5)Spiro[Chromane-2,4'-Piperidin]-4-One (6)Spiro[Indene-2,4'-Piperidin]-1(3H)-One (5)Spiro[Indoline-3,4'-Piperidin]-2-One (18)Spiro[Indoline-3,4'-Piperidine] (5)Tert-Butyl (2-Oxopiperidin-3-Yl)Carbamate (4)Tert-Butyl (6-Oxopiperidin-3-Yl)Carbamate (3)Tert-Butyl (Piperidin-3-Ylmethyl)Carbamate (5)Tert-Butyl 2-Oxopiperidine-1-Carboxylate (3)Tert-Butyl 3-Bromopiperidine-1-Carboxylate (4)Tert-Butyl 4-Oxopiperidine-1-Carboxylate (16)Tert-Butyl 4-Phenoxypiperidine-1-Carboxylate (12)Tert-Butyl 4-Phenylpiperidine-1-Carboxylate (16)Tert-Butyl Piperidin-3-Ylcarbamate (9)
Purines (2028)
Amides (7)Amines (12)Aryls (4)Chlorides (6)(R)-N-(9-(Tetrahydrofuran-2-Yl)-9H-Purin-6-Yl)Benzamide (5)2-(Acetoxymethyl)-5-(9H-Purin-9-Yl)Tetrahydrofuran-3,4-Diyl Diacetate (6)2-(Hydroxymethyl)-5-(9H-Purin-9-Yl)Tetrahydrofuran-3,4-Diol (28)2-Chloro-7H-Purine (3)2-Chloro-9H-Purine (5)7H-Purin-2-Amine (6)7H-Purin-6-Amine (9)9-((3As,4R,6Ar)-Tetrahydrofuro[3,4-D][1,3]Dioxol-4-Yl)-9H-Purine (7)9-(Tetrahydrofuran-2-Yl)-9H-Purin-6-Amine (30)9-(Tetrahydrofuran-2-Yl)-9H-Purin-6-Ol (6)9-(Tetrahydrofuran-2-Yl)-9H-Purine (2)9H-Purin-6-Amine (3)
Pyrans (1099)
Aryls (3)Carboxylic Acids (2)Esters (5)Ethers (5)Ketones (7)Thioglucopyranoside (4)(S)-2-Phenylchroman-4-One (17)1-(Tetrahydro-2H-Pyran-2-Yl)-1H-Indazole (17)2,2-Dimethylchromane (5)2-(2-Oxo-2H-Chromen-4-Yl)Acetic Acid (4)2-Oxo-2H-Chromene-3-Carbonitrile (4)2-Oxo-2H-Chromene-3-Carboxylic Acid (14)2H-Chromene (16)3,4-Dihydro-2H-Pyran (11)3,6-Dihydro-2H-Pyran (8)3-Acetyl-2H-Chromen-2-One (3)3-Hydroxy-2-Phenyl-4H-Chromen-4-One (21)4-(Trifluoromethyl)-2H-Chromen-2-One (6)4-Hydroxy-2H-Chromen-2-One (10)4-Methyl-2H-Chromen-2-One (27)5-Fluorochroman-4-One (3)5-Fluorochromane (6)5-Hydroxy-2-Phenyl-4H-Chromen-4-One (36)5-Methoxy-2-Phenyl-4H-Chromen-4-One (9)6-Bromochroman-4-One (6)6-Chlorochroman-4-One (6)6-Chlorochromane (6)6-Fluorochroman-4-One (5)6-Fluorochromane (6)6-Methoxychromane (5)7-(Diethylamino)-2H-Chromen-2-One (9)7-Bromochroman-4-One (4)7-Bromochromane (7)7-Chlorochromane (5)7-Fluorochroman-4-One (5)7-Hydroxy-2H-Chromen-2-One (23)7-Methoxy-2H-Chromen-2-One (12)7-Methoxychroman-4-One (3)8-Bromochroman-4-One (4)8-Bromochromane (6)8-Chlorochromane (5)8-Fluorochromane (5)8-Methylchroman-4-One (4)8-Methylchromane (11)Chroman-2-One (5)Chroman-2-Ylmethanamine (3)Chroman-3-Amine (3)Chroman-4-Amine (20)Chroman-4-Ol (4)Chromane-2-Carboxylic Acid (6)Chromane-3-Carboxylic Acid (4)Dihydro-2H-Pyran-2,6(3H)-Dione (5)Ethyl 2-Oxo-2H-Chromene-3-Carboxylate (5)Isochromane (11)Spiro[Chromane-2,4'-Piperidin]-4-One (6)Tetrahydro-2H-Pyran-2-One (15)Tetrahydro-2H-Thiopyran (10)Tetrahydro-4H-Pyran-4-One (12)Thiochroman-4-One (10)
Pyrazines (3356)
Alcohols (6)Aldehydes (4)Alkynyls (6)Amides (17)Amines (19)Aryls (13)Bromides (9)Carboxylic Acids (12)Chlorides (31)Esters (9)Ethers (19)Fluorinated Building Blocks (8)Trifluoromethyls (5)Dibromopyrazine (7)Dichloropyrazine (21)Dihydropyrazine (6)Dimethylpyrazine (30)Ethoxypyrazine (5)Ethylpyrazine (7)Fluoropyrazine (6)Hydroxypyrazine (4)Iodopyrazine (17)Methoxypyrazine (36)Phenylpyrazin (5)Pyrazino (5)Pyrazino-Nitrile (50)Piperidines (8)1,2,3,4-Tetrahydropyrido[2,3-B]Pyrazine (5)1-(Pyrazin-2-Yl)Ethan-1-One (5)1H-Pyrazolo[3,4-B]Pyrazine (5)2,3-Diphenylpyrazine (5)2,5-Dibromopyrazine (4)2,5-Dichloropyrazine (4)2,6-Dibromopyrazine (9)2,6-Dichloropyrazine (14)2-(Piperazin-1-Yl)Pyrazine (9)2-(Trifluoromethyl)Pyrazine (6)2-Bromo-3-Methoxypyrazine (5)2-Bromo-3-Methylpyrazine (5)2-Bromo-5-Chloropyrazine (6)2-Bromo-6-Chloropyrazine (5)2-Bromo-6-Methylpyrazine (5)2-Bromopyrazine (15)2-Chloro-3-Methylpyrazine (7)2-Chloro-6-Methylpyrazine (12)2-Chloropyrazine (24)2-Ethoxypyrazine (3)2-Ethylpyrazine (9)2-Fluoropyrazine (7)2-Iodopyrazine (25)2-Methoxypyrazine (45)2-Methylpyrazine (16)2-Phenylpyrazine (12)3-Chloropyrazin-2-Amine (6)3-Chloropyrazine-2-Carbonitrile (6)4,5,6,7-Tetrahydropyrazolo[1,5-A]Pyrazine (7)4-(Pyrazin-2-Yl)Morpholine (5)5,6,7,8-Tetrahydro-[1,2,4]Triazolo[4,3-A]Pyrazine (11)5,6,7,8-Tetrahydroimidazo[1,2-A]Pyrazine (15)5,6,7,8-Tetrahydroimidazo[1,5-A]Pyrazine (11)5-Bromopyrazin-2-Amine (13)5-Bromopyrazin-2-Ol (5)5-Chloropyrazin-2-Amine (9)5H-Pyrrolo[2,3-B]Pyrazine (14)6-Bromopyrazin-2-Amine (5)6-Chloropyrazin-2-Amine (11)[1,2,4]Triazolo[1,5-A]Pyrazine (9)[1,2,4]Triazolo[4,3-A]Pyrazine (11)Ethyl Pyrazine-2-Carboxylate (10)Imidazo[1,5-A]Pyrazine (9)Methyl 3-Bromopyrazine-2-Carboxylate (5)Methyl 6-Bromopyrazine-2-Carboxylate (7)Methyl 6-Chloropyrazine-2-Carboxylate (5)Methyl Pyrazine-2-Carboxylate (19)Pyrazin-2-Amine (22)Pyrazin-2-Ol (7)Pyrazin-2-Ylmethanamine (12)Pyrazine-2-Carbaldehyde (17)Pyrazine-2-Carbohydrazide (3)Pyrazine-2-Carbonitrile (15)Pyrazine-2-Carboxamide (10)Pyrazine-2-Carboxylic Acid (34)Pyrazolo[1,5-A]Pyrazine (5)Pyrido[2,3-B]Pyrazine (12)Pyrido[3,4-B]Pyrazine (11)Tert-Butyl Pyrazin-2-Ylcarbamate (8)
Pyrazoles (16147)
Alcohols (42)Aldehydes (41)Amides (46)Amines (159)Aryls (122)Bromides (38)Carboxylic Acids (71)Chlorides (27)Difluoromethyls (11)Esters (33)Ethers (39)Fluorinated Building Blocks (53)Iodides (18)Ketones (14)Nitriles (39)Nitroes (28)Sulfamides (4)Sulfonyl Chlorides (4)Trifluoromethyls (27)Bipyrazole (5)Chloropyrazolo (33)Dichloropyrazolo (11)Dihydropyrazolo (28)Dimethylpyrazolo (6)Ethylpyrazole (7)Fluoropyrazolo (9)Hydroxypyrazolo (10)Methoxypyrazolo (16)Methyl-Pyrazole-Carboxylic Acid (147)Methyl-Pyrazolone (87)Methylpyrazolo (32)Pyrazolopyridine-Carboxylic Acid (48)Tetrahydropyrazolo (23)(1-Methyl-1H-Pyrazol-3-Yl)Methanol (7)(1-Phenyl-1H-Pyrazol-4-Yl)Boronic Acid (4)1,3-Dimethyl-1H-Pyrazole (28)1,3-Diphenyl-1H-Pyrazole (6)1,4,5,6-Tetrahydropyrrolo[3,4-C]Pyrazole (6)1,5-Diphenyl-1H-Pyrazole (13)1-(2-Chlorophenyl)-1H-Pyrazole (3)1-(3-Chlorophenyl)-1H-Pyrazole (3)1-(4-Methoxybenzyl)-1H-Pyrazole (5)1-(Azetidin-3-Yl)-1H-Pyrazole (7)1-(Tetrahydro-2H-Pyran-2-Yl)-1H-Pyrazole (9)1-(Tetrahydro-2H-Pyran-4-Yl)-1H-Pyrazole (6)1-Benzyl-1H-Pyrazolo[3,4-B]Pyridine (9)1-Benzyl-4-Bromo-1H-Pyrazole (5)1-Benzyl-4-Iodo-1H-Pyrazole (5)1-Cyclopentyl-1H-Pyrazole (5)1-Cyclopropyl-1H-Pyrazole (9)1-Methyl-1H-Pyrazol-3-Amine (14)1-Methyl-3-Phenyl-1H-Pyrazole (21)1-Methyl-4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)-1H-Pyrazole (6)1-Methyl-5-Phenyl-1H-Pyrazole (7)1-Phenyl-1H-Pyrazol-3-Amine (3)1-Phenyl-1H-Pyrazol-3-Ol (3)1-Phenyl-1H-Pyrazol-5-Amine (4)1-Phenyl-1H-Pyrazole-3-Carboxylic Acid (10)1-Phenyl-1H-Pyrazole-4-Carbaldehyde (8)1-Phenyl-1H-Pyrazole-4-Carbonitrile (17)1-Phenyl-1H-Pyrazole-4-Carboxylic Acid (19)1-Phenyl-1H-Pyrazole-5-Carboxylic Acid (3)1-Phenyl-3-(Trifluoromethyl)-1H-Pyrazole (5)1-Trityl-1H-Pyrazole (5)1H-Pyrazol-4-Amine (6)1H-Pyrazol-4-Ol (3)1H-Pyrazol-5-Amine (19)1H-Pyrazole-4-Carbaldehyde (30)1H-Pyrazole-4-Carbonitrile (5)1H-Pyrazole-4-Carboxylic Acid (9)1H-Pyrazole-4-Sulfonyl Chloride (8)1H-Pyrazole-5-Carboxamide (3)1H-Pyrazole-5-Carboxylic Acid (10)1H-Pyrazolo[3,4-B]Pyrazine (5)1H-Pyrazolo[3,4-B]Pyridin-5-Amine (3)1H-Pyrazolo[3,4-D]Pyrimidin-4-Amine (5)1H-Pyrazolo[4,3-D]Pyrimidine (5)2,3-Dihydropyrazolo[5,1-B]Oxazole (7)2,4-Dihydro-3H-Pyrazol-3-One (10)2,6-Di(1H-Pyrazol-1-Yl)Pyridine (5)2-(1H-Pyrazol-1-Yl)Pyridine (16)2-(1H-Pyrazol-1-Yl)Pyrimidine (6)2-(1H-Pyrazol-3-Yl)Pyridine (5)2-Phenyl-2,4-Dihydro-3H-Pyrazol-3-One (24)3-(Tert-Butyl)-1-Phenyl-1H-Pyrazole (4)3-Bromo-1-Methyl-1H-Pyrazole (16)3-Bromopyrazolo[1,5-A]Pyridine (6)3-Bromopyrazolo[1,5-A]Pyrimidine (11)3-Chloro-1-Methyl-1H-Pyrazole (7)3-Cyclopropyl-1H-Pyrazole (5)3-Iodo-1-Methyl-1H-Pyrazole (6)3-Iodo-1H-Pyrazolo[3,4-B]Pyridine (4)3-Methyl-1-Phenyl-1H-Pyrazole (23)3-Phenyl-1H-Pyrazol-5-Amine (11)4,5,6,7-Tetrahydro-1H-Pyrazolo[3,4-C]Pyridine (7)4,5,6,7-Tetrahydro-1H-Pyrazolo[4,3-C]Pyridine (14)4,5,6,7-Tetrahydropyrazolo[1,5-A]Pyrazine (7)4,5,6,7-Tetrahydropyrazolo[1,5-A]Pyridine (6)4-(1H-Pyrazol-1-Yl)Benzoic Acid (3)4-(1H-Pyrazol-1-Yl)Piperidine (11)4-Bromo-1-Methyl-1H-Pyrazole (14)4-Bromo-1-Phenyl-1H-Pyrazole (11)4-Bromo-1H-Pyrazolo[3,4-B]Pyridine (4)4-Bromopyrazolo[1,5-A]Pyridine (8)4-Chloro-1H-Pyrazole (10)4-Chloro-1H-Pyrazolo[3,4-B]Pyridine (5)4-Chloro-1H-Pyrazolo[3,4-D]Pyrimidine (5)4-Iodo-1H-Pyrazole (9)4-Methyl-1-Phenyl-1H-Pyrazole (6)4-Methyl-1H-Pyrazole (11)4-Nitro-1H-Pyrazole (8)4-Phenyl-1H-Pyrazole (18)5,6-Dihydro-4H-Pyrrolo[1,2-B]Pyrazole (8)5-(Difluoromethyl)-1H-Pyrazole (6)5-Bromo-1H-Pyrazole (11)5-Bromo-1H-Pyrazolo[3,4-B]Pyridine (11)5-Bromo-1H-Pyrazolo[3,4-C]Pyridine (5)5-Chloro-1H-Pyrazole (8)5-Chloro-1H-Pyrazolo[3,4-B]Pyridine (3)5-Chloropyrazolo[1,5-A]Pyrimidine (24)5-Cyclopropyl-1H-Pyrazole (6)5-Iodo-1H-Pyrazole (4)5-Nitro-1H-Pyrazole (9)5-Nitro-1H-Pyrazolo[3,4-B]Pyridine (3)5-Phenyl-1H-Pyrazole-3-Carboxylic Acid (15)6,7-Dihydro-5H-Pyrazolo[5,1-B][1,3]Oxazine (13)6-Bromopyrazolo[1,5-A]Pyridine (8)6-Bromopyrazolo[1,5-A]Pyrimidine (7)6-Chloro-1H-Pyrazolo[3,4-D]Pyrimidine (12)7-Chloropyrazolo[1,5-A]Pyrimidine (8)Ethyl 1-Phenyl-1H-Pyrazole-3-Carboxylate (4)Ethyl 1-Phenyl-1H-Pyrazole-4-Carboxylate (13)Ethyl 1H-Pyrazole-4-Carboxylate (11)Ethyl Pyrazolo[1,5-A]Pyridine-3-Carboxylate (11)Ethyl Pyrazolo[1,5-A]Pyrimidine-3-Carboxylate (6)Methyl 1H-Pyrazole-4-Carboxylate (10)Methyl 1H-Pyrazolo[3,4-B]Pyridine-5-Carboxylate (3)Methyl Pyrazolo[1,5-A]Pyridine-3-Carboxylate (16)Pyrazolo[1,5-A]Pyrazine (5)Pyrazolo[1,5-A]Pyridine-2-Carboxylic Acid (5)Pyrazolo[1,5-A]Pyridine-3-Carboxylic Acid (13)Pyrazolo[1,5-A]Pyrimidine-3-Carboxylic Acid (7)Tert-Butyl 1H-Pyrazolo[3,4-B]Pyridine-1-Carboxylate (7)Tert-Butyl 4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)-1H-Pyrazole-1-Carboxylate (3)Tetrahydropyrans (4)Thiophenes (12)
Pyridazines (2753)
Alcohols (8)Amides (20)Amines (10)Aryls (15)Bromides (10)Carboxylic Acids (10)Chlorides (32)Esters (11)Ethers (7)Fluorinated Building Blocks (4)Ketones (4)Nitriles (5)Aminopyridazine (12)Dihydropyridazine (26)Dimethylpyridazine (9)Hexahydropyridazines (9)2-Methylpyridazin-3(2H)-One (7)3,6-Dichloropyridazine (20)3-(Piperazin-1-Yl)Pyridazine (7)3-(Trifluoromethyl)Pyridazine (3)3-Bromopyridazine (25)3-Chloro-4-Methylpyridazine (7)3-Chloropyridazine (44)3-Iodopyridazine (5)3-Methoxypyridazine (17)3-Methylpyridazine (6)3-Phenylpyridazine (8)4-Aminopyridazin-3(2H)-One (4)4-Bromopyridazin-3(2H)-One (3)4-Bromopyridazine (5)4-Chloropyridazin-3(2H)-One (4)4-Chloropyridazine (16)4-Methoxypyridazine (8)4-Methylpyridazine (3)6-Chloropyridazin-3(2H)-One (5)6-Chloropyridazin-3-Amine (5)[1,2,4]Triazolo[4,3-B]Pyridazine (10)Ethyl Pyridazine-3-Carboxylate (6)Methyl Pyridazine-3-Carboxylate (7)Pyridazin-3-Amine (18)Pyridazin-3-Ol (3)Pyridazin-4-Amine (10)Pyridazine-3-Carboxylic Acid (7)Morpholines (4)
Pyridines (70646)
Acyl Chlorides (13)Alcohols (160)Aldehydes (54)Alkenyls (36)Alkynyls (17)Amides (267)Amidines (13)Amines (394)Aryls (178)Bromides (398)Carboxylic Acids (172)Chlorides (372)Difluoromethyls (56)Esters (230)Ethers (239)Fluorinated Building Blocks (296)Hydrazides (17)Hydrazines (27)Iodides (78)Ketones (98)Nitriles (126)Nitroes (61)Oximes (11)Phenols (4)Sulfamides (19)Sulfides (18)Sulfones (14)Sulfonyl Chlorides (5)Thioureas (4)Trifluoromethyls (39)Acetylpyridin (5)Aminopicolinate (8)Benzyloxypyridine (36)Bipyridine (200)Bromopicolinate (19)Bromopicolinic (4)Bromopicolinic Acid (5)Bromopicolinonitrile (6)Bromopyrido (13)Butylpyridine (5)Chloroisonicotinic Acid (5)Chloronicotinaldehyde (23)Chloronicotinamide (11)Chloronicotinate (45)Chloronicotinic (41)Cyanopicolinate (7)Cyanopicolinic (6)Cyanopyridine (34)Cyclopropoxypyridine (6)Cyclopropylpyridine (30)Dibromopicolinate (4)Dibromopicolinic Acid (3)Dibromopyridine (36)Dichloropicolinate (9)Dichloropicolinonitrile (4)Dichloropyridine (120)Difluoropicolinic Acid (3)Difluoropyridine (58)Dihydropyrido (27)Diiodopyridine (13)Dimethoxypicolinic Acid (5)Dimethoxypyridine (33)Dimethylpyridine (143)Diphenylpyridine (6)Dipyridine (26)Ethoxypicolinic Acid (3)Ethoxypyridine (35)Ethylpyridine (36)Ethynylpicolinate (3)Fluoro-Pyridine--Boronate (79)Fluoropicolinaldehyde (8)Fluoropicolinamide (8)Fluoropicolinate (22)Fluoropyridine (287)Fluoropyrido (3)Formylpyridine (23)Hexahydropyrido (7)Hydrazinylpyridine (22)Hydroxymethylpyridine (26)Hydroxypicolinate (13)Hydroxypicolinic (9)Hydroxypicolinonitrile (7)Iodopicolinate (5)Iodopicolinic Acid (4)Iodopyridine (167)Isopropoxypyridine (20)Isopropylpyridine (32)Methoxyisonicotinaldehyde (10)Methoxynicotinaldehyde (18)Methoxypicolinaldehyde (13)Methoxypicolinamide (13)Methoxypicolinate (21)Methoxypicolinic (18)Methoxypicolinic Acid (16)Methoxypicolinimidamide (5)Methoxypicolinonitrile (13)Methoxypyridine (341)Methylisonicotinamide (8)Methylnicotinaldehyde (30)Methylnicotinamide (13)Methylpicolinaldehyde (16)Methylpicolinamide (15)Methylpicolinate (27)Methylpicolinic (23)Methylpicolinonitrile (28)Methylpyrido (13)Nitropyridine (392)Oxopyridin (2)Picolinate (41)Pyridinecarbonitrile (50)Pyridyl-Benzene-Boronic Acid/Ester (16)Pyridyl-Phenyl-Boronate (2)Pyridylmethyl (6)Terpyridine (32)Tetrachloropyridine (5)Tetrafluoropyridine (21)Tetrahydropyridine (49)Tetrahydropyrido (32)Tribromopyridine (9)Trichloropyridine (17)Trifluoropyridine (8)Trimethylpyridine (5)Azetidines (4)Benzimidazoles (6)Imidazoles (4)Indoles (5)Morpholines (24)Piperazines (16)Piperidines (15)Pyrroles (7)Pyrrolidines (2)Thiadiazoles (7)Thiophenes (17)Thiophenols (6)Triazoles (13)Other Aromatic Heterocycles (2)(2-Chloropyridin-4-Yl)Boronic Acid (6)(3-Bromopyridin-2-Yl)Methanol (7)(3-Chloropyridin-2-Yl)Methanamine (6)(3-Fluoropyridin-2-Yl)Methanamine (5)(3-Methylpyridin-2-Yl)Methanamine (5)(3-Methylpyridin-2-Yl)Methanol (10)(3Ar,8As)-2-(Pyridin-2-Yl)-3A,8A-Dihydro-8H-Indeno[1,2-D]Oxazole (6)(3As,8Ar)-2-(Pyridin-2-Yl)-3A,8A-Dihydro-8H-Indeno[1,2-D]Oxazole (7)(4R,5S)-4,5-Diphenyl-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (5)(5-(Trifluoromethyl)Pyridin-3-Yl)Boronic Acid (4)(5-Bromopyridin-2-Yl)Methanamine (5)(5-Bromopyridin-2-Yl)Methanol (5)(5-Chloropyridin-2-Yl)Methanamine (5)(6-Methylpyridin-2-Yl)Methanamine (6)(6-Methylpyridin-2-Yl)Methanol (5)(6-Phenoxypyridin-3-Yl)Boronic Acid (5)(R)-3-(Pyrrolidin-2-Yl)Pyridine (9)(R)-4-Benzyl-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (7)(R)-4-Phenyl-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (7)(S)-4-Benzyl-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (8)(S)-4-Phenyl-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (6)1,1'-Diphenyl-[4,4'-Bipyridine]-1,1'-Diium (7)1,10-Phenanthrolin-4-Ol (3)1,2,3,4-Tetrahydropyrido[2,3-B]Pyrazine (5)1,2,3,6-Tetrahydropyridine (22)1,3-Dihydro-2H-Pyrrolo[2,3-B]Pyridin-2-One (19)1,3-Dihydro-2H-Pyrrolo[2,3-C]Pyridin-2-One (5)1,3-Dihydro-2H-Pyrrolo[3,2-B]Pyridin-2-One (11)1,4-Di(Pyridin-4-Yl)Benzene (5)1-(5-Bromopyridin-2-Yl)Piperazine (6)1-(Phenylsulfonyl)-1H-Pyrrolo[2,3-B]Pyridine (30)1-(Pyridin-2-Yl)Ethan-1-Amine (15)1-(Pyridin-2-Yl)Ethan-1-One (36)1-(Pyridin-2-Yl-L2-Azaneyl)Ethan-1-One (12)1-(Pyridin-3-Yl)Ethan-1-Amine (17)1-(Pyridin-3-Yl)Ethan-1-One (29)1-(Pyridin-3-Yl)Piperazine (13)1-(Pyridin-3-Ylmethyl)Piperazine (6)1-(Pyridin-4-Yl)Ethan-1-Amine (23)1-(Pyridin-4-Yl)Ethan-1-One (14)1-Benzyl-1,2,3,6-Tetrahydropyridine (9)1-Benzyl-1H-Pyrazolo[3,4-B]Pyridine (9)1-Methyl-4-(Pyridin-2-Yl)Piperazine (5)1-Methylpyridin-1-Ium Iodide (5)1-Phenyl-3-(4-(Pyridin-4-Yloxy)Phenyl)Urea (8)1H-Imidazo[4,5-B]Pyridine (14)1H-Imidazo[4,5-C]Pyridine (12)1H-Pyrazolo[3,4-B]Pyridin-5-Amine (3)1H-Pyrrolo[2,3-C]Pyridin-7-Ol (3)1H-Pyrrolo[2,3-C]Pyridine-2-Carboxylic Acid (4)1H-Pyrrolo[3,2-B]Pyridine-3-Carboxylic Acid (6)2,2':6',2''-Terpyridine (15)2,2-Dimethyl-1-(Pyridin-2-Yl-L2-Azaneyl)Propan-1-One (7)2,3'-Bipyridine (11)2,3,4,5-Tetrahydro-1H-Pyrido[4,3-B]Indole (12)2,3,4,9-Tetrahydro-1H-Pyrido[3,4-B]Indole (13)2,3,5-Trichloropyridine (13)2,3,6-Trichloropyridine (16)2,3-Dibromopyridine (21)2,3-Dichloropyridine (38)2,3-Difluoropyridine (13)2,3-Dihydro-1H-Pyrrolo[2,3-B]Pyridine (10)2,3-Dihydro-1H-Pyrrolo[2,3-C]Pyridine (5)2,3-Dihydro-1H-Pyrrolo[3,2-C]Pyridine (7)2,3-Dihydro-1H-Pyrrolo[3,4-C]Pyridine (6)2,3-Dihydro-[1,4]Dioxino[2,3-B]Pyridine (5)2,3-Dihydropyridin-2-One (5)2,3-Dimethoxypyridine (7)2,3-Dimethylpyridine (8)2,4,6-Trichloropyridine (5)2,4,6-Triphenylpyridine (7)2,4-Dibromopyridine (5)2,4-Dichloro-6-Methylpyridine (8)2,4-Dichloropyridine (50)2,4-Dimethylpyridine (5)2,5-Dibromopyridine (26)2,5-Dichloropyridine (32)2,5-Difluoropyridine (8)2,6-Bis(4,5-Dihydrooxazol-2-Yl)Pyridine (12)2,6-Di(1H-Pyrazol-1-Yl)Pyridine (5)2,6-Dibromo-3-Fluoropyridine (3)2,6-Dibromopyridin-4-Amine (3)2,6-Dibromopyridine (28)2,6-Dichloro-3-Fluoropyridine (10)2,6-Dichloro-3-Methylpyridine (5)2,6-Dichloro-3-Nitropyridine (6)2,6-Dichloro-4-Iodopyridine (4)2,6-Dichloro-4-Methylpyridine (5)2,6-Dichloronicotinonitrile (8)2,6-Dichloropyridin-3-Amine (11)2,6-Dichloropyridine (49)2,6-Difluoropyridine (29)2,6-Dimethoxypyridine (11)2,6-Dimethylpyridin-4-Ol (4)2,6-Dimethylpyridine (26)2-(1H-Pyrazol-1-Yl)Pyridine (16)2-(1H-Pyrazol-3-Yl)Pyridine (5)2-(2,2,2-Trifluoroethoxy)Pyridine (8)2-(2-Chlorophenyl)Pyridine (3)2-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (6)2-(4-Fluorophenyl)Pyridine (11)2-(6-Benzhydrylpyridin-2-Yl)-4,5-Dihydrooxazole (10)2-(6-Cyclopropylpyridin-2-Yl)-4,5-Dihydrooxazole (10)2-(Benzyloxy)Pyridine (18)2-(Bromomethyl)Pyridine (27)2-(Chloromethyl)-3-Methylpyridine (12)2-(Chloromethyl)Pyridine (12)2-(Chloromethyl)Pyridine 1-Oxide (4)2-(Difluoromethyl)Pyridine (21)2-(Methylsulfonyl)Pyridine (11)2-(Methylthio)Pyridine (5)2-(P-Tolyl)Pyridine (6)2-(Phenoxymethyl)Pyridine (5)2-(Piperidin-1-Yl)Pyridine (11)2-(Piperidin-4-Yl)Pyridine (5)2-(Pyridin-2-Yl)-1H-Benzo[D]Imidazole (5)2-(Pyridin-2-Yl)Acetonitrile (8)2-(Pyridin-2-Yl)Pyrimidine (8)2-(Pyridin-2-Ylmethyl)-4,5-Dihydrooxazole (14)2-(Pyridin-3-Yl)Acetic Acid (14)2-(Pyridin-3-Yl)Acetonitrile (6)2-(Pyrrolidin-2-Yl)Pyridine (5)2-(Thiophen-2-Yl)Pyridine (5)2-(Trifluoromethoxy)Pyridine (6)2-(Trifluoromethyl)Pyridine (6)2-Aminonicotinonitrile (6)2-Benzylpyridine (12)2-Bromo-3-(Trifluoromethyl)Pyridine (4)2-Bromo-3-Chloropyridine (25)2-Bromo-3-Fluoropyridine (26)2-Bromo-3-Iodopyridine (6)2-Bromo-3-Methoxypyridine (7)2-Bromo-3-Methylpyridine (25)2-Bromo-3-Nitropyridine (5)2-Bromo-4-Chloropyridine (7)2-Bromo-4-Methylpyridine (6)2-Bromo-5-(Trifluoromethyl)Pyridine (6)2-Bromo-5-Chloropyridine (22)2-Bromo-5-Fluoropyridine (33)2-Bromo-5-Methoxypyridine (11)2-Bromo-5-Methylpyridine (19)2-Bromo-5-Nitropyridine (11)2-Bromo-6-(Trifluoromethyl)Pyridine (6)2-Bromo-6-Chloropyridine (23)2-Bromo-6-Fluoropyridine (21)2-Bromo-6-Iodopyridine (5)2-Bromo-6-Methoxypyridine (15)2-Bromo-6-Methylpyridine (17)2-Bromo-6-Phenylpyridine (4)2-Bromonicotinic Acid (5)2-Bromopyridin-3-Amine (10)2-Bromopyridin-3-Ol (7)2-Bromopyridine (10)2-Bromopyridine 1-Oxide (6)2-Chloro-3,6-Dimethylpyridine (4)2-Chloro-3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (4)2-Chloro-3-(Trifluoromethyl)Pyridine (12)2-Chloro-3-Fluoropyridin-4-Amine (3)2-Chloro-3-Fluoropyridine (20)2-Chloro-3-Iodopyridin-4-Amine (3)2-Chloro-3-Iodopyridine (13)2-Chloro-3-Methoxypyridine (12)2-Chloro-3-Methylpyridine (17)2-Chloro-3-Nitropyridine (10)2-Chloro-4,6-Dimethylpyridine (7)2-Chloro-4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (14)2-Chloro-4-(Trifluoromethyl)Pyridine (9)2-Chloro-4-Ethoxypyridine (5)2-Chloro-4-Fluoropyridine (10)2-Chloro-4-Iodopyridine (19)2-Chloro-4-Methoxypyridine (22)2-Chloro-4-Methylpyridine (31)2-Chloro-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (6)2-Chloro-5-(Trifluoromethyl)Pyridine (4)2-Chloro-5-Fluoropyridin-4-Amine (3)2-Chloro-5-Fluoropyridine (16)2-Chloro-5-Iodopyridine (18)2-Chloro-5-Methylpyridine (11)2-Chloro-5-Nitropyridine (8)2-Chloro-6-(Trifluoromethyl)Pyridine (16)2-Chloro-6-Fluoropyridine (14)2-Chloro-6-Iodopyridine (9)2-Chloro-6-Methoxypyridine (24)2-Chloro-6-Methyl-3-Nitropyridine (4)2-Chloro-6-Methylnicotinonitrile (10)2-Chloro-6-Methylpyridine (49)2-Chloroisonicotinaldehyde (19)2-Chloroisonicotinic Acid (17)2-Chloroisonicotinonitrile (7)2-Chloronicotinic Acid (7)2-Chloronicotinonitrile (9)2-Chloropyridin-3-Amine (11)2-Chloropyridin-3-Ol (9)2-Chloropyridin-4-Amine (28)2-Chloropyridine 1-Oxide (10)2-Cyclopropyl-4-Phenylquinoline (12)2-Cyclopropylpyridine (20)2-Ethoxypyridine (20)2-Ethylpyridine (9)2-Ethynylpyridine (31)2-Fluoro-3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (7)2-Fluoro-3-Iodopyridine (8)2-Fluoro-3-Methylpyridine (14)2-Fluoro-4-Methylpyridine (6)2-Fluoro-5-Methoxypyridine (4)2-Fluoro-5-Methylpyridine (6)2-Fluoro-6-Methoxypyridine (6)2-Fluoro-6-Methylpyridine (20)2-Fluoropyridine (15)2-Hydrazineylpyridine (32)2-Iodo-6-Methylpyridine (5)2-Iodopyridine (27)2-Isopropoxypyridine (16)2-Isopropylpyridine (14)2-Methoxy-3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (7)2-Methoxy-3-(Trifluoromethyl)Pyridine (6)2-Methoxy-4-Methylpyridine (5)2-Methoxy-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (5)2-Methoxy-5-(Trifluoromethyl)Pyridine (4)2-Methoxy-5-Methylpyridine (5)2-Methoxy-5-Nitropyridine (8)2-Methoxy-6-Methylpyridine (14)2-Methoxyisonicotinic Acid (13)2-Methoxypyridin-3-Amine (6)2-Methoxypyridine (434)2-Methyl-1,10-Phenanthroline (3)2-Methyl-3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (5)2-Methyl-3-Nitropyridine (9)2-Methyl-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (3)2-Methylimidazo[1,2-A]Pyridine (8)2-Methylpyridin-3-Ol (9)2-Methylpyridin-4-Amine (7)2-Methylpyridine 1-Oxide (9)2-Nitropyridine (40)2-Phenoxy-5-(Trifluoromethyl)Pyridine (7)2-Phenylimidazo[1,2-A]Pyridine (11)2-Propoxypyridine (9)2H-Pyrido[3,2-B][1,4]Oxazin-3(4H)-One (8)3,3'-Bipyridine (7)3,4-Dihydro-2H-Pyrido[3,2-B][1,4]Oxazine (5)3,5-Dibromopyridine (12)3,5-Dichloropyridine (16)3,5-Difluoropyridine (12)3,5-Dimethoxypyridine (4)3,5-Dimethylpyridine (8)3,5-Diphenylpyridine (5)3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)-5-(Trifluoromethyl)Pyridine (3)3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (74)3-(Benzyloxy)Pyridine (19)3-(Bromomethyl)Pyridine (18)3-(Chloromethyl)Pyridine (22)3-(Cyclopropylmethyl)Pyridine (6)3-(Difluoromethyl)Pyridine (5)3-(Methylsulfonyl)Pyridine (5)3-(Piperidin-1-Yl)Pyridine (5)3-(Piperidin-4-Yl)Pyridine (5)3-(Pyrrolidin-2-Yl)Pyridine (6)3-(Tert-Butyl)Pyridine (5)3-(Trifluoromethyl)-1H-Pyrrolo[2,3-B]Pyridine (3)3-(Trifluoromethyl)Pyridin-2-Ol (5)3-(Trifluoromethyl)Pyridine (51)3-Bromo-1H-Pyrrolo[3,2-B]Pyridine (6)3-Bromo-2-Chloropyridin-4-Amine (3)3-Bromo-2-Chloropyridine (20)3-Bromo-2-Fluoropyridine (21)3-Bromo-2-Methoxypyridine (20)3-Bromo-2-Methylpyridine (27)3-Bromo-4-Methylpyridine (13)3-Bromo-5-Chloropyridine (14)3-Bromo-5-Fluoropyridine (18)3-Bromo-5-Methylpyridine (14)3-Bromo-5-Nitropyridine (10)3-Bromopicolinic Acid (9)3-Bromopicolinonitrile (13)3-Bromopyrazolo[1,5-A]Pyridine (6)3-Bromopyridin-2-Amine (30)3-Bromopyridin-2-Ol (17)3-Bromopyridine 1-Oxide (8)3-Chloro-2-Fluoropyridine (13)3-Chloro-2-Methoxypyridine (16)3-Chloro-2-Methylpyridine (4)3-Chloro-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (5)3-Chloro-5-Fluoropyridine (10)3-Chloro-5-Methylpyridine (7)3-Chloro-5-Nitropyridine (5)3-Chloroimidazo[1,2-A]Pyridine (3)3-Chloropicolinic Acid (9)3-Chloropicolinonitrile (6)3-Chloropyridin-2-Amine (17)3-Chloropyridin-2-Ol (10)3-Cyclopropylpyridine (28)3-Ethoxypyridine (12)3-Ethylpyridine (11)3-Ethynylpyridine (22)3-Fluoro-2-Methoxypyridine (14)3-Fluoro-2-Methylpyridine (15)3-Fluoro-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (7)3-Fluoropicolinic Acid (6)3-Fluoropicolinonitrile (5)3-Fluoropyridin-2-Amine (12)3-Fluoropyridin-4-Amine (4)3-Hydrazineylpyridine (8)3-Iodo-1H-Pyrazolo[3,4-B]Pyridine (4)3-Iodo-1H-Pyrrolo[3,2-B]Pyridine (6)3-Iodo-1H-Pyrrolo[3,2-C]Pyridine (4)3-Iodopyridin-2-Amine (12)3-Iodopyridin-2-Ol (4)3-Iodopyridin-4-Amine (6)3-Iodopyridin-4-Ol (4)3-Iodopyridine (37)3-Methoxypyridin-2-Amine (5)3-Methoxypyridine (51)3-Methyl-1H-Pyrrolo[2,3-B]Pyridine (5)3-Methyl-2-Phenylpyridine (3)3-Methyl-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (6)3-Methyl-5-Nitropyridine (4)3-Methylimidazo[1,2-A]Pyridine (3)3-Methylpicolinic Acid (13)3-Methylpyridin-2-Amine (7)3-Methylpyridine (49)3-Methylpyridine 1-Oxide (7)3-Nitropyridin-2-Ol (11)3-Nitropyridin-4-Amine (5)3-Nitropyridine (54)3-Phenoxypyridine (12)4'-Phenyl-2,2':6',2''-Terpyridine (13)4,4'-Bipyridine (10)4,5,6,7-Tetrahydro-1H-Pyrazolo[3,4-C]Pyridine (7)4,5,6,7-Tetrahydro-1H-Pyrazolo[4,3-C]Pyridine (14)4,5,6,7-Tetrahydro-3H-Imidazo[4,5-C]Pyridine (5)4,5,6,7-Tetrahydrothiazolo[5,4-C]Pyridine (10)4,5,6,7-Tetrahydrothieno[2,3-C]Pyridine (10)4,5,6,7-Tetrahydrothieno[3,2-C]Pyridine (9)4,6-Dichloropyridin-2-Amine (6)4,6-Dichloropyridin-3-Amine (4)4,7-Diphenyl-1,10-Phenanthroline (3)4-(2,4-Diphenyl-1H-Imidazol-5-Yl)Pyridine (5)4-(Benzyloxy)Pyridine (8)4-(Bromomethyl)Pyridine (8)4-(Pyridin-2-Yl)Morpholine (21)4-(Pyridin-3-Yl)Pyrimidine (5)4-(Pyridin-4-Yl)Morpholine (5)4-(Tert-Butyl)-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (16)4-(Tert-Butyl)Pyridine (5)4-(Trifluoromethyl)Nicotinic Acid (3)4-(Trifluoromethyl)Pyridine (14)4-Bromo-2-Chloropyridine (29)4-Bromo-2-Fluoropyridine (4)4-Bromo-2-Methoxypyridine (4)4-Bromo-3-Methylpyridine (5)4-Bromopyrazolo[1,5-A]Pyridine (8)4-Bromopyridine (31)4-Chloro-1H-Pyrazolo[4,3-C]Pyridine (7)4-Chloro-1H-Pyrrolo[2,3-B]Pyridine (13)4-Chloro-1H-Pyrrolo[3,2-C]Pyridine (10)4-Chloro-2,6-Dimethylpyridine (4)4-Chloro-2-Methoxypyridine (6)4-Chloro-3-Methylpyridine (7)4-Chloronicotinic Acid (4)4-Chloropyridin-2-Amine (6)4-Chloropyridine (31)4-Cyclopropylpyridine (7)4-Ethoxypyridine (7)4-Ethynylpyridine (10)4-Iodopyridine (17)4-Isopropyl-2-(Pyridin-2-Yl)-4,5-Dihydrooxazole (16)4-Methoxypyridine (136)4-Methyl-2,2'-Bipyridine (8)4-Methyl-2-Phenylpyridine (3)4-Methyl-3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (5)4-Methyl-3-Nitropyridine (8)4-Methylnicotinic Acid (5)4-Methylpyridine (24)4-Nitropyridine (11)4-Phenoxypyridine (7)4-Phenyl-1,2,3,6-Tetrahydropyridine (7)4-Phenyl-1,4-Dihydropyridine (10)5,6,7,8-Tetrahydroimidazo[1,2-A]Pyridine (12)5,6,7,8-Tetrahydropyrido[3,4-D]Pyrimidine (7)5,6,7,8-Tetrahydropyrido[4,3-D]Pyrimidine (17)5,6-Dichloropyridin-2-Amine (4)5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Nicotinonitrile (4)5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridin-2-Amine (9)5-(Trifluoromethyl)Pyridin-2-Amine (5)5-(Trifluoromethyl)Pyridin-2-Ol (8)5-Benzyl-4,5,6,7-Tetrahydrothieno[3,2-C]Pyridine (7)5-Bromo-2-Chloro-4-Methylpyridine (5)5-Bromo-2-Chloropyridine (40)5-Bromo-2-Fluoropyridine (22)5-Bromo-2-Iodopyridine (5)5-Bromo-2-Methoxypyridine (29)5-Bromo-2-Methylpyridine (23)5-Bromo-2-Phenylpyridine (3)5-Bromo-4-Methylpyridin-2-Amine (5)5-Bromo-6-Chloropyridin-2-Amine (3)5-Bromonicotinaldehyde (12)5-Bromonicotinic Acid (6)5-Bromonicotinonitrile (6)5-Bromopicolinic Acid (11)5-Bromopicolinonitrile (13)5-Bromopyridin-2-Amine (35)5-Bromopyridin-2-Ol (18)5-Bromopyridin-3-Amine (10)5-Bromopyridin-3-Ol (6)5-Chloro-1H-Pyrazolo[3,4-B]Pyridine (3)5-Chloro-1H-Pyrazolo[4,3-B]Pyridine (6)5-Chloro-1H-Pyrrolo[2,3-C]Pyridine (5)5-Chloro-1H-Pyrrolo[3,2-B]Pyridine (6)5-Chloro-2-Fluoropyridine (13)5-Chloro-2-Methoxypyridine (10)5-Chloro-2-Methylpyridine (10)5-Chloroimidazo[1,2-A]Pyridine (3)5-Chloropicolinic Acid (7)5-Chloropicolinonitrile (6)5-Chloropyridin-2-Amine (26)5-Chloropyridin-2-Ol (15)5-Chloropyridin-3-Amine (7)5-Fluoro-2-Methoxypyridine (15)5-Fluoro-2-Methylpyridine (8)5-Fluoropicolinonitrile (5)5-Fluoropyridin-2-Amine (15)5-Iodopyridin-2-Amine (17)5-Iodopyridin-2-Ol (8)5-Methoxy-1H-Pyrrolo[2,3-C]Pyridine (4)5-Methoxypyridin-3-Amine (4)5-Methylimidazo[1,2-A]Pyridine (4)5-Methylpyridin-2-Amine (7)5-Methylpyridin-3-Amine (4)5-Nitronicotinic Acid (5)5-Nitropyridin-2-Amine (6)5-Phenylpicolinic Acid (4)6,7-Dihydro-5H-Cyclopenta[B]Pyridine (18)6,7-Dihydro-5H-Cyclopenta[C]Pyridine (7)6,7-Dihydro-5H-Pyrrolo[3,4-B]Pyridine (5)6-(Trifluoromethyl)Pyridin-2-Amine (11)6-(Trifluoromethyl)Pyridin-3-Amine (5)6-Bromo-1H-Pyrazolo[4,3-B]Pyridine (4)6-Bromo-1H-Pyrrolo[3,2-B]Pyridine (7)6-Bromo-1H-Pyrrolo[3,2-C]Pyridine (5)6-Bromoimidazo[1,2-A]Pyridine (14)6-Bromonicotinaldehyde (10)6-Bromonicotinic Acid (9)6-Bromonicotinonitrile (7)6-Bromopicolinic Acid (5)6-Bromopyrazolo[1,5-A]Pyridine (8)6-Bromopyridin-2-Amine (8)6-Bromopyridin-3-Amine (12)6-Bromopyridin-3-Ol (13)6-Chloro-1H-Pyrazolo[4,3-C]Pyridine (8)6-Chloro-1H-Pyrrolo[3,2-C]Pyridine (13)6-Chloro-3-Nitropyridin-2-Amine (4)6-Chloro-5-(Trifluoromethyl)Pyridin-2-Amine (3)6-Chloro-5-Iodopyridin-2-Amine (3)6-Chloroimidazo[1,2-A]Pyridine (4)6-Chloronicotinaldehyde (24)6-Chloronicotinic Acid (4)6-Chloropicolinic Acid (12)6-Chloropicolinonitrile (9)6-Chloropyridin-2-Amine (21)6-Chloropyridin-2-Ol (9)6-Chloropyridin-3-Amine (4)6-Chloropyridine-3-Sulfonyl Chloride (5)6-Fluoro-1H-Pyrrolo[2,3-B]Pyridine (3)6-Fluoropyridin-2-Amine (8)6-Fluoropyridin-3-Amine (4)6-Methoxy-1H-Pyrrolo[2,3-B]Pyridine (3)6-Methoxynicotinaldehyde (4)6-Methoxypicolinic Acid (9)6-Methoxypyridin-2-Amine (14)6-Methoxypyridin-3-Amine (8)6-Methylnicotinic Acid (6)6-Methylnicotinonitrile (7)6-Methylpicolinic Acid (12)6-Methylpicolinonitrile (10)6-Methylpyridin-2-Amine (46)6-Methylpyridin-2-Ol (23)6-Methylpyridin-3-Amine (5)6-Methylpyridin-3-Ol (7)6-Phenoxypyridin-3-Amine (5)6-Phenylnicotinic Acid (8)6-Phenylpicolinic Acid (3)7-Bromo-1H-Pyrrolo[2,3-C]Pyridine (4)7-Chloro-1H-Pyrrolo[2,3-C]Pyridine (5)7-Methoxy-1H-Pyrrolo[2,3-C]Pyridine (5)7-Methylimidazo[1,2-A]Pyridine (3)9H-Pyrido[3,4-B]Indole (5)Bis(Pyridin-2-Ylmethyl)Amine (6)Dimethyl Pyridine-2,6-Dicarboxylate (7)Dimethyl(Pyridin-2-Yl)Phosphine Oxide (8)Dimethyl(Pyridin-3-Yl)Phosphine Oxide (7)Ethyl 2-(Pyridin-2-Yl)Acetate (15)Ethyl 2-Methylnicotinate (6)Ethyl 4,6-Dichloronicotinate (4)Ethyl 6-Chloropicolinate (7)Ethyl Isonicotinate (12)Ethyl Nicotinate (25)Ethyl Picolinate (41)Ethyl Pyrazolo[1,5-A]Pyridine-3-Carboxylate (11)Furo[3,2-B]Pyridine (7)Furo[3,2-C]Pyridine (7)Imidazo[1,2-A]Pyridin-2-Ylmethanol (4)Imidazo[1,2-A]Pyridine-5-Carboxylic Acid (3)Imidazo[1,2-A]Pyridine-6-Carbonitrile (3)Isonicotinaldehyde (68)Isonicotinamide (12)Isonicotinic Acid (23)Isonicotinohydrazide (6)Isonicotinonitrile (16)Methyl 1H-Pyrazolo[3,4-B]Pyridine-5-Carboxylate (3)Methyl 1H-Pyrrolo[2,3-B]Pyridine-2-Carboxylate (3)Methyl 2-(Pyridin-2-Yl)Acetate (9)Methyl 2-Chloroisonicotinate (11)Methyl 2-Chloronicotinate (6)Methyl 3-Bromopicolinate (9)Methyl 3-Chloropicolinate (9)Methyl 5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Nicotinate (4)Methyl 5-Hydroxypicolinate (5)Methyl 6-Bromonicotinate (6)Methyl 6-Chloropicolinate (11)Methyl 6-Fluoronicotinate (4)Methyl 6-Methoxynicotinate (4)Methyl Isonicotinate (16)Methyl Nicotinate (52)Methyl Pyrazolo[1,5-A]Pyridine-3-Carboxylate (16)N,N-Dimethylpyridin-2-Amine (10)N,N-Dimethylpyridin-4-Amine (6)N-(Pyridin-2-Yl)Benzamide (9)N-Benzylpyridin-2-Amine (9)N-Methylpicolinamide (10)N-Methylpyridin-2-Amine (47)N-Methylpyridin-3-Amine (15)N-Phenyl-4-(Pyridin-3-Yl)Pyrimidin-2-Amine (5)N-Phenylpicolinamide (8)N-Phenylpyridin-2-Amine (9)Nicotinaldehyde (152)Nicotinamide (64)Nicotinic Acid (53)Nicotinonitrile (82)Oxazolo[4,5-B]Pyridine (7)Oxazolo[5,4-B]Pyridine (5)Phenyl(Pyridin-2-Yl)Methanone (10)Phenyl(Pyridin-3-Yl)Methanone (9)Picolinaldehyde (98)Picolinamide (26)Picolinic Acid (22)Picolinimidamide (11)Picolinohydrazide (5)Pyrazolo[1,5-A]Pyridine-2-Carboxylic Acid (5)Pyrazolo[1,5-A]Pyridine-3-Carboxylic Acid (13)Pyridin-2-Amine (29)Pyridin-2-Ol (170)Pyridin-2-Ylboronic Acid (9)Pyridin-2-Ylmethanamine (38)Pyridin-2-Ylmethanol (65)Pyridin-3-Amine (21)Pyridin-3-Ol (13)Pyridin-3-Ylboronic Acid (49)Pyridin-3-Ylmethanamine (29)Pyridin-3-Ylmethanol (39)Pyridin-4-Amine (22)Pyridin-4-Ol (16)Pyridin-4-Ylboronic Acid (21)Pyridin-4-Ylmethanamine (12)Pyridin-4-Ylmethanol (16)Pyridine 1-Oxide (33)Pyridine-2,3-Diamine (5)Pyridine-2,6-Diamine (12)Pyridine-2,6-Dicarboxylic Acid (9)Pyridine-2,6-Diol (6)Pyridine-2-Sulfonamide (6)Pyridine-2-Sulfonyl Chloride (9)Pyridine-2-Thiol (8)Pyridine-3-Sulfonic Acid (6)Pyridine-3-Sulfonyl Chloride (25)Pyrido[2,3-B]Pyrazine (12)Pyrido[2,3-D]Pyrimidine (8)Pyrido[3,2-D]Pyrimidine (14)Pyrido[3,4-B]Pyrazine (11)Pyrido[3,4-D]Pyrimidine (10)Tert-Butyl (2-Chloropyridin-4-Yl)Carbamate (4)Tert-Butyl 1H-Pyrazolo[3,4-B]Pyridine-1-Carboxylate (7)Tert-Butyl 4-(Pyridin-2-Yl)Piperazine-1-Carboxylate (12)Tert-Butyl Pyridin-2-Ylcarbamate (37)Tert-Butyl Pyridin-3-Ylcarbamate (16)Tert-Butyl Pyridin-4-Ylcarbamate (6)Thiazolo[4,5-B]Pyridine (12)Thiazolo[5,4-B]Pyridine (13)Thiazolo[5,4-C]Pyridine (5)Thieno[2,3-B]Pyridine (22)Thieno[2,3-C]Pyridine (14)Thieno[3,2-B]Pyridine (18)Thieno[3,2-C]Pyridine (13)
Pyrimidines (14593)
Alcohols (63)Aldehydes (11)Alkenyls (10)Alkynyls (7)Amides (120)Amidines (4)Amines (191)Aryls (79)Bromides (71)Carboxylic Acids (46)Chlorides (209)Difluoromethyls (9)Esters (63)Ethers (113)Fluorinated Building Blocks (38)Hydrazines (8)Iodides (15)Ketones (8)Nitriles (15)Nitroes (13)Sulfamides (1)Sulfides (53)Sulfones (7)Trifluoromethyls (6)Bipyrimidine (4)Dibromopyrimidine (15)Methoxypyrimidine (107)Pyrimidinone (3)Tetraoxatetradecan (10)Trichloropyrimidine (11)Azetidines (4)Indoles (7)Morpholines (4)Piperazines (25)Piperidines (4)Pyrrolidines (31)Thiophenols (4)(R)-7-(Tetrahydrofuran-2-Yl)-7H-Pyrrolo[2,3-D]Pyrimidine (8)(R)-N-(Piperidin-3-Yl)-7H-Pyrrolo[2,3-D]Pyrimidin-4-Amine (5)1,4,5,6-Tetrahydropyrimidine (4)1-(3-Fluorotetrahydrofuran-2-Yl)Pyrimidin-2(1H)-One (10)1-(5-(Hydroxymethyl)Tetrahydrofuran-2-Yl)Pyrimidine-2,4(1H,3H)-Dione (31)1-(Pyrimidin-2-Yl)Ethan-1-One (5)1-(Pyrimidin-4-Yl)Ethan-1-One (6)1-(Tetrahydrofuran-2-Yl)Pyrimidin-2(1H)-One (50)1H-Pyrazolo[3,4-D]Pyrimidin-4-Amine (5)1H-Pyrazolo[4,3-D]Pyrimidine (5)2,4,5-Trichloropyrimidine (6)2,4,6-Trichloropyrimidine (14)2,4,6-Triphenylpyrimidine (9)2,4-Dichloro-6-Methylpyrimidine (7)2,4-Dichloropyrimidin-5-Amine (6)2,4-Dimethoxypyrimidine (13)2,4-Dimethylpyrimidine (7)2-(1H-Pyrazol-1-Yl)Pyrimidine (6)2-(Chloromethyl)Pyrimidine (5)2-(Methylsulfonyl)Pyrimidine (25)2-(Methylthio)Pyrimidin-4-Amine (10)2-(Methylthio)Pyrimidine (19)2-(Piperazin-1-Yl)Pyrimidine (11)2-(Piperidin-1-Yl)Pyrimidine (16)2-(Propylthio)Pyrimidine (5)2-(Pyridin-2-Yl)Pyrimidine (8)2-(Pyrrolidin-1-Yl)Pyrimidine (8)2-(Trifluoromethyl)Pyrimidine (27)2-Aminopyrimidin-4(3H)-One (11)2-Bromopyrimidine (22)2-Chloro-4-(Trifluoromethyl)Pyrimidine (6)2-Chloro-4-Methoxypyrimidine (14)2-Chloro-4-Methylpyrimidine (16)2-Chloro-4-Phenylpyrimidine (7)2-Chloro-5-Fluoropyrimidine (6)2-Chloro-5-Methylpyrimidine (5)2-Chloro-5-Nitropyrimidine (5)2-Chloro-7H-Pyrrolo[2,3-D]Pyrimidine (13)2-Chloropyrimidin-4-Amine (22)2-Cyclopropylpyrimidine (10)2-Ethoxypyrimidine (7)2-Ethylpyrimidine (6)2-Fluoropyrimidine (9)2-Hydrazineylpyrimidine (6)2-Iodopyrimidine (8)2-Methoxypyrimidin-4-Amine (6)2-Methoxypyrimidine (59)2-Methylpyrimidin-4-Amine (22)2-Methylpyrimidin-4-Ol (11)2-Phenoxypyrimidine (8)2-Phenylpyrimidin-5-Amine (5)3-Methylpyrimidin-4(3H)-One (6)4,5-Dichloropyrimidine (7)4,6-Dichloro-2-(Methylthio)Pyrimidine (9)4,6-Dichloro-2-Methylpyrimidine (7)4,6-Dichloropyrimidin-2-Amine (9)4,6-Dichloropyrimidine (14)4,6-Diphenylpyrimidine (7)4-(Dimethoxymethyl)Pyrimidine (5)4-(Piperazin-1-Yl)Pyrimidine (13)4-(Piperidin-1-Yl)Pyrimidine (9)4-(Pyridin-3-Yl)Pyrimidine (5)4-(Pyrimidin-2-Yl)Morpholine (9)4-(Pyrimidin-4-Yl)Morpholine (13)4-(Trifluoromethyl)Pyrimidin-2-Amine (6)4-(Trifluoromethyl)Pyrimidine (12)4-Amino-1-(Tetrahydrofuran-2-Yl)Pyrimidin-2(1H)-One (22)4-Bromopyrimidine (24)4-Chloro-2-(Methylthio)Pyrimidine (29)4-Chloro-2-Methoxypyrimidine (12)4-Chloro-2-Phenylpyrimidine (5)4-Chloro-5-Fluoro-2-Methylpyrimidine (5)4-Chloro-5-Iodopyrimidine (6)4-Chloro-5-Methoxypyrimidine (7)4-Chloro-5-Methylpyrimidine (8)4-Chloro-6-Methoxypyrimidine (5)4-Chloropyrimidin-2-Amine (23)4-Chloropyrimidin-5-Amine (7)4-Chloropyrimidine-5-Carbonitrile (6)4-Ethoxypyrimidine (6)4-Fluoropyrimidine (8)4-Iodopyrimidine (9)4-Methoxy-2-Methylpyrimidine (8)4-Methoxypyrimidin-2-Amine (6)4-Methoxypyrimidine (102)4-Methyl-2-(Methylthio)Pyrimidine (7)4-Methylpyrimidin-2-Amine (9)4-Methylpyrimidine (22)4-Phenoxypyrimidine (5)4-Phenylpyrimidin-2-Amine (15)5,6,7,8-Tetrahydropyrido[3,4-D]Pyrimidine (7)5,6,7,8-Tetrahydropyrido[4,3-D]Pyrimidine (17)5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyrimidine (27)5-(Benzyloxy)Pyrimidine (5)5-Benzylpyrimidine (7)5-Bromo-1-(Tetrahydrofuran-2-Yl)Pyrimidine-2,4(1H,3H)-Dione (4)5-Bromo-2-(Methylthio)Pyrimidine (6)5-Bromo-2-Chloropyrimidine (6)5-Bromo-2-Methylpyrimidine (5)5-Bromo-4-Chloropyrimidine (4)5-Bromo-7H-Pyrrolo[2,3-D]Pyrimidine (3)5-Bromopyrimidin-4(3H)-One (3)5-Chloropyrimidin-4(3H)-One (4)5-Chloropyrimidine (8)5-Chloropyrimidine-2,4(1H,3H)-Dione (3)5-Ethylpyrimidine (5)5-Fluoro-1-(Tetrahydrofuran-2-Yl)Pyrimidin-2(1H)-One (7)5-Fluoro-1-(Tetrahydrofuran-2-Yl)Pyrimidine-2,4(1H,3H)-Dione (3)5-Fluoropyrimidin-4(3H)-One (3)5-Fluoropyrimidine (11)5-Fluoropyrimidine-2,4(1H,3H)-Dione (3)5-Iodo-7H-Pyrrolo[2,3-D]Pyrimidine (4)5-Iodopyrimidine (5)5-Methylpyrimidine (3)5-Phenylpyrimidine (16)5H-Pyrrolo[3,2-D]Pyrimidine (16)6,7-Dihydro-5H-Cyclopenta[D]Pyrimidine (5)6,7-Dihydro-5H-Pyrrolo[3,4-D]Pyrimidine (8)6-Chloro-1H-Pyrazolo[3,4-D]Pyrimidine (12)6-Chloropyrimidin-4-Amine (8)7-(Phenylsulfonyl)-7H-Pyrrolo[2,3-D]Pyrimidine (7)7H-Pyrrolo[2,3-D]Pyrimidin-2-Amine (5)7H-Pyrrolo[2,3-D]Pyrimidin-4-Amine (3)[1,2,4]Triazolo[1,5-A]Pyrimidine (14)Ethyl 4-Chloropyrimidine-5-Carboxylate (5)Ethyl Pyrazolo[1,5-A]Pyrimidine-3-Carboxylate (6)Ethyl Pyrimidine-4-Carboxylate (7)Ethyl Pyrimidine-5-Carboxylate (5)Imidazo[1,2-C]Pyrimidine (6)Methyl Pyrimidine-2-Carboxylate (10)Methyl Pyrimidine-4-Carboxylate (11)Methyl Pyrimidine-5-Carboxylate (5)N,N-Dimethylpyrimidin-2-Amine (10)N-(9-((2R,5S)-5-((Trityloxy)Methyl)Tetrahydrofuran-2-Yl)-9H-Purin-6-Yl)Benzamide (5)N-Benzyl-9H-Purin-6-Amine (6)N-Cyclopropylpyrimidin-2-Amine (5)N-Methyl-N-(4-Phenylpyrimidin-2-Yl)Methanesulfonamide (7)N-Methylpyrimidin-2-Amine (10)N-Methylpyrimidin-4-Amine (10)N-Phenyl-4-(Pyridin-3-Yl)Pyrimidin-2-Amine (5)N-Phenylpyrimidin-2-Amine (5)N-Phenylpyrimidin-4-Amine (7)N2,N4-Diphenylpyrimidine-2,4-Diamine (5)Pyrido[2,3-D]Pyrimidine (8)Pyrido[3,2-D]Pyrimidine (14)Pyrido[3,4-D]Pyrimidine (10)Pyrimidin-2-Ol (21)Pyrimidin-2-Ylmethanamine (7)Pyrimidin-2-Ylmethanol (5)Pyrimidin-4-Amine (29)Pyrimidin-4-Ol (25)Pyrimidin-5-Amine (6)Pyrimidin-5-Ol (5)Pyrimidin-5-Ylboronic Acid (9)Pyrimidine-2,4-Diamine (14)Pyrimidine-2-Carbonitrile (14)Pyrimidine-2-Carboxylic Acid (11)Pyrimidine-2-Thiol (6)Pyrimidine-4,6(1H,5H)-Dione (3)Pyrimidine-4-Carbonitrile (9)Pyrimidine-4-Carboxylic Acid (17)Pyrimidine-5-Carbaldehyde (38)Pyrimidine-5-Carbonitrile (6)Pyrimidine-5-Carboxylic Acid (7)Tetrahydropyrimidin-2(1H)-One (6)Thiazolo[5,4-D]Pyrimidine (5)Thieno[2,3-D]Pyrimidine (30)Thieno[3,2-D]Pyrimidine (24)
Pyrroles (4887)
Alcohols (2)Aldehydes (12)Amides (15)Amines (10)Aryls (30)Bromides (5)Carboxylic Acids (28)Chlorides (13)Esters (21)Fluorinated Building Blocks (8)Ketones (16)Nitriles (6)Nitroes (8)Trifluoromethyls (4)Bromopyrrolo (8)Chloropyrrolo (10)Dihydropyrrolo (15)Octahydropyrrolo (6)Pyrrolopyridine-Methylamine (6)(9H-Fluoren-9-Yl)Methyl Pyrrolidine-1-Carboxylate (26)(Z)-3-((1H-Pyrrol-2-Yl)Methylene)Indolin-2-One (8)1,3-Dihydro-2H-Pyrrolo[2,3-B]Pyridin-2-One (19)1,3-Dihydro-2H-Pyrrolo[3,2-B]Pyridin-2-One (11)1,4,5,6-Tetrahydropyrrolo[3,4-C]Pyrazole (6)1-(1H-Pyrrolo[2,3-B]Pyridin-3-Yl)Ethan-1-One (4)1-(Phenylsulfonyl)-1H-Pyrrole (24)1-(Phenylsulfonyl)-1H-Pyrrolo[2,3-B]Pyridine (30)1-Benzylpyrrolidin-2-One (18)1-Methyl-1H-Pyrrole (43)1H-Pyrrol-1-Amine (7)1H-Pyrrole-2-Carbaldehyde (21)1H-Pyrrole-2-Carbonitrile (7)1H-Pyrrole-2-Carboxylic Acid (30)1H-Pyrrole-3-Carbaldehyde (9)1H-Pyrrole-3-Carbonitrile (9)1H-Pyrrolo[2,3-B]Pyridin-6-Amine (4)1H-Pyrrolo[2,3-B]Pyridine-3-Carboxylic Acid (5)1H-Pyrrolo[2,3-C]Pyridin-7-Ol (3)2,3-Dihydro-1H-Pyrrolo[2,3-B]Pyridine (10)2,3-Dihydro-1H-Pyrrolo[3,4-C]Pyridine (6)2-Bromo-1H-Pyrrole (11)2-Methyl-1H-Pyrrole (49)2-Nitro-1H-Pyrrole (5)2-Phenyl-1H-Pyrrole (24)3,6-Di(Thiophen-2-Yl)-2,5-Dihydropyrrolo[3,4-C]Pyrrole-1,4-Dione (17)3-Bromo-1H-Pyrrole (16)3-Bromo-1H-Pyrrolo[2,3-B]Pyridine (7)3-Iodo-1H-Pyrrolo[3,2-C]Pyridine (4)3-Methyl-1H-Pyrrolo[2,3-B]Pyridine (5)3-Phenyl-1H-Pyrrole (4)4-Bromo-1H-Pyrrolo[2,3-B]Pyridine (9)4-Chloro-1H-Pyrrolo[2,3-B]Pyridine (13)4H-Thieno[3,2-B]Pyrrole (6)5-Bromo-1H-Pyrrolo[2,3-B]Pyridine (13)5-Chloro-1H-Pyrrolo[2,3-C]Pyridine (5)5-Methoxy-1H-Pyrrolo[2,3-C]Pyridine (4)5H-Pyrrolo[2,3-B]Pyrazine (14)5H-Pyrrolo[3,2-D]Pyrimidine (16)7-(Phenylsulfonyl)-7H-Pyrrolo[2,3-D]Pyrimidine (7)7-Methoxy-1H-Pyrrolo[2,3-C]Pyridine (5)Ethyl 1H-Pyrrole-2-Carboxylate (24)Ethyl 1H-Pyrrole-3-Carboxylate (8)Methyl 1H-Pyrrole-2-Carboxylate (30)Methyl 1H-Pyrrole-3-Carboxylate (5)Methyl 1H-Pyrrolo[2,3-B]Pyridine-3-Carboxylate (4)Methyl 1H-Pyrrolo[2,3-B]Pyridine-6-Carboxylate (3)Pyrrolo[2,1-F][1,2,4]Triazine (14)Tert-Butyl 1H-Pyrrole-1-Carboxylate (8)Tert-Butyl 1H-Pyrrole-2-Carboxylate (3)Pyridines (7)Thiophenes (5)
Pyrrolidines (14354)
Alcohols (82)Alkenyls (14)Alkynyls (8)Amides (307)Amines (81)Aryls (142)Bromides (25)Carboxylic Acids (26)Chlorides (49)Esters (32)Ethers (56)Fluorinated Building Blocks (38)Hydrazines (3)Ketones (23)Nitriles (11)Nitroes (12)Sulfamides (10)Sulfides (7)Sulfonates (4)Trifluoromethyls (4)Ureas (2)Acetylpyrrolidine (9)Benzylpyrrolidine (31)Bipyrrolidine (8)Bromopyrrolidine (7)Carbamoylpyrrolidine (5)Difluoropyrrolidine (16)Dimethylpyrrolidine (31)Dioxopyrrolidin (131)Ethylpyrrolidine (9)Fluoropyrrolidine (26)Formylpyrrolidine (10)Mercaptopyrrolidine (4)Methoxypyrrolidine (19)Methylenepyrrolidine (2)Methylpyrrolidine (128)Phenoxypyrrolidine (3)Pyrrolidine-Benzene-Boronic Acid/Ester (20)(3Ar,6As)-Octahydrocyclopenta[C]Pyrrole (5)(9H-Fluoren-9-Yl)Methyl Pyrrolidine-1-Carboxylate (26)(R)-3-(Pyrrolidin-2-Yl)Pyridine (9)(R)-3-Benzylpyrrolidine (6)(S)-N-(((S)-Tetrahydro-2H-Pyran-2-Yl)Methyl)Pyrrolidine-2-Carboxamide (4)(S)-N-(4-(Thiazol-5-Yl)Benzyl)Pyrrolidine-2-Carboxamide (7)(S)-N-Phenethyl-3-(Pyrrolidin-2-Yl)Propanamide (5)1-((Benzyloxy)Carbonyl)Pyrrolidine-3-Carboxylic Acid (6)1-(3-Chlorophenyl)Pyrrolidine (3)1-(Phenylsulfonyl)Pyrrolidine (15)1-(Pyrrolidin-1-Yl)Ethan-1-One (9)1-Benzyl 2-Methyl Pyrrolidine-1,2-Dicarboxylate (7)1-Benzylpyrrolidin-2-One (18)1-Benzylpyrrolidine (26)1-Methylpyrrolidin-2-One (13)1-Phenylpyrrolidin-2-One (18)2,2-Dimethylpyrrolidine (4)2,5-Dioxopyrrolidin-1-Yl ((Benzyloxy)Carbonyl)Glycinate (8)2,5-Dioxopyrrolidin-1-Yl Benzoate (8)2,8-Diazaspiro[4.5]Decan-3-One (6)2-((3-Phenylpropyl)Amino)-1-(Pyrrolidin-1-Yl)Ethan-1-One (2)2-(2-Chlorophenyl)Pyrrolidine (17)2-(2-Fluorophenyl)Pyrrolidine (16)2-(3-Fluorophenyl)Pyrrolidine (5)2-(Methoxymethyl)Pyrrolidine (6)2-(Pyrrolidin-1-Yl)Benzoic Acid (3)2-(Pyrrolidin-1-Yl)Pyridine (11)2-(Pyrrolidin-1-Yl)Pyrimidine (8)2-(Pyrrolidin-2-Yl)Acetic Acid (8)2-(Pyrrolidin-2-Yl)Pyridine (5)2-(Pyrrolidin-3-Yl)Acetic Acid (6)2-Amino-1-(Pyrrolidin-1-Yl)Ethan-1-One (5)2-Azabicyclo[2.2.1]Heptane (6)2-Methyl-1-Phenyl-1H-Pyrrole (8)2-Methylpyrrolidine (21)3,3-Difluoropyrrolidine (29)3-(Pyrrolidin-2-Yl)Pyridine (6)3-Aminopyrrolidin-2-One (5)3-Bromopyrrolidine (6)3-Hydroxypyrrolidin-2-One (4)3-Phenoxypyrrolidine (5)3-Phenyl-1-(Pyrrolidin-1-Yl)Propan-1-One (1)4-(Hydroxymethyl)Pyrrolidin-2-One (4)4-(Pyrrolidin-1-Yl)Aniline (3)4-(Pyrrolidin-1-Yl)Piperidine (6)4-Hydroxypyrrolidin-2-One (7)4H-Thieno[3,4-C]Pyrrole-4,6(5H)-Dione (7)5-(Aminomethyl)Pyrrolidin-2-One (3)5-(Hydroxymethyl)Pyrrolidin-2-One (7)5-Azaspiro[2.4]Heptane (13)5-Oxopyrrolidine-2-Carboxylic Acid (4)6-Azaspiro[3.4]Octane (4)Benzyl (S)-Pyrrolidin-3-Ylcarbamate (7)Benzyl 3-Hydroxypyrrolidine-1-Carboxylate (5)Benzyl-2-(Hydroxymethyl)Pyrrolidine-1-Carboxylate (9)Benzyl-3-Aminopyrrolidine-1-Carboxylate (7)Ethyl 5-Oxopyrrolidine-2-Carboxylate (3)Hexahydro-1H-Pyrrolizine (12)Methyl 5-Oxopyrrolidine-2-Carboxylate (3)Methyl Prolinate (43)Methyl Pyrrolidine-3-Carboxylate (9)N,N-Dimethylpyrrolidin-3-Amine (7)N-Methylpyrrolidin-3-Amine (6)Octahydrocyclopenta[C]Pyrrole (6)Phenyl(Pyrrolidin-1-Yl)Methanone (12)Proline (13)Pyrrolidin-2-Ylmethanamine (12)Pyrrolidin-2-Ylmethanol (26)Pyrrolidin-3-Ol (59)Pyrrolidin-3-Ylmethanamine (6)Pyrrolidin-3-Ylmethanol (13)Pyrrolidine-2-Carbonitrile (9)Pyrrolidine-2-Carboxamide (12)Pyrrolidine-3-Carbonitrile (6)Pyrrolidine-3-Carboxylic Acid (14)Tert-Butyl (2-Oxopyrrolidin-3-Yl)Carbamate (3)Tert-Butyl (5-Oxopyrrolidin-3-Yl)Carbamate (3)Tert-Butyl 2-Oxopyrrolidine-1-Carboxylate (15)Tert-Butyl 3-((Methylsulfonyl)Oxy)Pyrrolidine-1-Carboxylate (5)Tert-Butyl 3-Hydroxypyrrolidine-1-Carboxylate (20)Tert-Butyl 3-Methoxypyrrolidine-1-Carboxylate (6)Tert-Butyl Pyrrolidin-3-Ylcarbamate (12)Tert-Butyl Pyrrolidine-1-Carboxylate (21)Azetidines (12)Imidazoles (2)Oxetanes (4)Piperidines (36)Pyridines (2)Pyrimidines (31)Spiroes (86)Tetrahydropyrans (6)
Pyrrolines (1759)
Amides (24)Aryls (14)Bromides (6)Esters (2)1,3-Dihydro-2H-Pyrrolo[2,3-C]Pyridin-2-One (5)1-Phenyl-1H-Pyrrole-2,5-Dione (12)2,3-Dihydro-1H-Pyrrolo[2,3-C]Pyridine (5)2,3-Dihydro-1H-Pyrrolo[3,2-C]Pyridine (7)2,5-Dihydro-1H-Pyrrol-2-One (5)2,5-Dihydro-1H-Pyrrole (13)2-Phenyl-2,5,6,7-Tetrahydropyrrolo[2,1-C][1,2,4]Triazol-4-Ium (6)3,4-Dihydro-2H-Pyrrole (5)3-Bromo-1H-Pyrrole-2,5-Dione (3)5,6-Dihydro-4H-Pyrrolo[1,2-B]Pyrazole (8)6,7-Dihydro-5H-Pyrrolo[1,2-A]Imidazole (9)6,7-Dihydro-5H-Pyrrolo[3,4-B]Pyridine (5)6,7-Dihydro-5H-Pyrrolo[3,4-D]Pyrimidine (8)
Quinazolines (3145)
Alcohols (12)Amides (42)Amines (9)Aryls (7)Bromides (13)Carboxylic Acids (4)Chlorides (20)Esters (5)Ethers (17)Fluorinated Building Blocks (5)Nitroes (4)Dichloroquinazoline (14)Iodoquinazoline (10)Methylquinazoline (46)Nitroquinazoline (20)Trichloroquinazoline (5)2,4-Dichloroquinazoline (19)2-(Trifluoromethyl)Quinazoline (4)2-Bromoquinazoline (4)2-Chloroquinazolin-4(3H)-One (4)2-Cyclopropylquinazoline (5)2-Mercaptoquinazolin-4(3H)-One (3)2-Methylquinazoline (20)4-Chloroquinazoline (39)4-Methylquinazoline (4)5,6,7,8-Tetrahydroquinazoline (8)5-Chloroquinazoline (4)6-Bromoquinazoline (7)7-Bromoquinazolin-4(3H)-One (4)7-Fluoroquinazolin-4(3H)-One (4)7-Methoxy-N-Phenylquinazolin-4-Amine (12)8-Bromoquinazoline (8)8-Chloroquinazoline (5)8-Fluoroquinazoline (4)8-Methoxyquinazoline (3)8-Methylquinazoline (3)Ethyl Quinazoline-2-Carboxylate (4)N-(3-Chlorophenyl)Quinazolin-4-Amine (10)Quinazolin-2-Amine (13)Quinazolin-2-Ol (7)Quinazolin-4-Amine (8)Quinazolin-4-Ol (10)
Quinolines (11915)
Alcohols (79)Aldehydes (23)Alkenyls (10)Amides (33)Amines (79)Aryls (24)Bromides (106)Carboxylic Acids (90)Chlorides (110)Esters (74)Ethers (82)Fluorinated Building Blocks (50)Hydrazines (4)Iodides (12)Ketones (50)Nitriles (19)Nitroes (19)Sulfonyl Chlorides (6)Trifluoromethyls (7)Iodoquinoline (32)Furans (3)Quinuclidines (8)1,2-Dihydroquinoline (5)1-Cyclopropyl-3-(4-(Quinolin-4-Yloxy)Phenyl)Urea (6)2,2'-Biquinoline (3)2,3,6,7-Tetrahydro-1H,5H-Pyrido[3,2,1-Ij]Quinoline (7)2-(Quinolin-2-Yl)-4,5-Dihydrooxazole (7)2-Hydroxyquinolin-4(1H)-One (3)2-Phenylquinoline (19)3,4-Dihydroisoquinoline (11)4-Oxo-1,4-Dihydroquinoline-3-Carboxylic Acid (3)4-Phenoxyquinoline (5)4-Phenylquinoline (9)6-Bromo-3,4-Dihydroquinolin-2(1H)-One (4)7,8-Dihydroquinolin-5(6H)-One (5)7-(Benzyloxy)Quinoline (6)7-Methoxy-3,4-Dihydroquinolin-2(1H)-One (3)N-Phenyl-N-(4-(Quinolin-4-Yloxy)Phenyl)Cyclopropane-1,1-Dicarboxamide (5)N-Phenylquinolin-4-Amine (11)
Quinoxalines (1405)
Alcohols (6)Amides (15)Amines (7)Bromides (9)Carboxylic Acids (5)Chlorides (9)Fluorinated Building Blocks (5)Nitroes (7)Dichloroquinoxaline (8)Dihydroquinoxalin (7)1,2,3,4-Tetrahydroquinoxaline (6)2-Chloroquinoxaline (39)2-Methylquinoxaline (5)2-Phenylquinoxaline (5)3,4-Dihydroquinoxalin-2(1H)-One (7)5-Bromoquinoxaline (6)6-Bromoquinoxaline (9)6-Chloroquinoxaline (3)6-Methoxyquinoxaline (6)Quinoxalin-2-Amine (3)Quinoxalin-2-Ol (4)
Spiroes (4761)
Alcohols (15)Aliphatic Cyclic Hydrocarbons (19)Alkenyls (4)Amides (190)Amines (14)Aryls (36)Bromides (11)Carboxylic Acids (20)Chlorides (4)Esters (19)Fluorinated Building Blocks (9)Ketones (47)Nitriles (2)Aminospiro (4)Azaspiro (246)Bromospiro (11)Diazaspiro (182)Dihydrospiro (18)Dioxaspiro (39)Fluorospiro (4)Oxospiro (22)Triazaspiro (13)1,3-Dihydrospiro[Indene-2,4'-Piperidine] (5)1,4-Dioxaspiro[4.5]Decane (17)2,8-Diazaspiro[4.5]Decane (5)2-Oxa-8-Azaspiro[4.5]Decane (5)3-Phenyloxetane (11)6-Azaspiro[3.4]Octane (4)7-Azaspiro[3.5]Nonane (7)9,9'-Spirobi[Fluorene] (19)Spiro[Cyclopropane-1,3'-Indolin]-2'-One (6)Spiro[Indene-2,4'-Piperidin]-1(3H)-One (5)Spiro[Indoline-3,4'-Piperidine] (5)Azetidines (85)Benzofurans (6)Dioxolanes (23)Epoxides (5)Imidazolidines (4)Imidazolines (4)Indolines (5)Morpholines (33)Oxazolidines (6)Oxetanes (26)Piperazines (14)Piperidines (139)Pyrrolidines (86)Tetrahydrofurans (18)Tetrahydropyrans (24)Other Aliphatic Heterocycles (14)Other Aromatic Heterocycles (12)
Tetrahydrofurans (2277)
Alcohols (11)Amides (19)Amines (8)Anhydrides (3)Aryls (12)Bromides (4)Carboxylic Acids (7)Chlorides (3)Esters (22)Ethers (1)Ketones (6)Hydroxytetrahydrofuran (16)Iodotetrahydrofuran (3)Methoxytetrahydrofuran (11)Oxotetrahydrofuran (26)(2R,3S)-2-((Benzoyloxy)Methyl)Tetrahydrofuran-3-Yl Benzoate (8)(3Ar,6Ar)-Hexahydrofuro[3,2-B]Furan (5)(3As,6As)-Tetrahydrofuro[3,4-D][1,3]Dioxole (6)(R)-7-(Tetrahydrofuran-2-Yl)-7H-Pyrrolo[2,3-D]Pyrimidine (8)(R)-N-(9-(Tetrahydrofuran-2-Yl)-9H-Purin-6-Yl)Benzamide (5)(Tetrahydrofuran-2-Yl)Methanamine (4)(Tetrahydrofuran-2-Yl)Methanol (6)(Tetrahydrofuran-3-Yl)Methanamine (3)1,3-Dihydrobenzo[C][1,2]Oxaborole (20)1-(3-Fluorotetrahydrofuran-2-Yl)Pyrimidin-2(1H)-One (10)1-(5-(Hydroxymethyl)Tetrahydrofuran-2-Yl)Pyrimidine-2,4(1H,3H)-Dione (31)1-(Tetrahydrofuran-2-Yl)Pyrimidin-2(1H)-One (50)1-Oxa-8-Azaspiro[4.5]Decane (5)2-(Acetoxymethyl)-5-(9H-Purin-9-Yl)Tetrahydrofuran-3,4-Diyl Diacetate (6)2-(Hydroxymethyl)-5-(9H-Purin-9-Yl)Tetrahydrofuran-3,4-Diol (28)2-Oxa-8-Azaspiro[4.5]Decane (5)2-Oxabicyclo[2.1.1]Hexane (6)3-Aminodihydrofuran-2(3H)-One (4)3-Iodotetrahydrofuran (3)4-Amino-1-(Tetrahydrofuran-2-Yl)Pyrimidin-2(1H)-One (22)4-Fluoro-2-(Hydroxymethyl)-5-(9H-Purin-9-Yl)Tetrahydrofuran-3-Ol (4)4-Hydroxydihydrofuran-2(3H)-One (14)5-(Hydroxymethyl)Dihydrofuran-2(3H)-One (7)5-Bromo-1-(Tetrahydrofuran-2-Yl)Pyrimidine-2,4(1H,3H)-Dione (4)5-Fluoro-1-(Tetrahydrofuran-2-Yl)Pyrimidin-2(1H)-One (7)5-Fluoro-1-(Tetrahydrofuran-2-Yl)Pyrimidine-2,4(1H,3H)-Dione (3)5-Oxotetrahydrofuran-2-Carboxylic Acid (4)9-(Tetrahydrofuran-2-Yl)-9H-Purin-6-Amine (30)9-(Tetrahydrofuran-2-Yl)-9H-Purin-6-Ol (6)9-(Tetrahydrofuran-2-Yl)-9H-Purine (2)Dihydrofuran-3(2H)-One (8)Tert-Butyl (Tetrahydrofuran-3-Yl)Carbamate (4)Tetrahydrofuran-2-Carboxylic Acid (4)Tetrahydrofuran-3-Amine (6)Tetrahydrofuran-3-Carboxylic Acid (4)Tetrahydrofuran-3-Ol (6)Piperidines (9)Spiroes (18)
Tetrahydroisoquinolines (1289)
Amides (7)Amines (3)Aryls (3)Chlorides (4)Tetrahydroisoquinoline-Carboxylic Acid (48)(9H-Fluoren-9-Yl)Methyl 3,4-Dihydroisoquinoline-2(1H)-Carboxylate (7)1,2,3,4-Tetrahydroisoquinolin-5-Amine (4)1,2,3,4-Tetrahydroisoquinolin-5-Ol (3)1,2,3,4-Tetrahydroisoquinolin-6-Amine (3)1,2,3,4-Tetrahydroisoquinolin-6-Ol (6)1,2,3,4-Tetrahydroisoquinolin-7-Amine (3)1,2,3,4-Tetrahydroisoquinoline-1-Carboxylic Acid (19)1,2,3,4-Tetrahydroisoquinoline-3-Carboxylic Acid (12)1,4-Dihydroisoquinolin-3(2H)-One (5)1-Methyl-1,2,3,4-Tetrahydroisoquinoline (4)2-Methyl-1,2,3,4-Tetrahydroisoquinoline (13)4,4-Dimethyl-1,2,3,4-Tetrahydroisoquinoline (3)5,6,7,8-Tetrahydroisoquinoline (5)5-Bromo-1,2,3,4-Tetrahydroisoquinoline (4)5-Chloro-1,2,3,4-Tetrahydroisoquinoline (3)5-Fluoro-1,2,3,4-Tetrahydroisoquinoline (3)5-Methyl-1,2,3,4-Tetrahydroisoquinoline (4)6-Bromo-1,2,3,4-Tetrahydroisoquinoline (4)6-Chloro-1,2,3,4-Tetrahydroisoquinoline (3)6-Fluoro-1,2,3,4-Tetrahydroisoquinoline (3)6-Methoxy-1,2,3,4-Tetrahydroisoquinoline (3)6-Nitro-1,2,3,4-Tetrahydroisoquinoline (3)7-Bromo-1,2,3,4-Tetrahydroisoquinoline (4)7-Chloro-1,2,3,4-Tetrahydroisoquinoline (5)7-Fluoro-1,2,3,4-Tetrahydroisoquinoline (3)7-Methoxy-1,2,3,4-Tetrahydroisoquinoline (6)7-Nitro-1,2,3,4-Tetrahydroisoquinoline (3)Isoquinoline-1,3(2H,4H)-Dione (7)
Tetrahydropyrans (3363)
Alcohols (36)Aldehydes (6)Alkenyls (7)Amides (15)Amines (3)Aryls (28)Bromides (6)Carboxylic Acids (12)Chlorides (5)Esters (25)Ethers (24)Fluorinated Building Blocks (8)Hydrazines (4)Iodides (4)Ketones (7)Nitriles (9)Sulfides (3)Difluorochroman (16)(1S,4Ar,7Ar)-1-(((S)-Tetrahydro-2H-Pyran-2-Yl)Oxy)-1,4A,5,7A-Tetrahydrocyclopenta[C]Pyran (4)(6-Phenoxytetrahydro-2H-Pyran-2-Yl)Methanol (18)(R)-2-Phenoxytetrahydro-2H-Pyran (7)(S)-2-(Phenylthio)Tetrahydro-2H-Pyran (5)(S)-7-(((S)-Tetrahydro-2H-Pyran-2-Yl)Oxy)-7,8,9,10-Tetrahydrotetracene-5,12-Dione (5)(S)-N-(((S)-Tetrahydro-2H-Pyran-2-Yl)Methyl)Pyrrolidine-2-Carboxamide (4)1-(Tetrahydro-2H-Pyran-4-Yl)-1H-Pyrazole (6)2-(Phenoxymethyl)Tetrahydro-2H-Pyran (5)2-Phenoxytetrahydro-2H-Pyran (6)2-Phenoxytetrahydro-2H-Pyran-4-Yl Acetate (6)4-Phenoxytetrahydro-2H-Pyran (5)9-Bromoanthracene (5)Azetidines (6)Piperidines (5)Pyrazoles (4)Pyrrolidines (6)Spiroes (24)
Tetrahydroquinolines (1465)
Amides (14)Amines (5)Bromides (6)Fluorinated Building Blocks (6)Tetrahydroquinoline-Carboxylic Acid (18)1-Ethyl-1,2,3,4-Tetrahydroquinoline (6)1-Methyl-1,2,3,4-Tetrahydroquinoline (6)2,3-Dihydroquinolin-4(1H)-One (19)2-Methyl-1,2,3,4-Tetrahydroquinoline (3)4,4-Dimethyl-1,2,3,4-Tetrahydroquinoline (3)4-Methyl-1,2,3,4-Tetrahydroquinoline (2)5,6,7,8-Tetrahydroquinoline (17)6-Bromo-1,2,3,4-Tetrahydroquinoline (6)6-Chloro-1,2,3,4-Tetrahydroquinoline (3)6-Fluoro-1,2,3,4-Tetrahydroquinoline (3)7-Fluoro-1,2,3,4-Tetrahydroquinoline (3)Tert-Butyl 3,4-Dihydroquinoline-1(2H)-Carboxylate (11)
Thiadiazoles (2120)
Amides (11)Amines (24)Aryls (17)Chlorides (4)Esters (4)Ethers (7)Sulfamides (4)Sulfides (7)1,2,3-Thiadiazole (8)1,2,4-Thiadiazole (16)1,2,5-Thiadiazole (6)1,3,4-Thiadiazol-2-Amine (9)2-Bromo-1,3,4-Thiadiazole (11)2-Methyl-1,3,4-Thiadiazole (12)2-Phenyl-1,3,4-Thiadiazole (19)4,7-Diphenylbenzo[C][1,2,5]Thiadiazole (6)4-Bromobenzo[C][1,2,5]Thiadiazole (14)Benzo[D][1,2,3]Thiadiazole (7)Pyridines (7)Thiophenols (6)
Thiazoles (6687)
Alcohols (32)Aldehydes (15)Amides (40)Amines (102)Aryls (97)Bromides (70)Carboxylic Acids (62)Chlorides (29)Esters (70)Ethers (16)Fluorinated Building Blocks (9)Iodides (4)Ketones (19)Nitriles (7)Nitroes (8)Sulfides (8)Acetylthiazole (7)Bromothiazolo (9)Chlorothiazole (32)Chlorothiazolo (18)Cyanothiazole (7)Cyclopropylthiazole (11)Dibromothiazole (7)Dichlorothiazole (8)Dihydrothiazole (16)Dihydrothiazolo (14)Dimethylthiazole (22)Ethylthiazole (8)Formylthiazole (7)Methylthiazole (109)Nitrothiazol (8)Tetrahydrothiazolo (8)1-(Thiazol-2-Yl)Ethan-1-One (9)2,4-Dibromothiazole (5)2,4-Dichlorothiazole (4)2,4-Dimethylthiazole (7)2,5-Dibromothiazole (5)2,5-Dimethylthiazole (4)2-(Chloromethyl)Thiazole (5)2-(Methylthio)Thiazole (5)2-(Thiazol-4-Yl)Acetic Acid (9)2-(Trifluoromethyl)Thiazole (11)2-Bromo-4-Methylthiazole (7)2-Bromo-5-Methylthiazole (7)2-Chloro-4-Methylthiazole (5)2-Chloro-5-Methylthiazole (5)2-Chlorothiazole (32)2-Imino-N-((6R,7R)-8-Oxo-5-Thia-1-Azabicyclo[4.2.0]Oct-2-En-7-Yl)-2-(Thiazol-4-Yl)Acetamide (3)2-Isopropylthiazole (11)2-Methyl-4-Phenylthiazole (6)2-Phenoxythiazole (5)2-Phenylbenzo[D]Thiazole (16)2-Phenylthiazole-4-Carbaldehyde (9)4,5,6,7-Tetrahydrobenzo[D]Thiazole (14)4,5,6,7-Tetrahydrothiazolo[5,4-C]Pyridine (10)4,5-Dihydrothiazole (8)4-(4-Fluorophenyl)Thiazole (9)4-(Trifluoromethyl)Thiazole (7)4-Bromothiazole (10)4-Cyclopropylthiazole (6)4-Methyl-2-Phenylthiazole (6)4-Methylthiazol-2-Amine (7)4-Methylthiazole (5)4-Phenylthiazol-2-Amine (37)5,6-Dihydrobenzo[D]Thiazol-7(4H)-One (5)5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Thiazole (7)5-Bromothiazol-2-Amine (6)5-Bromothiazole (8)5-Methyl-4-Phenylthiazole (5)5-Methylthiazol-2-Amine (6)5-Phenylthiazole (20)Ethyl 2-Aminothiazole-4-Carboxylate (6)Ethyl 2-Aminothiazole-5-Carboxylate (5)Ethyl 2-Phenylthiazole-4-Carboxylate (12)Ethyl 2-Phenylthiazole-5-Carboxylate (9)Ethyl Thiazole-2-Carboxylate (7)Ethyl Thiazole-4-Carboxylate (7)Ethyl Thiazole-5-Carboxylate (7)Methyl 2-Aminothiazole-4-Carboxylate (6)Methyl Thiazole-2-Carboxylate (6)Methyl Thiazole-4-Carboxylate (6)N-(Thiazol-2-Yl)Acetamide (9)N-(Thiazol-2-Yl)Benzamide (9)N-Phenylthiazol-2-Amine (9)Tert-Butyl Thiazol-2-Ylcarbamate (22)Thiazol-2-Ylmethanamine (5)Thiazol-2-Ylmethanol (9)Thiazol-5-Amine (6)Thiazole-2-Carbaldehyde (9)Thiazole-2-Carboxylic Acid (9)Thiazole-4-Carboxylic Acid (5)Thiazole-5-Carbaldehyde (19)Thiazole-5-Carbonitrile (7)Thiazole-5-Carboxylic Acid (10)Thiazolo[4,5-B]Pyridine (12)Thiazolo[5,4-B]Pyridine (13)Thiazolo[5,4-C]Pyridine (5)Thiazolo[5,4-D]Pyrimidine (5)Morpholines (5)Thiophenols (4)
Thiophenes (1233)
Acetylthiophene (5)Aminothiophene (23)Bromothieno (29)Chlorothieno (41)Chlorothiophene (61)Cyanothiophene (5)Dibromothieno (5)Dibromothiophene (17)Dichlorothieno (10)Dichlorothiophene (13)Dihydrothieno (24)Dihydrothiophene (1)Dihydroxythiophene (7)Dimethylthieno (10)Dimethylthiophene (21)Dithiophene (40)Fluorothiophene (18)Formylthiophene (16)Hydroxythiophene (7)Iodothieno (6)Iodothiophene (13)Methylthieno (27)Nitrothiophene (27)Terthiophene (10)Tetrahydrothieno (17)Thiophenecarboxaldehyde (10)Thiphen-Amine-Hydrochloride (53)Trithiophene (5)(9H-Fluoren-9-Yl)Methyl (2-(Thiophen-2-Yl)Ethyl)Carbamate (5)1-(Thiophen-2-Yl)Ethan-1-One (17)2,2'-Bithiophene (25)2,2':5',2''-Terthiophene (9)2,3-Dihydrothieno[3,4-B][1,4]Dioxine (9)2,5-Dibromothiophene (18)2,5-Diphenylthiophene (5)2-(Thiophen-2-Yl)Pyridine (5)2-(Trifluoromethyl)Thiophene (10)2-Chlorothiophene (20)2-Ethynylthiophene (8)2-Iodothiophene (14)2-Methoxythiophene (7)2-Nitrothiophene (20)3-Bromothiophene (6)3-Chlorothiophene (17)3-Fluorothiophene (6)3-Methoxythiophene (9)3-Methylthiophene (14)3-Nitrothiophene (6)3-Phenylthiophene (13)4,4,5,5-Tetramethyl-2-(Thiophen-2-Yl)-1,3,2-Dioxaborolane (23)4,4,5,5-Tetramethyl-2-(Thiophen-3-Yl)-1,3,2-Dioxaborolane (10)4,5,6,7-Tetrahydrothieno[2,3-C]Pyridine (10)4,5,6,7-Tetrahydrothieno[3,2-C]Pyridine (9)4,7-Di(Thiophen-2-Yl)Benzo[C][1,2,5]Thiadiazole (9)4,8-Di(Thiophen-2-Yl)Benzo[1,2-B:4,5-B']Dithiophene (8)4-Phenyl-6H-Thieno[3,2-F][1,2,4]Triazolo[4,3-A][1,4]Diazepine (5)4H-Cyclopenta[2,1-B:3,4-B']Dithiophene (9)4H-Thieno[3,2-B]Pyrrole (6)4H-Thieno[3,4-C]Pyrrole-4,6(5H)-Dione (7)5,6-Dihydro-4H-Cyclopenta[B]Thiophene (7)5-(Thiophen-2-Yl)Isoxazole (5)5-Benzyl-4,5,6,7-Tetrahydrothieno[3,2-C]Pyridine (7)5-Phenylthiophen-2-Amine (5)Benzo[1,2-B:4,5-B']Dithiophene (7)Dibenzo[B,D]Thiophene (22)Dihydrothiophen-3(2H)-One (4)Dithieno[3,2-B:2',3'-D]Thiophene (5)Ethyl Thiophene-2-Carboxylate (19)Ethyl Thiophene-3-Carboxylate (11)Methyl Thiophene-3-Carboxylate (34)N,N-Diphenyl-4-(Thiophen-2-Yl)Aniline (5)Phenyl(Thiophen-2-Yl)Methanone (6)Thieno[2,3-B]Pyridine (22)Thieno[2,3-C]Pyridine (14)Thieno[2,3-D]Pyrimidine (30)Thieno[3,2-B]Pyridine (18)Thieno[3,2-B]Thiophene (19)Thieno[3,2-C]Pyridine (13)Thieno[3,2-D]Pyrimidine (24)Thieno[3,4-B]Thiophene (5)Thiophen-2-Ylboronic Acid (20)Thiophen-2-Ylmethanol (11)Thiophen-3-Amine (18)Thiophen-3-Ol (6)Thiophene-2-Carbaldehyde (43)Thiophene-2-Carbonitrile (23)Thiophene-3-Carbaldehyde (17)Thiophene-3-Carbonitrile (13)Thiophene-3-Carboxamide (5)Thiophene-3-Carboxylic Acid (8)
Triazines (1388)
Amides (11)Amines (14)Aryls (12)Chlorides (13)1,2,4-Triazine (10)1,3,5-Triazin-2-Amine (3)2,4,6-Triphenoxy-1,3,5-Triazine (4)2,4,6-Triphenyl-1,3,5-Triazine (24)2,4-Diphenyl-1,3,5-Triazine (6)2-Chloro-1,3,5-Triazine (10)2-Methoxy-1,3,5-Triazine (10)2-Phenyl-1,3,5-Triazine (6)N2,N4,N6-Triphenyl-1,3,5-Triazine-2,4,6-Triamine (4)Pyrrolo[2,1-F][1,2,4]Triazine (14)
Triazoles (7725)
Amides (8)Amines (21)Aryls (24)Carboxylic Acids (10)Esters (9)Ethers (5)Hydrazines (3)Sulfides (3)(5As,10Br)-2-Phenyl-2,5A,6,10B-Tetrahydro-4H-Indeno[2,1-B][1,2,4]Triazolo[4,3-D][1,4]Oxazin-11-Ium (6)1-(Phenylsulfonyl)-1H-1,2,4-Triazole (6)1-Benzyl-1H-1,2,3-Triazole (8)1-Benzyl-1H-1,2,4-Triazole (6)1-Methyl-1H-1,2,3-Triazole (9)1-Methyl-1H-1,2,4-Triazole (17)1-Methyl-1H-Benzo[D][1,2,3]Triazole (10)1-Phenyl-1H-1,2,3-Triazole (7)1-Phenyl-1H-1,2,4-Triazole (20)1H-1,2,4-Triazol-3-Amine (7)1H-1,2,4-Triazol-5-Amine (6)2-Bromo-[1,2,4]Triazolo[1,5-A]Pyridine (6)2-Phenyl-2,5,6,7-Tetrahydropyrrolo[2,1-C][1,2,4]Triazol-4-Ium (6)2-Phenyl-2H-1,2,3-Triazole (7)2-Phenyl-2H-Benzo[D][1,2,3]Triazole (8)2H-1,2,3-Triazole (7)4-(4-Cyclopropylnaphthalen-1-Yl)-4H-1,2,4-Triazole (7)4-Phenyl-4H-1,2,4-Triazole (6)4-Phenyl-6H-Thieno[3,2-F][1,2,4]Triazolo[4,3-A][1,4]Diazepine (5)4H-1,2,4-Triazole (13)5-Bromo-1H-1,2,4-Triazole (6)5-Bromo-1H-Benzo[D][1,2,3]Triazole (5)5-Fluoro-1H-Benzo[D][1,2,3]Triazole (4)6-Bromo-[1,2,4]Triazolo[1,5-A]Pyridine (12)6-Bromo-[1,2,4]Triazolo[4,3-A]Pyridine (8)8-Bromo-[1,2,4]Triazolo[1,5-A]Pyridine (6)[1,2,4]Triazolo[1,5-A]Pyrazine (9)[1,2,4]Triazolo[1,5-A]Pyridin-2-Amine (17)[1,2,4]Triazolo[4,3-A]Pyridin-3-Ylmethanamine (5)Ethyl 1H-1,2,4-Triazole-3-Carboxylate (3)Pyridines (13)Thiophenols (8)
Other Aliphatic Heterocycles (6214)
Alcohols (39)Alkenyls (19)Amides (103)Amines (17)Anhydrides (10)Aryls (55)Carboxylic Acids (12)Chlorides (3)Esters (69)Ethers (5)Fluorinated Building Blocks (5)Ketones (59)Sulfamides (11)Sulfides (8)Sulfones (9)1,3-Dioxane (20)1,4-Diazepane (12)3-Azabicyclo[3.1.0]Hexane (14)Epoxides (11)Spiroes (14)
Other Aromatic Heterocycles (37790)
Alcohols (110)Aldehydes (40)Alkenyls (5)Amides (181)Amines (134)Anhydrides (33)Aryls (110)Bromides (92)Carboxylic Acids (65)Chlorides (149)Esters (146)Ethers (48)Fluorinated Building Blocks (47)Iodides (28)Ketones (131)Nitriles (31)Nitroes (22)Sulfamides (17)Sulfides (8)Sulfones (5)Trifluoromethyls (19)Indolizine (15)Phenazine (8)Piperidines (14)Pyridines (2)Spiroes (12)
Organic Building Blocks
Alcohols (45612)
Aldehydes (19)Aliphatic Chain Hydrocarbons (404)Aliphatic Cyclic Hydrocarbons (117)Alkenyls (101)Alkynyls (34)Amides (371)Amines (287)Aryls (633)Azetidines (18)Benzimidazoles (11)Benzodioxans (9)Benzofurans (18)Benzothiazoles (6)Bromides (147)Carboxylic Acid Salts (22)Carboxylic Acids (171)Chlorides (157)Difluoromethyls (10)Dioxolanes (4)Esters (239)Ethers (224)Fluorinated Building Blocks (169)Furans (8)Hydrazides (5)Hydrazines (5)Imidazoles (27)Indazoles (17)Indoles (26)Indolines (13)Iodides (16)Isoquinolines (4)Isoxazoles (8)Ketones (64)Morpholines (26)Nitriles (45)Nitroes (42)Oxazoles (9)Oxazolidines (6)Oxetanes (24)Phenols (15)Piperazines (26)Piperidines (132)Pyrazines (6)Pyrazoles (42)Pyridazines (8)Pyridines (160)Pyrimidines (63)Pyrroles (2)Pyrrolidines (82)Quinazolines (12)Quinolines (79)Quinoxalines (6)Quinuclidines (5)Spiroes (15)Sulfamides (28)Sulfides (26)Sulfones (7)Tetrahydrofurans (11)Tetrahydropyrans (36)Thiazoles (32)Thiols (5)Thiophenes (23)Trifluoromethyls (71)Ureas (5)Other Aliphatic Heterocycles (39)Other Aromatic Heterocycles (110)Amino-Propanol (6)Cyclopropylethanol (6)Diethanol (23)Diethanol (23)Dihydroxybenzylidene (4)Dihydroxynaphthalene (7)Dihydroxypropanoate (7)Dihydroxyterephthalic Acid (3)Hydroxyacetamide (1)Hydroxyacetimidamide (3)Hydroxyadamantan (4)Hydroxyadamantan (4)Hydroxyazetidine (7)Hydroxybenzyl (17)Hydroxybenzylidene (5)Hydroxybicyclo (7)Hydroxybutan (4)Hydroxybutan (4)Hydroxybutanoate (36)Hydroxybutanoic (33)Hydroxycarbamimidoyl (2)Hydroxycinnamic Acid (2)Hydroxycyclobutyl (3)Hydroxycyclohexyl (7)Hydroxycyclopentyl (1)Hydroxydiphenylmethyl (2)Hydroxyethoxy (22)Hydroxyethoxy (22)Hydroxyhexanoate (8)Hydroxyhexanoic (6)Hydroxyimino (11)Hydroxyisonicotinaldehyde (7)Hydroxyisonicotinate (5)Hydroxyisonicotinic (5)Hydroxyisonicotinic (5)Hydroxyisonicotinic Acid (4)Hydroxymethyl-Phenyl-Boronic-Acid (32)Hydroxynaphthalene (27)Hydroxynicotinaldehyde (7)Hydroxynicotinate (13)Hydroxynicotinonitrile (6)Hydroxyoctadec (1)Hydroxyoctadec (1)Hydroxyoctadecanoic (7)Hydroxypentanoate (6)Hydroxypropanoic (35)Pentahydroxyhexanal (9)Phenyl-Methanol (1)Propanediol (9)Propanediol (9)Tetrahydroxy (25)Tetrahydroxy (25)Tetrahydroxyhexanal (9)Tetrahydroxypentanal (5)Trihydroxy (246)Trihydroxy (246)(1-Methyl-1H-Pyrazol-3-Yl)Methanol (7)(1-Phenylcyclopropyl)Methanol (3)(1H-Benzo[D]Imidazol-2-Yl)Methanol (14)(1H-Indol-2-Yl)Methanol (8)(2-(Trifluoromethyl)Phenyl)Methanol (10)(2-Bromo-6-Fluorophenyl)Methanol (4)(2-Bromophenyl)Methanol (39)(2-Chlorophenyl)Methanol (40)(2-Fluorophenyl)Methanol (60)(2-Iodophenyl)Methanol (12)(2-Nitrophenyl)Methanol (29)(3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Phenyl)Methanol (7)(3-(Trifluoromethoxy)Phenyl)Methanol (4)(3-(Trifluoromethyl)Phenyl)Methanol (22)(3-Chlorophenyl)Methanol (38)(3-Fluorophenyl)Methanol (58)(3-Iodophenyl)Methanol (13)(3-Methylpyridin-2-Yl)Methanol (10)(3-Nitrophenyl)Methanol (31)(3-Phenoxyphenyl)Methanol (3)(3-Phenylisoxazol-5-Yl)Methanol (7)(4-Fluorophenyl)Methanol (29)(4-Nitrophenyl)Methanol (17)(6-Methylpyridin-2-Yl)Methanol (5)(Tetrahydrofuran-2-Yl)Methanol (6)1,2-Diphenylethan-1-Ol (8)1-Phenyl-1H-Pyrazol-3-Ol (3)1-Phenylethan-1-Ol (103)2,2,2-Trifluoro-1-Phenylethan-1-Ol (27)2-(1H-Indol-3-Yl)Ethan-1-Ol (10)2-(2-Chlorophenyl)Ethan-1-Ol (8)2-(Piperazin-1-Yl)Ethan-1-Ol (10)2-(Piperidin-1-Yl)Ethan-1-Ol (10)2-(Piperidin-2-Yl)Ethan-1-Ol (8)2-(Piperidin-4-Yl)Ethan-1-Ol (5)2-Amino-2-(2-Fluorophenyl)Ethan-1-Ol (7)2-Amino-2-(Anthracen-9-Yl)Ethan-1-Ol (4)2-Amino-2-Phenylethan-1-Ol (196)2-Hydroxy-1,2-Diphenylethan-1-One (3)2-Hydroxy-1-Phenylethan-1-One (8)2-Hydroxyethyl Benzoate (3)2-Phenoxyethan-1-Ol (14)2-Phenylethan-1-Ol (78)2-Phenylpropan-2-Ol (21)3-Phenylpropan-1-Ol (24)4-Fluoro-2-(Hydroxymethyl)-5-(9H-Purin-9-Yl)Tetrahydrofuran-3-Ol (4)4-Phenylpiperidin-4-Ol (10)Azetidin-2-Ylmethanol (7)Azetidin-3-Ol (11)Benzyl 3-(Hydroxymethyl)Piperazine-1-Carboxylate (6)Benzyl-2-(Hydroxymethyl)Pyrrolidine-1-Carboxylate (9)Bicyclo[1.1.1]Pentan-1-Ylmethanol (3)Cyclobutanol (46)Cyclobutylmethanol (15)Cyclohexanol (71)Cyclohexylmethanol (31)Cyclopentanol (51)Diphenylmethanol (20)Isoxazol-3-Ylmethanol (3)Isoxazol-5-Ylmethanol (5)M-Tolylmethanol (34)Morpholin-2-Ylmethanol (12)Morpholin-3-Ylmethanol (14)Naphthalen-1-Ylmethanol (8)O-Tolylmethanol (35)Oxiran-2-Ylmethanol (3)Piperidin-2-Ylmethanol (11)Piperidin-3-Ol (27)Piperidin-3-Ylmethanol (15)Piperidin-4-Ylmethanol (11)Pyridin-2-Ylmethanol (65)Pyridin-3-Ylmethanol (39)Pyridin-4-Ylmethanol (16)Pyrimidin-2-Ylmethanol (5)Pyrrolidin-2-Ylmethanol (26)Pyrrolidin-3-Ylmethanol (13)Quinazolin-2-Ol (7)Tert-Butyl 3-(Hydroxymethyl)-1H-Indole-1-Carboxylate (10)Thiazol-2-Ylmethanol (9)Thiophen-2-Ylmethanol (11)Triphenylmethanol (13)
Aldehydes (18934)
Alcohols (19)Aliphatic Chain Hydrocarbons (55)Aliphatic Cyclic Hydrocarbons (12)Alkenyls (29)Alkynyls (6)Amides (56)Amines (29)Aryls (346)Benzimidazoles (5)Benzofurans (4)Benzothiazoles (5)Bromides (78)Carboxylic Acids (18)Chlorides (88)Esters (47)Ethers (130)Fluorinated Building Blocks (79)Furans (15)Imidazoles (21)Indazoles (9)Indoles (21)Isoxazoles (9)Ketones (19)Morpholines (9)Nitriles (12)Nitroes (22)Phenols (13)Piperidines (17)Pyrazines (4)Pyrazoles (41)Pyridines (54)Pyrimidines (11)Pyrroles (12)Quinolines (23)Sulfamides (6)Sulfides (8)Tetrahydropyrans (6)Thiazoles (15)Thiophenes (13)Trifluoromethyls (10)Other Aromatic Heterocycles (40)Aminonicotinaldehyde (5)Aminonicotinaldehyde (5)Carboxyaldehyde (1093)Dicarbaldehyde (37)Dicarbaldehyde (37)Formylisonicotinate (7)Tetracarbaldehyde (8)Tetracarbaldehyde (8)Tricarbaldehyde (10)1-Methyl-1H-Indole-3-Carbaldehyde (9)1-Naphthaldehyde (23)1-Phenyl-1H-Pyrazole-4-Carbaldehyde (8)1H-Benzo[D]Imidazole-2-Carbaldehyde (5)1H-Indazole-3-Carbaldehyde (19)1H-Indole-2-Carbaldehyde (20)1H-Pyrazole-4-Carbaldehyde (30)1H-Pyrrole-2-Carbaldehyde (21)1H-Pyrrole-3-Carbaldehyde (9)1H-Pyrrolo[2,3-B]Pyridine-3-Carbaldehyde (14)1H-Pyrrolo[2,3-B]Pyridine-5-Carbaldehyde (5)2,3-Dichlorobenzaldehyde (7)2,3-Difluorobenzaldehyde (18)2,4-Dichlorobenzaldehyde (8)2,4-Difluorobenzaldehyde (19)2,6-Dibromobenzaldehyde (4)2,6-Difluorobenzaldehyde (25)2-(Benzyloxy)Benzaldehyde (13)2-(Phenylethynyl)Benzaldehyde (9)2-(Trifluoromethoxy)Benzaldehyde (7)2-(Trifluoromethyl)Benzaldehyde (27)2-Bromo-3-Hydroxybenzaldehyde (4)2-Bromo-3-Methoxybenzaldehyde (4)2-Bromo-6-Fluorobenzaldehyde (15)2-Chloro-3-Hydroxybenzaldehyde (4)2-Chloro-3-Methoxybenzaldehyde (7)2-Chloro-4-Fluorobenzaldehyde (10)2-Fluoro-3-Methylbenzaldehyde (8)2-Fluorobenzaldehyde (175)2-Hydroxy-3-Iodobenzaldehyde (3)2-Iodobenzaldehyde (17)2-Methyl-1H-Indole-3-Carbaldehyde (3)2-Naphthaldehyde (12)2-Nitrobenzaldehyde (53)2-Oxo-2-Phenylacetaldehyde (12)2-Phenylacetaldehyde (20)3-(Benzyloxy)Benzaldehyde (13)3-(Trifluoromethoxy)Benzaldehyde (10)3-(Trifluoromethyl)Benzaldehyde (31)3-Bromo-2-Chlorobenzaldehyde (4)3-Bromo-2-Fluorobenzaldehyde (9)3-Bromo-2-Hydroxybenzaldehyde (10)3-Bromo-4-Fluorobenzaldehyde (7)3-Bromo-4-Hydroxybenzaldehyde (9)3-Bromo-4-Methoxybenzaldehyde (9)3-Chloro-2-Fluorobenzaldehyde (9)3-Chloro-2-Hydroxybenzaldehyde (5)3-Chloro-4-Methoxybenzaldehyde (7)3-Chlorobenzaldehyde (106)3-Fluorobenzaldehyde (137)3-Iodobenzaldehyde (28)3-Methoxy-2-Nitrobenzaldehyde (3)3-Nitrobenzaldehyde (49)3-Phenoxybenzaldehyde (7)4-(1H-Imidazol-1-Yl)Benzaldehyde (7)4-(Benzyloxy)Benzaldehyde (23)4-(Piperazin-1-Yl)Benzaldehyde (5)4-Fluorobenzaldehyde (78)4-Hydroxy-3-Nitrobenzaldehyde (5)4-Morpholinobenzaldehyde (8)4-Nitrobenzaldehyde (18)4-Phenoxybenzaldehyde (12)5-Phenylfuran-2-Carbaldehyde (9)6-Methoxynicotinaldehyde (4)[1,1'-Biphenyl]-3-Carbaldehyde (20)[1,1'-Biphenyl]-4-Carbaldehyde (31)Benzo[B]Thiophene-2-Carbaldehyde (7)Benzo[B]Thiophene-3-Carbaldehyde (6)Benzo[D][1,3]Dioxole-5-Carbaldehyde (6)Benzofuran-2-Carbaldehyde (5)Furan-2-Carbaldehyde (22)Imidazo[1,2-A]Pyridine-2-Carbaldehyde (8)Imidazo[1,2-A]Pyridine-3-Carbaldehyde (9)Isonicotinaldehyde (68)Methyl 3-Formylbenzoate (23)Nicotinaldehyde (152)Picolinaldehyde (98)Pyrazine-2-Carbaldehyde (17)Pyrimidine-5-Carbaldehyde (38)Thiazole-2-Carbaldehyde (9)Thiazole-5-Carbaldehyde (19)Thiophene-2-Carbaldehyde (43)Thiophene-3-Carbaldehyde (17)
Aliphatic Chain Hydrocarbons (12641)
Acyl Chlorides (24)Alcohols (404)Aldehydes (55)Alkenyls (383)Alkynyls (23)Amides (456)Amidines (21)Amines (304)Anhydrides (11)Bromides (148)Carboxylic Acid Salts (64)Carboxylic Acids (159)Chlorides (143)Difluoromethyls (13)Epoxides (7)Esters (450)Ethers (199)Fluorinated Building Blocks (169)Guanidines (18)Hydrazides (48)Hydrazines (61)Hydroxylamines (11)Iodides (38)Ketones (182)Nitriles (92)Nitroes (7)Sulfamides (37)Sulfides (27)Sulfonates (4)Sulfones (19)Sulfonyl Chlorides (19)Thioesters (4)Thiols (27)Thioureas (19)Trifluoromethyls (68)Ureas (28)Acetylthio (7)Acetylthio (7)Azanonadecan (7)Azanylylidene (1)Azapentadecan (10)Azapentadecan (10)Azatridecan (4)Butyloctyl (4)Butyloctyl (4)Carbonylbis (1)Carbonylbis (1)Diazirine (13)Diazirine (13)Didodecyl (7)Didodecyl (7)Diethyl (73)Diethyl (73)Dihydroxypropan (6)Diisobutyl (2)Diisopropyl (27)Diisopropyl (27)Dimethoxy (305)Dimethyladamantane (14)Dimethyladamantane (14)Dimethylalkanes (7)Dioctyl (1)Dioxide (445)Dioxide (445)Ethoxy (708)Ethoxyethoxy (5)Ethoxyethoxy (5)Ethoxyethyl (15)Ethoxyethyl (15)Ethoxymethyl (11)Ethoxymethylene (1)Ethoxymethylene (1)Isopropylidenebis (2)Isopropylsulfonyl (10)Isopropylsulfonyl (10)Methoxybut (2)Methoxybut (2)Methoxyethoxy (67)Methoxyethoxy (67)Methoxyethyl (66)Methoxyisonicotinic (11)Methoxyisonicotinic (11)Methoxymethoxy (23)Methoxymethoxy (23)Methoxymethyl (117)Methoxynicotinic (26)Methoxynicotinic (26)Methoxynicotinic Acid (20)Methoxypropan (24)Methoxypropan (24)Methoxypropoxy (8)Methoxypropoxy (8)Methoxypropyl (10)Methoxytetrahydro (14)Methoxytetrahydro (14)Methyladamantan (4)Methylisonicotinic (13)Oxoethoxy (1)Oxopropane (10)Oxopropyl (32)Oxopropyl (32)Pentyloxy (7)Tetramethoxy (6)Trimethoxy (15)Trimethoxy (15)Yldisulfanyl (7)
Aliphatic Cyclic Hydrocarbons (11010)
Acyl Chlorides (10)Alcohols (117)Aldehydes (12)Alkenyls (57)Amides (132)Amines (135)Aryls (10)Bromides (31)Carboxylic Acids (65)Chlorides (19)Esters (109)Ethers (32)Fluorinated Building Blocks (56)Hydrazides (5)Hydrazines (11)Iodides (10)Ketones (156)Nitriles (29)Spiroes (19)Sulfamides (8)Sulfonic Acids (4)Sulfonyl Chlorides (8)Trifluoromethyls (19)Ureas (6)(1-Phenylcyclobutyl)Methanamine (9)(1-Phenylcyclopropyl)Methanamine (12)(1-Phenylcyclopropyl)Methanol (3)(3Ar,E)-4-((Z)-2-(2-Methylenecyclohexylidene)Ethylidene)Octahydro-1H-Indene (7)(4-Cyclohexylphenyl)Boronic Acid (7)(4-Pentylcyclohexyl)Benzene (5)(4-Propylcyclohexyl)Benzene (5)(9H-Fluoren-2-Yl)Boronic Acid (6)(9H-Fluoren-9-Yl)Methyl (4-Methoxybenzyl)Carbamate (7)(Cyclopropylmethoxy)Benzene (18)1,1-Difluorocyclobutane (14)1,1-Difluorocyclohexane (18)1,1-Difluorocyclopentane (5)1-Bromo-2-Cyclopropylbenzene (3)1-Bromo-3-Cyclopropylbenzene (6)1-Chloro-2-Cyclopropylbenzene (7)1-Cyclohexyl-2-Fluorobenzene (4)1-Cyclopropyl-3-Methylbenzene (3)1-Methylcyclohex-1-Ene (26)1-Phenylcyclobutan-1-Amine (5)1-Phenylcyclobutane-1-Carbonitrile (7)1-Phenylcyclobutane-1-Carboxylic Acid (16)1-Phenylcyclohexane-1-Carboxylic Acid (6)1-Phenylcyclopropan-1-Amine (26)1-Phenylcyclopropane-1-Carbonitrile (19)1-Phenylcyclopropane-1-Carboxylic Acid (23)2-((((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Amino)-2-Phenylacetic Acid (5)2-Bromo-9,9-Diphenyl-9H-Fluorene (5)2-Bromo-9H-Fluorene (15)2-Cyclohexylacetic Acid (14)2-Cyclopropyl-4-Phenylquinoline (12)2-Cyclopropylpyridine (20)2-Cyclopropylpyrimidine (10)2-Iodo-9H-Fluorene (8)2-Methylcyclohexan-1-One (3)2-Phenylcyclopropan-1-Amine (20)3,3-Dimethylcyclohexan-1-One (4)3-Cyclopropylpyridine (28)3-Methylcyclohexan-1-One (9)4-Cyclohexylbenzoic Acid (6)4-Cyclohexylbenzonitrile (10)4-Cyclohexylphenol (7)5-Azaspiro[2.4]Heptane (13)6-Azaspiro[2.5]Octane (10)9,9-Dimethyl-9H-Fluorene (16)Bicyclo[1.1.1]Pentan-1-Ylmethanol (3)Bicyclo[1.1.1]Pentane-1-Carboxylic Acid (6)Bicyclo[2.2.1]Hept-2-Ene (18)Bicyclo[2.2.1]Heptane (15)Bromocyclohexane (7)Cyclobutanamine (90)Cyclobutanecarboxylic Acid (23)Cyclobutanol (46)Cyclobutylmethanol (15)Cycloheptane (12)Cyclohex-3-Ene-1-Carboxylic Acid (7)Cyclohexanamine (59)Cyclohexanecarboxylic Acid (83)Cyclohexanol (71)Cyclohexylmethanol (31)Cyclopentanamine (5)Cyclopentanecarboxylic Acid (26)Cyclopentanol (51)Cyclopropoxybenzene (13)Ethyl 1-Phenylcyclopropane-1-Carboxylate (4)Ethyl 2-Phenylcyclopropane-1-Carboxylate (5)Ethyl Cyclohexanecarboxylate (33)Isopropylcyclohexane (19)Methoxycyclohexane (7)Methyl 1-Phenylcyclopropane-1-Carboxylate (10)Methyl 4-Oxocyclohexane-1-Carboxylate (5)Methyl Bicyclo[1.1.1]Pentane-1-Carboxylate (10)Methyl Cyclobutanecarboxylate (24)Methyl Cyclohexanecarboxylate (45)Methyl Cyclopentanecarboxylate (18)N-Methylcyclohexanamine (7)Naphthalene-1,4-Dione (23)Spiro[3.3]Heptane (11)Tert-Butyl (1-Phenylcyclopropyl)Carbamate (6)Tert-Butyl (2-Phenylcyclopropyl)Carbamate (3)Tert-Butyl Bicyclo[1.1.1]Pentan-1-Ylcarbamate (5)Tert-Butyl Cyclobutylcarbamate (17)Tert-Butyl Cyclohexylcarbamate (9)Tert-Butyl Cyclopentylcarbamate (28)Aminobicyclo (24)Aminocyclobutanol (5)Aminocyclobutyl (5)Aminocyclohexane (55)Aminocyclohexanol (21)Aminocyclopentanol (17)Aminocyclopentyl (6)Aminocyclopropyl (4)Azabicyclo (290)Azabicyclo (290)Azatricyclo (5)Azatricyclo (5)Azetidine (177)Azetidine (177)Benzylazetidin (9)Bicyclo (61)Bicyclohexyl (6)Bromobicyclo (9)Butylcyclohexyl (9)Butylsulfonyl (6)Chlorocyclopropyl (2)Cyanoazetidine (5)Cyanocyclopropyl (4)Cyclobutanecarbonitrile (2)Cyclobutylacetic (4)Cyclobutylmethyl (4)Cyclobutylmethyl (4)Cyclohexa (168)Cyclohexa (168)Cyclohexane (126)Cyclohexane (126)Cyclohexanecarboxylic (19)Cyclohexanediamine (8)Cyclohexanone (93)Cyclohexylacetic (2)Cyclohexylamino (9)Cyclohexylbutanoic (4)Cyclohexylcarbamoyl (5)Cyclohexylethanamine (11)Cyclohexylmethoxy (4)Cyclohexylmethoxy (4)Cyclohexylmethyl (7)Cyclohexylmethyl (7)Cyclohexyloxy (10)Cyclohexylpropan (75)Cyclohexylpropanoic (3)Cyclopentadienyl (2)Cyclopentane (169)Cyclopentane (169)Cyclopentanecarboxylic (7)Cyclopentyl (56)Cyclopentyl (56)Cyclopentylacetic (3)Cyclopentylamino (1)Cyclopentylmethyl (4)Cyclopentyloxy (15)Cyclopentyloxy (15)Cyclopropanamine (32)Cyclopropanamine (32)Cyclopropanecarbonitrile (14)Cyclopropanecarbonyl (6)Cyclopropanecarbonyl (6)Cyclopropanecarboxamide (14)Cyclopropanol (4)Cyclopropoxyphenyl (7)Cyclopropyl (356)Cyclopropyl (356)Cyclopropylacetate (6)Cyclopropylacetic (3)Cyclopropylamino (24)Cyclopropylcarbamoyl (5)Cyclopropylethan (5)Cyclopropylethan (5)Cyclopropylethynyl (1)Cyclopropylmethoxy (32)Cyclopropylmethoxy (32)Cyclopropylmethyl (21)Cyclopropylmethyl (21)Cyclopropylpropanoic (5)Cyclopropylsulfonyl (6)Cyclopropylsulfonyl (6)Diazabicyclo (77)Diazabicyclo (77)Dicyclohexyl (6)Dicyclohexyl (6)Dicyclohexylphosphino (18)Dicyclohexylphosphino (18)Difluorocyclobutyl (6)Difluorocyclohexyl (6)Dihydroinden (8)Dihydroindeno (5)Dihydroindeno (5)Dihydropteridin (6)Dimethylbicyclo (5)Dimethylbicyclo (5)Dimethylcyclohex (5)Dimethylcyclohexane (16)Dimethylcyclohexanone (7)Dioxathiolane (1)Dioxathiolane (1)Dioxol (174)Dithiolan (1)Dithiolan (1)Ethylcyclohexyl (4)Ethylcyclohexyl (4)Ethylsulfonyl (42)Fluoroazetidine (1)Fluorobicyclo (4)Hexahydrocyclohepta (6)Hexahydrocyclohepta (6)Hexahydrocyclopenta (4)Hydroxycyclobutanecarboxylic (4)Hydroxycyclohexanecarboxylic (6)Methanesulfonyl (314)Methanesulfonyl (314)Methoxycyclohexanamine (6)Methylazetidine (30)Methylbicyclo (6)Methylbicyclo (6)Methylcyclobutanamine (5)Methylcyclobutanecarboxylic (5)Methylcyclohexanamine (11)Methylcyclohexane (8)Methylcyclohexane (8)Methylcyclohexanecarboxylic (5)Methylcyclohexanecarboxylic Acid (4)Methylcyclohexanol (21)Methylcyclohexanone (5)Methylcyclohexyl (33)Methylcyclopentane (2)Methylcyclopropanamine (4)Methylcyclopropanamine (4)Methylcyclopropane (16)Methylsulfinyl (24)Methylsulfonimidoyl (6)Methylsulfonimidoyl (6)Methylsulphonyl (10)Octahydrocyclopenta (14)Oxabicyclo (40)Oxabicyclo (40)Oxazepane (13)Oxetanes (228)Oxoazetidine (8)Oxoazetidine (8)Oxobicyclo (11)Oxobicyclo (11)Oxocyclobutyl (2)Oxocyclohex (3)Oxocyclohexane (4)Oxocyclohexane (4)Oxocyclohexanecarboxylic (6)Oxocyclohexyl (3)Oxocyclohexyl (3)Oxocyclopentane (2)Oxocyclopentyl (5)Pentamethylcyclopentadienyl (11)Pentylcyclohexyl (10)Pentylcyclohexyl (10)Phenylcyclopropanamine (8)Propylcyclohexyl (15)Propylcyclohexyl (15)Tetraazacyclododecane (13)Tetraazacyclododecane (13)Tetraazacyclotetradecane (7)Tetraazacyclotetradecane (7)Tetramethylcyclohexane (4)Tetraoxatridecan (3)Tetraoxatridecan (3)Thiazepine (2)Tricyclohexylphosphine (9)Trimethylbicyclo (21)Trimethylcyclohex (20)
Alkenyls (16087)
Acyl Chlorides (7)Alcohols (101)Aldehydes (29)Aliphatic Chain Hydrocarbons (383)Aliphatic Cyclic Hydrocarbons (57)Amides (98)Amines (112)Anhydrides (4)Aryls (281)Benzyl Chlorides (1)Bromides (45)Carboxylic Acid Salts (9)Carboxylic Acids (129)Chlorides (57)Esters (245)Ethers (79)Fluorinated Building Blocks (42)Furans (9)Imidazoles (7)Indoles (4)Indolines (3)Iodides (6)Ketones (85)Nitriles (62)Nitroes (21)Oxetanes (4)Phenols (13)Piperazines (10)Piperidines (18)Pyridines (36)Pyrimidines (10)Pyrrolidines (14)Quinolines (10)Quinuclidines (6)Spiroes (4)Sulfamides (12)Sulfones (12)Tetrahydropyrans (7)Thiophenes (15)Trifluoromethyls (5)Other Aliphatic Heterocycles (19)Other Aromatic Heterocycles (5)(1E,4E)-1,5-Diphenylpenta-1,4-Dien-3-One (7)(1E,6E)-1,7-Diphenylhepta-1,6-Diene-3,5-Dione (7)(2,2-Dibromovinyl)Benzene (6)(2-(4-Bromophenyl)Ethene-1,1,2-Triyl)Tribenzene (11)(2-Nitrovinyl)Benzene (28)(Allyloxy)Benzene (17)(E)-5-Benzylidenethiazolidine-2,4-Dione (6)(E)-[3,3'-Biindolinylidene]-2,2'-Dione (5)(E)-N-(3-Oxo-1-Phenyl-3-(Piperidin-1-Yl)Prop-1-En-2-Yl)Benzamide (6)(Z)-3-(Phenyl(Phenylamino)Methylene)Indolin-2-One (5)(Z)-3-Benzylideneindolin-2-One (7)1,2-Diphenylethene (5)1,3,3-Trimethylcyclohex-1-Ene (12)1-Bromo-4-Styrylbenzene (5)1-Methylcyclohex-1-Ene (26)2-Benzalacetophenone (36)2-Methoxycyclohexa-2,5-Diene-1,4-Dione (7)2-Methylcyclohexa-2,5-Diene-1,4-Dione (5)3-(2-Fluorophenyl)Acrylic Acid (17)3-(3-Chlorophenyl)Acrylic Acid (10)3-Methylene-2,3-Dihydro-1H-Inden-1-One (5)3-Methyleneazetidine (8)3-Phenylacrylaldehyde (29)3-Styrylphenol (11)4-(1,2,2-Triphenylvinyl)Benzaldehyde (5)4-(Prop-1-En-2-Yl)Cyclohex-1-Ene (5)5H-Dibenzo[B,F]Azepine (5)Allyl Benzoate (5)Cyclohex-1-En-1-Yl Trifluoromethanesulfonate (3)Cyclohex-1-En-1-Ylboronic Acid (4)Cyclohex-1-Ene-1-Carboxylic Acid (4)Cyclohexa-1,3-Diene (5)Cyclopent-2-En-1-Amine (4)Ethyl Cyclohex-1-Ene-1-Carboxylate (5)Methyl Cinnamate (37)Methyl Cyclohex-3-Ene-1-Carboxylate (3)Methylenecyclobutane (8)Methylenecyclohexane (8)N-Phenylacrylamide (7)Styrene (78)Tert-Butyl-Cyclopent-2-En-1-Ylcarbamate (6)Vinylcyclopropane (8)Acrylaldehyde (11)Acryloyl (8)Acryloyl (8)Annulene (2)Cyclohexene (89)Cyclooctene (9)Cyclopentene (3)Diacrylamide (6)Diallyl (5)Diazene (21)Divinyl (1)Divinyl (1)Enamide (6)Enamide (6)Enecarbaldehyde (2)Enoyl (7)Methacrylamide (3)Methacrylamide (3)Methoxycinnamaldehyde (3)Nitroethylene (37)Tetrahydrocyclopenta (19)
Alkynyls (2695)
Alcohols (34)Aldehydes (6)Aliphatic Chain Hydrocarbons (23)Amides (30)Amines (19)Aryls (116)Bromides (11)Carboxylic Acids (13)Chlorides (18)Esters (19)Ethers (29)Fluorinated Building Blocks (6)Ketones (8)Pyrazines (6)Pyridines (17)Pyrimidines (7)Pyrrolidines (8)(Bromoethynyl)Benzene (5)(Prop-2-Yn-1-Yloxy)Benzene (12)1,3,5-Tris(Phenylethynyl)Benzene (6)1,4-Bis(Phenylethynyl)Benzene (7)1,4-Diphenylbuta-1,3-Diyne (9)1-Ethynyl-2-Fluorobenzene (10)1-Methyl-4-(Phenylethynyl)Benzene (7)2-(Phenylethynyl)Benzaldehyde (9)2-Ethynylpyridine (31)2-Ethynylthiophene (8)3-Ethynylpyridine (22)4-(Phenylethynyl)-1,1'-Biphenyl (5)4-(Phenylethynyl)Phenol (7)4-Ethynylpyridine (10)9,10-Bis(Phenylethynyl)Anthracene (5)Ethynylbenzene (145)Trimethyl(Phenylethynyl)Silane (23)
Amides (89669)
Acyl Chlorides (13)Alcohols (371)Aldehydes (56)Aliphatic Chain Hydrocarbons (456)Aliphatic Cyclic Hydrocarbons (132)Alkenyls (98)Alkynyls (30)Amidines (8)Amines (614)Anhydrides (33)Aryls (1193)Azetidines (102)Benzimidazoles (26)Benzisoxazoles (4)Benzofurans (7)Benzothiazoles (23)Benzoxazoles (33)Benzyl Bromides (6)Benzyl Chlorides (8)Bromides (311)Carboxylic Acid Salts (6)Carboxylic Acids (226)Chlorides (372)Epoxides (10)Esters (300)Ethers (254)Fluorinated Building Blocks (240)Furans (30)Guanidines (12)Hydrazides (169)Hydrazines (176)Hydrazones (9)Hydroxylamines (12)Imidazoles (26)Imidazolidines (19)Imidazolines (16)Indazoles (6)Indoles (42)Indolines (73)Iodides (36)Isoquinolines (10)Isoxazoles (16)Ketones (185)Morpholines (99)Nitriles (92)Oxadiazoles (10)Oxazoles (10)Oxazolidines (50)Oxazolines (4)Oxetanes (14)Phenols (44)Phthalazines (8)Piperazines (273)Piperidines (436)Purines (7)Pyrazines (17)Pyrazoles (46)Pyridazines (20)Pyridines (267)Pyrimidines (120)Pyrroles (15)Pyrrolidines (307)Pyrrolines (24)Quinazolines (42)Quinolines (33)Quinoxalines (15)Spiroes (190)Sulfamides (43)Sulfides (40)Sulfonates (15)Sulfones (13)Tetrahydrofurans (19)Tetrahydroisoquinolines (7)Tetrahydropyrans (15)Tetrahydroquinolines (14)Thiadiazoles (11)Thiazoles (40)Thiazolidines (16)Thioesters (25)Thiols (4)Thiophenes (44)Thiophenols (6)Thioureas (61)Triazines (11)Triazoles (8)Trifluoromethyls (74)Ureas (82)Other Aliphatic Heterocycles (103)Other Aromatic Heterocycles (181)(3-(Phenylcarbamoyl)Phenyl)Boronic Acid (3)(4-(Phenylcarbamoyl)Phenyl)Boronic Acid (11)(9H-Fluoren-9-Yl)Methyl (3-Phenylpropyl)Carbamate (24)(S)-N-Phenethyl-3-(Pyrrolidin-2-Yl)Propanamide (5)(S)-N-Phenylpiperidine-2-Carboxamide (1)1-(Pyridin-2-Yl-L2-Azaneyl)Ethan-1-One (12)1-Methylindoline-2,3-Dione (8)1-Phenylurea (11)1H-Pyrazole-5-Carboxamide (3)2,2-Dimethyl-1-(Pyridin-2-Yl-L2-Azaneyl)Propan-1-One (7)2-((Tert-Butoxycarbonyl)Amino)Benzoic Acid (11)2-(Trifluoromethyl)Benzamide (6)2-Amino-N-Phenylbenzamide (21)2-Aminobenzamide (30)2-Bromo-N-Phenylacetamide (5)2-Chloro-N-Phenylacetamide (26)2-Chloro-N-Phenylbenzamide (4)2-Chlorobenzamide (16)2-Fluoro-N-Methylbenzamide (12)2-Fluorobenzamide (32)2-Hydroxy-N-Phenylbenzamide (3)2-Hydroxybenzamide (14)2-Iodobenzamide (4)2-Methoxybenzamide (9)3-Benzamidopropanoic Acid (3)3-Bromo-4-Fluorobenzamide (3)3-Fluorobenzamide (26)3-Nitrobenzamide (17)3-Oxo-N-Phenylbutanamide (10)4-Amino-N-Phenylbenzamide (7)4-Benzamidobenzoic Acid (3)4-Fluorobenzamide (19)4-Oxo-4-(Phenylamino)Butanoic Acid (7)Benzoylglycine (15)Benzyl Benzylcarbamate (14)Benzyl Cyclohexylcarbamate (9)Benzyl Phenylcarbamate (15)Ethyl 2-Oxo-2-(Phenylamino)Acetate (8)Isonicotinamide (12)N,N-Diethylbenzamide (17)N,N-Dimethylbenzamide (34)N-((5R,6R)-7-Oxo-4-Thia-1-Azabicyclo[3.2.0]Heptan-6-Yl)-3-Phenylisoxazole-4-Carboxamide (1)N-((6R,7R)-8-Oxo-5-Thia-1-Azabicyclo[4.2.0]Oct-2-En-7-Yl)-2-Phenylacetamide (5)N-(2-(1H-Indol-3-Yl)Ethyl)Acetamide (5)N-(2-Benzoylphenyl)-2-Bromoacetamide (3)N-(2-Chlorophenyl)Acetamide (16)N-(2-Iodophenyl)Acetamide (8)N-(2-Nitrophenyl)Acetamide (11)N-(3-Fluorophenyl)Acetamide (14)N-(4-Aminophenyl)Benzamide (2)N-(4-Chlorophenyl)Acetamide (9)N-(4-Chlorophenyl)Benzamide (6)N-(4-Fluorophenyl)Benzamide (3)N-(4-Phenoxyphenyl)Acetamide (4)N-(O-Tolyl)Benzamide (9)N-(P-Tolyl)Benzamide (6)N-(Thiazol-2-Yl)Acetamide (9)N-Isopropylbenzamide (16)N-Methoxy-N-Methylbenzamide (24)N-Methylbenzamide (58)N-Methylpicolinamide (10)N-Phenylacrylamide (7)N-Phenylpivalamide (11)Nicotinamide (64)Phenyl Dimethylcarbamate (5)Phenyl Ethyl(Methyl)Carbamate (5)Picolinamide (26)Piperidine-3-Carboxamide (5)Pyrazine-2-Carboxamide (10)Pyrrolidine-2-Carboxamide (12)Tert-Butyl (1-Phenylethyl)Carbamate (19)Tert-Butyl (4-Fluorophenyl)Carbamate (9)Tert-Butyl Benzylcarbamate (52)Tert-Butyl Methyl(Phenyl)Carbamate (9)Tert-Butyl O-Tolylcarbamate (11)Tert-Butyl Phenethylcarbamate (1)Tert-Butyl-Cyclopent-2-En-1-Ylcarbamate (6)Thiophene-3-Carboxamide (5)Acetamide (645)Acetamide (645)Acetamido (150)Carboxamide (919)Carboxamide (919)Diacetamide (3)Dicarboxamide (19)Diethylacetamide (4)Diethylacetamide (4)Dimethylacetamide (9)Dimethylnicotinamide (7)Dimethylnicotinamide (7)Formamidophenylboronic Acid/Ester (11)Isobutyramide (7)Oxobutanamide (10)Oxobutanamide (10)Pentanamide (18)Pentanamide (18)Propanamide (1)Propanamido (5)Propanamido (5)
Amines (139022)
Alcohols (287)Aldehydes (29)Aliphatic Chain Hydrocarbons (304)Aliphatic Cyclic Hydrocarbons (135)Alkenyls (112)Alkynyls (19)Amides (614)Amidines (6)Aryls (1269)Azetidines (17)Benzimidazoles (19)Benzisoxazoles (4)Benzodioxans (5)Benzofurans (13)Benzothiazoles (30)Benzothiophenes (6)Benzoxazoles (7)Bromides (216)Carboxylic Acid Salts (6)Carboxylic Acids (178)Chlorides (309)Difluoromethyls (19)Dioxolanes (4)Esters (302)Ethers (403)Fluorinated Building Blocks (284)Furans (22)Hydrazides (8)Hydrazines (26)Hydroxylamines (14)Imidazoles (38)Imidazolines (4)Indazoles (18)Indoles (33)Indolines (9)Iodides (36)Isoquinolines (12)Isoxazoles (25)Ketones (88)Morpholines (54)Nitriles (142)Nitroes (114)Oxadiazoles (16)Oxazoles (16)Oxazolidines (4)Oxetanes (29)Oximes (5)Phenols (26)Piperazines (36)Piperidines (177)Purines (12)Pyrazines (19)Pyrazoles (159)Pyridazines (10)Pyridines (394)Pyrimidines (191)Pyrroles (10)Pyrrolidines (81)Quinazolines (9)Quinolines (79)Quinoxalines (7)Quinuclidines (5)Spiroes (14)Sulfamides (50)Sulfides (42)Sulfonates (6)Sulfones (20)Sulfonic Acids (12)Tetrahydrofurans (8)Tetrahydroisoquinolines (3)Tetrahydropyrans (3)Tetrahydroquinolines (5)Thiadiazoles (24)Thiazoles (102)Thioesters (5)Thiophenes (54)Thiophenols (5)Triazines (14)Triazoles (21)Trifluoromethyls (95)Ureas (6)Other Aliphatic Heterocycles (17)Other Aromatic Heterocycles (134)(1-Phenylcyclobutyl)Methanamine (9)(1-Phenylcyclopropyl)Methanamine (12)(1H-Benzo[D]Imidazol-2-Yl)Methanamine (7)(2,3-Dihydrobenzo[B][1,4]Dioxin-2-Yl)Methanamine (3)(2-Aminophenyl)(Phenyl)Methanone (30)(2-Bromophenyl)Methanamine (12)(2-Chlorophenyl)Methanamine (28)(2-Fluorophenyl)Methanamine (49)(3-Chlorophenyl)Methanamine (19)(3-Fluoropyridin-2-Yl)Methanamine (5)(3-Methylpyridin-2-Yl)Methanamine (5)(4-Aminophenyl)(Phenyl)Methanone (4)(4-Fluorophenyl)Methanamine (27)(6-Methylpyridin-2-Yl)Methanamine (6)(Tetrahydrofuran-2-Yl)Methanamine (4)(Tetrahydrofuran-3-Yl)Methanamine (3)1,2,3,4-Tetrahydroisoquinolin-5-Amine (4)1,2,3,4-Tetrahydroisoquinolin-6-Amine (3)1,2,3,4-Tetrahydroisoquinolin-7-Amine (3)1,3,4-Thiadiazol-2-Amine (9)1,3,5-Triazin-2-Amine (3)1-(2,3-Dihydro-1H-Inden-5-Yl)Ethan-1-Amine (3)1-(2-Aminophenyl)Ethan-1-One (29)1-(2-Bromophenyl)Ethan-1-Amine (10)1-(2-Chlorophenyl)-1H-Pyrazole (3)1-(2-Fluorophenyl)Ethan-1-Amine (80)1-(2-Fluorophenyl)Propan-1-Amine (8)1-(3-(Trifluoromethoxy)Phenyl)Ethan-1-Amine (6)1-(3-(Trifluoromethyl)Phenyl)Ethan-1-Amine (25)1-(3-Aminophenyl)Ethan-1-One (18)1-(3-Chlorophenyl)-N-Methylmethanamine (8)1-(4-(Trifluoromethoxy)Phenyl)Ethan-1-Amine (6)1-(Anthracen-9-Yl)Ethan-1-Amine (3)1-(Pyridin-2-Yl)Ethan-1-Amine (15)1-(Pyridin-3-Yl)Ethan-1-Amine (17)1-(Pyridin-4-Yl)Ethan-1-Amine (23)1-Aminoanthracene-9,10-Dione (8)1-Benzyl-N-Methylpiperidin-3-Amine (6)1-Benzylpiperidin-4-Amine (8)1-Methyl-1H-Pyrazol-3-Amine (14)1-Phenyl-1H-Pyrazol-3-Amine (3)1-Phenyl-1H-Pyrazol-5-Amine (4)1-Phenylcyclobutan-1-Amine (5)1-Phenylcyclopropan-1-Amine (26)1-Phenylethan-1-Amine (436)1H-1,2,4-Triazol-3-Amine (7)1H-1,2,4-Triazol-5-Amine (6)1H-Benzo[D]Imidazol-2-Amine (7)1H-Benzo[D]Imidazol-5-Amine (4)1H-Benzo[D]Imidazol-6-Amine (6)1H-Imidazol-2-Amine (6)1H-Indazol-3-Amine (23)1H-Indazol-4-Amine (12)1H-Indazol-5-Amine (3)1H-Indazol-6-Amine (5)1H-Indazol-7-Amine (6)1H-Indol-4-Amine (10)1H-Indol-6-Amine (21)1H-Indol-7-Amine (10)1H-Pyrazol-4-Amine (6)1H-Pyrazol-5-Amine (19)1H-Pyrrol-1-Amine (7)1H-Pyrrolo[2,3-B]Pyridin-6-Amine (4)2,2,2-Trifluoro-1-Phenylethan-1-Amine (48)2,3-Dichloroaniline (18)2,3-Difluoroaniline (34)2,3-Dihydrobenzofuran-3-Amine (11)2,3-Dihydrobenzofuran-5-Amine (3)2,4-Dibromoaniline (18)2,4-Dichloropyrimidin-5-Amine (6)2,4-Difluoroaniline (44)2,5-Difluoroaniline (34)2,6-Dibromoaniline (17)2,6-Dichloroaniline (27)2,6-Difluoroaniline (23)2,6-Diiodoaniline (8)2-((2-Nitrophenyl)Amino)Ethan-1-Ol (5)2-((3-Phenylpropyl)Amino)-1-(Pyrrolidin-1-Yl)Ethan-1-One (2)2-(1H-Benzo[D]Imidazol-2-Yl)Ethan-1-Amine (6)2-(1H-Indol-3-Yl)Ethan-1-Amine (4)2-(Diethylamino)Ethyl Benzoate (3)2-(Methylsulfonyl)Aniline (3)2-(Phenylamino)Benzoic Acid (11)2-(Piperazin-1-Yl)Ethan-1-Amine (8)2-(Piperidin-1-Yl)Ethan-1-Amine (8)2-(Trifluoromethoxy)Aniline (18)2-(Trifluoromethyl)Aniline (58)2-Amino-2-(2-Fluorophenyl)Ethan-1-Ol (7)2-Amino-2-Phenylethan-1-Ol (196)2-Amino-3-Chlorophenol (4)2-Amino-3-Fluorobenzoic Acid (11)2-Amino-6-Bromophenol (3)2-Amino-6-Chlorobenzoic Acid (5)2-Amino-6-Chlorobenzonitrile (3)2-Amino-6-Chlorophenol (5)2-Aminobenzamide (30)2-Aminobenzenesulfonic Acid (9)2-Aminophenol (107)2-Aminopyrimidin-4(3H)-One (11)2-Bromo-3-Chloroaniline (4)2-Bromo-3-Fluoroaniline (13)2-Bromo-3-Methylaniline (6)2-Bromo-4-Methylaniline (14)2-Bromo-6-Methylaniline (11)2-Bromo-N,N-Dimethylaniline (5)2-Bromo-N-Methylaniline (8)2-Chloro-3-Methylaniline (4)2-Chloro-4-Methylaniline (7)2-Chloro-6-Methylaniline (11)2-Chloro-N-Methylaniline (4)2-Chloro-N-Phenylaniline (8)2-Chloroaniline (214)2-Chlorobenzene-1,3-Diamine (3)2-Chlorobenzene-1,4-Diamine (6)2-Chloropyrimidin-4-Amine (22)2-Fluoro-6-Iodoaniline (7)2-Fluoro-N-Phenylaniline (4)2-Fluoroaniline (262)2-Iodo-4-Methylaniline (5)2-Iodo-N,N-Dimethylaniline (3)2-Iodoaniline (87)2-Methoxypyridin-3-Amine (6)2-Methyl-6-Nitroaniline (10)2-Methyl-N-Phenylaniline (7)2-Methylpyridin-4-Amine (7)2-Methylpyrimidin-4-Amine (22)2-Nitro-N-Phenylaniline (3)2-Phenoxyaniline (4)2-Phenylcyclopropan-1-Amine (20)2-Phenylpyrimidin-5-Amine (5)3,4-Dibromoaniline (4)3,4-Dichloroaniline (21)3,4-Difluoroaniline (44)3-(1,3,2-Dioxaborinan-2-Yl)Aniline (3)3-(Benzyloxy)Aniline (12)3-(Methylsulfonyl)Aniline (7)3-(Piperazin-1-Yl)Aniline (3)3-(Trifluoromethoxy)Aniline (22)3-(Trifluoromethyl)Aniline (102)3-Aminobenzenesulfonic Acid (6)3-Aminodihydrofuran-2(3H)-One (4)3-Bromo-2-Fluoroaniline (15)3-Bromo-4-Chloroaniline (13)3-Bromo-4-Fluoroaniline (22)3-Bromo-4-Methoxyaniline (7)3-Bromo-4-Methylaniline (14)3-Bromoaniline (187)3-Bromobenzene-1,2-Diamine (11)3-Chloro-2-Iodoaniline (3)3-Chloro-2-Methylaniline (11)3-Chloro-4-Fluoroaniline (18)3-Chloro-4-Iodoaniline (3)3-Chloro-4-Methoxyaniline (6)3-Chloro-4-Methylaniline (12)3-Chlorobenzene-1,2-Diamine (6)3-Chloropyrazin-2-Amine (6)3-Fluoro-2-Methylaniline (9)3-Fluoroaniline (288)3-Iodo-2-Methylaniline (4)3-Iodoaniline (32)3-Methoxypyridin-2-Amine (5)3-Methyl-N-Phenylaniline (4)3-Methylpyridin-2-Amine (7)3-Nitroaniline (98)3-Phenoxyaniline (2)3-Phenyl-1H-Pyrazol-5-Amine (11)3-Phenylisoxazol-5-Amine (8)4,6-Dichloropyrimidin-2-Amine (9)4-(Benzyloxy)Aniline (8)4-(Oxazol-5-Yl)Aniline (3)4-(Pyrrolidin-1-Yl)Aniline (3)4-(Trifluoromethoxy)Aniline (26)4-Amino-2-Chlorobenzonitrile (8)4-Amino-2-Fluorophenol (9)4-Amino-3-Chlorobenzoic Acid (6)4-Amino-3-Chlorophenol (5)4-Amino-N-Phenylbenzenesulfonamide (3)4-Aminopyridazin-3(2H)-One (4)4-Benzylaniline (7)4-Bromo-3-Chloroaniline (21)4-Bromo-3-Fluoroaniline (29)4-Bromo-3-Methylaniline (19)4-Bromo-3-Nitroaniline (8)4-Bromo-N,N-Diphenylaniline (10)4-Bromo-N-Phenylaniline (6)4-Chloro-2-Iodoaniline (9)4-Chloro-3-Fluoroaniline (17)4-Chloro-3-Iodoaniline (3)4-Chloro-3-Methylaniline (11)4-Chloro-N-Phenylaniline (6)4-Chloropyrimidin-2-Amine (23)4-Chloropyrimidin-5-Amine (7)4-Fluoro-N-Phenylaniline (4)4-Fluoroaniline (225)4-Iodo-N,N-Diphenylaniline (3)4-Methoxy-N-Phenylaniline (5)4-Methyl-3-Nitroaniline (8)4-Methylpyrimidin-2-Amine (9)4-Methylthiazol-2-Amine (7)4-Morpholinoaniline (4)4-Nitroaniline (65)4-Phenoxyaniline (6)4-Phenylpyrimidin-2-Amine (15)4-Phenylthiazol-2-Amine (37)5-Bromopyrazin-2-Amine (13)5-Bromothiazol-2-Amine (6)5-Chloropyrazin-2-Amine (9)5-Methoxypyridin-3-Amine (4)5-Methyl-2-Nitroaniline (6)5-Methylpyridin-2-Amine (7)5-Methylpyridin-3-Amine (4)5-Methylthiazol-2-Amine (6)5-Phenylthiophen-2-Amine (5)5H-Dibenzo[B,F]Azepine (5)6-Amino-2H-Benzo[B][1,4]Oxazin-3(4H)-One (3)6-Bromopyrazin-2-Amine (5)6-Chloropyrazin-2-Amine (11)6-Chloropyridazin-3-Amine (5)6-Chloropyrimidin-4-Amine (8)6-Methoxypyridin-2-Amine (14)6-Methoxypyridin-3-Amine (8)6-Methylpyridin-2-Amine (46)6-Methylpyridin-3-Amine (5)6-Phenoxypyridin-3-Amine (5)7-Methoxy-N-Phenylquinazolin-4-Amine (12)7H-Purin-2-Amine (6)7H-Purin-6-Amine (9)7H-Pyrrolo[2,3-D]Pyrimidin-2-Amine (5)7H-Pyrrolo[2,3-D]Pyrimidin-4-Amine (3)9H-Purin-6-Amine (3)[1,1'-Biphenyl]-2-Amine (8)[1,1'-Biphenyl]-3-Amine (11)[1,2,4]Triazolo[1,5-A]Pyridin-2-Amine (17)[1,2,4]Triazolo[4,3-A]Pyridin-3-Ylmethanamine (5)Azetidin-3-Amine (4)Azetidin-3-Ylmethanamine (3)Aziridine (9)Benzo[B]Thiophen-2-Amine (5)Benzo[D]Oxazol-2-Amine (16)Benzo[D]Thiazol-2-Amine (30)Benzo[D]Thiazol-5-Amine (3)Benzo[D]Thiazol-6-Amine (3)Benzyl-3-Aminopiperidine-1-Carboxylate (6)Benzyl-3-Aminopyrrolidine-1-Carboxylate (7)Bis(Pyridin-2-Ylmethyl)Amine (6)Chroman-2-Ylmethanamine (3)Chroman-3-Amine (3)Chroman-4-Amine (20)Cyclobutanamine (90)Cyclohexanamine (59)Cyclopent-2-En-1-Amine (4)Cyclopentanamine (5)Cyclopropyl(Phenyl)Methanamine (22)Diphenylmethanamine (26)Ethyl 2-Aminobenzoate (23)Ethyl 3-Aminobenzoate (19)Ethyl 4-Aminobenzoate (23)Imidazo[1,2-A]Pyridin-3-Amine (7)Imidazo[1,2-A]Pyridin-8-Amine (7)Isoquinolin-1-Amine (18)Isoquinolin-3-Amine (13)Isoquinolin-4-Amine (16)Isoquinolin-5-Amine (9)Isoquinolin-8-Amine (6)Isoxazol-5-Amine (15)Methyl 2-Amino-2-Phenylacetate (38)Methyl 2-Aminobenzoate (95)Methyl 3-Amino-3-Phenylpropanoate (27)Methyl 3-Aminobenzoate (88)Methyl 4-Aminobenzoate (71)Morpholin-2-Ylmethanamine (8)N,N-Diethylaniline (18)N,N-Dimethyl-2-Phenoxyethan-1-Amine (16)N,N-Dimethylaniline (117)N,N-Dimethylnaphthalen-1-Amine (4)N,N-Dimethylpyridin-2-Amine (10)N,N-Dimethylpyridin-4-Amine (6)N,N-Dimethylpyrimidin-2-Amine (10)N,N-Dimethylpyrrolidin-3-Amine (7)N,N-Diphenyl-4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Aniline (6)N,N-Diphenyl-[1,1'-Biphenyl]-4-Amine (8)N-(4-Aminophenyl)Benzamide (2)N-(Furan-2-Ylmethyl)Aniline (5)N-(Prop-2-Yn-1-Yl)-2,3-Dihydro-1H-Inden-1-Amine (3)N-Benzyl-1-Phenylethan-1-Amine (7)N-Benzyl-2-Phenylethan-1-Amine (1)N-Benzylpiperidin-4-Amine (3)N-Cyclopropylpyrimidin-2-Amine (5)N-Methyl-1-Phenylmethanamine (65)N-Methyl-2-Nitroaniline (20)N-Methyl-4-Nitroaniline (6)N-Methylaniline (140)N-Methylcyclohexanamine (7)N-Methylpiperidin-3-Amine (5)N-Methylpyridin-2-Amine (47)N-Methylpyridin-3-Amine (15)N-Methylpyrimidin-2-Amine (10)N-Methylpyrimidin-4-Amine (10)N-Methylpyrrolidin-3-Amine (6)N-Phenyl-3-(Trifluoromethyl)Aniline (3)N-Phenyl-4-(Trifluoromethyl)Aniline (3)N-Phenylnaphthalen-1-Amine (5)N-Phenylpyridin-2-Amine (9)N-Phenylpyrimidin-2-Amine (5)N-Phenylpyrimidin-4-Amine (7)N1,N1-Diphenylbenzene-1,4-Diamine (2)N1-Phenylbenzene-1,4-Diamine (3)N2,N4,N6-Triphenyl-1,3,5-Triazine-2,4,6-Triamine (4)N2,N4-Diphenylpyrimidine-2,4-Diamine (5)Naphthalen-2-Amine (12)O-Benzylhydroxylamine (16)Oxazol-2-Amine (8)Phenylmethanamine (315)Piperidin-2-Ylmethanamine (6)Piperidin-3-Amine (17)Piperidin-3-Ylmethanamine (5)Piperidin-4-Amine (14)Piperidin-4-Ylmethanamine (7)Pyrazin-2-Amine (22)Pyrazin-2-Ylmethanamine (12)Pyridazin-3-Amine (18)Pyridazin-4-Amine (10)Pyridin-2-Amine (29)Pyridin-2-Ylmethanamine (38)Pyridin-3-Amine (21)Pyridin-3-Ylmethanamine (29)Pyridin-4-Amine (22)Pyridin-4-Ylmethanamine (12)Pyridine-2,3-Diamine (5)Pyridine-2,6-Diamine (12)Pyrimidin-2-Ylmethanamine (7)Pyrimidin-4-Amine (29)Pyrimidin-5-Amine (6)Pyrimidine-2,4-Diamine (14)Pyrrolidin-2-Ylmethanamine (12)Pyrrolidin-3-Ylmethanamine (6)Quinazolin-2-Amine (13)Quinazolin-4-Amine (8)Quinoxalin-2-Amine (3)Tetrahydrofuran-3-Amine (6)Thiazol-2-Ylmethanamine (5)Thiazol-5-Amine (6)Thiophen-3-Amine (18)Tri([1,1'-Biphenyl]-4-Yl)Amine (9)Amino-Propanol (6)Aminoacetamido (4)Aminoacetyl (2)Aminoadamantan (5)Aminoadamantan (5)Aminoazepane (10)Aminobutane (23)Aminobutane (23)Aminobutyl (15)Aminobutyl (15)Aminoethoxy (18)Aminoethoxy (18)Aminoethyl (152)Aminoethyl (152)Aminohexan (6)Aminohexyl (3)Aminohexyl (3)Aminoisonicotinic Acid (12)Aminomethyl (402)Aminomethyl (402)Aminooxy (6)Aminopentyl (10)Aminopentyl (10)Aminopropanamido (1)Aminopropane (20)Aminopropane (20)Aminopropoxy (2)Aminopropoxy (2)Aminotetrahydro (12)Aminotetrahydro (12)Benzylazetidine (4)Bromobenzylamine (6)Bromobenzylcarbamate (28)Butoxycarbonylamino (7)Butylcarbamoyl (5)Carbamothioyl (2)Carbamoyl (114)Chlorobenzylamine (4)Diamino (88)Diamino (88)Diaminopteridin (3)Diaminopteridin (3)Dibutylamino (6)Dibutylamino (6)Dichlorobenzylamine (5)Diethylamino (91)Diethylamino (91)Diisopropylamino (1)Dimethyl-Ethylamine (2)Dimethylamino (484)Dimethylamino (484)Dimethylcarbamoyl (14)Dimethylmethanamine (16)Dipropylamino (1)Dipropylamino (1)Ethylamino (37)Ethylamino (37)Fluindamine (32)Hydroxyamino (2)Hydroxyamino (2)Imidazol-Amine (1)Methoxyimino (2)Methylamino (236)Methylamino (236)Methylcarbamoyl (18)Methylcarbamoyl (18)Propanediamine (6)Propylamino (5)Propylamino (5)Pyrrolopyridine-Methylamine (6)Silanamine (3)Triamine (25)Triamine (25)
Anhydrides (655)
Aliphatic Chain Hydrocarbons (11)Alkenyls (4)Amides (33)Aryls (14)Benzofurans (13)Bromides (5)Chlorides (8)Esters (81)Ethers (8)Fluorinated Building Blocks (8)Nitroes (5)Oxazolidines (5)Tetrahydrofurans (3)Other Aliphatic Heterocycles (10)Other Aromatic Heterocycles (33)Dianhydride (3)1H,3H-Benzo[De]Isochromene-1,3-Dione (8)Furan-2,5-Dione (8)
Aryls (186252)
Acyl Chlorides (28)Alcohols (633)Aldehydes (346)Aliphatic Cyclic Hydrocarbons (10)Alkenyls (281)Alkynyls (116)Amides (1193)Amidines (20)Amines (1269)Anhydrides (14)Azetidines (44)Azoes (12)Benzimidazoles (40)Benzofurans (12)Benzothiazoles (10)Benzothiophenes (4)Benzyl Bromides (82)Benzyl Chlorides (53)Bromides (936)Carboxylic Acid Salts (16)Carboxylic Acids (849)Chlorides (1047)Difluoromethyls (43)Dioxolanes (16)Epoxides (25)Esters (893)Ethers (1461)Fluorinated Building Blocks (868)Furans (39)Guanidines (13)Hydrazides (84)Hydrazines (121)Hydrazones (10)Hydroxylamines (8)Imidazoles (50)Imidazolidines (10)Imidazolines (14)Indazoles (8)Indoles (51)Indolines (20)Iodides (87)Isocyanates and Isothiocyanates (8)Isocyanides (2)Isoxazoles (54)Ketones (582)Morpholines (137)Nitriles (353)Nitroes (356)Oxadiazoles (12)Oxazoles (48)Oxazolidines (25)Oxazolines (8)Oxetanes (21)Oximes (24)Phenols (289)Phosphoruses (10)Piperazines (139)Piperidines (311)Purines (4)Pyrans (3)Pyrazines (13)Pyrazoles (122)Pyridazines (15)Pyridines (178)Pyrimidines (79)Pyrroles (30)Pyrrolidines (142)Pyrrolines (14)Quinazolines (7)Quinolines (24)Quinuclidines (2)Spiroes (36)Sulfamides (218)Sulfides (143)Sulfonates (83)Sulfones (121)Sulfonic Acids (23)Sulfonyl Chlorides (57)Sulfoxides (19)Tetrahydrofurans (12)Tetrahydroisoquinolines (3)Tetrahydropyrans (28)Tetrazoles (5)Thiadiazoles (17)Thiazoles (97)Thiazolidines (8)Thioesters (9)Thiols (8)Thiophenes (46)Thiophenols (29)Thioureas (28)Triazines (12)Triazoles (24)Trifluoromethyls (260)Ureas (30)Other Aliphatic Heterocycles (55)Other Aromatic Heterocycles (110)(6-Phenoxytetrahydro-2H-Pyran-2-Yl)Methanol (18)(8S,9S,14R)-1,2,6,7,8,9,10,11,12,13,14,15,16,17-Tetradecahydro-3H-Cyclopenta[A]Phenanthren-3-One (1)(9H-Fluoren-9-Yl)Methyl (2-(Tritylthio)Ethyl)Carbamate (9)(9H-Fluoren-9-Yl)Methyl Cyclohexylcarbamate (8)(Benzylsulfonyl)Benzene (11)(Cyclopentyloxy)Benzene (12)1,1-Dimethylcyclopropane (5)1,3,4,5-Tetrahydro-2H-Benzo[B]Azepin-2-One (15)1,3-Diphenylurea (24)1-(2-Hydroxyphenyl)-2-Phenylethan-1-One (6)1-(2-Hydroxyphenyl)-3-Phenylprop-2-En-1-One (11)1-([1,1'-Biphenyl]-4-Yl)Ethan-1-One (5)1-(Naphthalen-2-Yl)Ethan-1-One (3)1-Benzyl-4-Chlorobenzene (6)1-Benzyl-4-Fluorobenzene (2)1-Bromonaphthalene (4)1-Chloronaphthalene (24)1-Fluoronaphthalene (23)1-Methoxynaphthalene (18)1-Methyl-4-(Phenylethynyl)Benzene (7)1-Methylnaphthalene (10)1-Naphthaldehyde (23)1-Naphthoic Acid (23)1-Nitronaphthalene (3)10H-Phenothiazine (20)2,2',3,3',5,5',6,6'-Octafluoro-1,1'-Biphenyl (4)2,5-Diphenylthiophene (5)2-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Phenol (7)2-(Trifluoromethyl)-1,1'-Biphenyl (8)2-Benzylaniline (5)2-Bromo-1,1'-Biphenyl (12)2-Bromonaphthalene (4)2-Chloro-1,1'-Biphenyl (8)2-Chloronaphthalene (6)2-Ethoxynaphthalene (4)2-Fluorocyclopropane-1-Carboxylic Acid (7)2-Hydroxycyclohexa-2,5-Diene-1,4-Dione (3)2-Iodonaphthalene (5)2-Methoxy-1,1'-Biphenyl (7)2-Methoxynaphthalene (22)2-Methoxytetrahydro-2H-Pyran (9)2-Methyl-1,1'-Biphenyl (26)2-Methylnaphthalene (13)2-Naphthaldehyde (12)2-Naphthoic Acid (14)2-Naphthonitrile (15)2-Nitronaphthalene (4)2-Phenoxytetrahydro-2H-Pyran-4-Yl Acetate (6)2-Phenylnaphthalene (12)3,3'-Diphenyl-5,5',6,6',7,7',8,8'-Octahydro-1,1'-Binaphthalene (13)3,3-Diphenyl-3H-Benzo[C][1,2]Oxathiole 1,1-Dioxide (19)3-Bromo-1,1'-Biphenyl (12)3-Chloro-1,1'-Biphenyl (12)3-Fluoro-1,1'-Biphenyl (16)3-Methyl-1,1'-Biphenyl (10)3-Methyl-1,1':4',1''-Terphenyl (3)3-Phenethylphenol (5)3-Phenyl-1,2,4,5-Tetrazine (9)3-Styrylphenol (11)4-(Bromomethyl)-1,1'-Biphenyl (13)4-(Phenyldiazenyl)Benzoic Acid (4)4-(Phenylsulfonyl)Morpholine (14)4-(Tert-Butyl)-1,1'-Biphenyl (5)4-Cyclohexylphenol (7)4-Iodo-1,1'-Biphenyl (9)4-Methoxy-1,1'-Biphenyl (4)9H-Xanthene (7)[1,1'-Biphenyl]-2-Amine (8)[1,1'-Biphenyl]-2-Carbonitrile (12)[1,1'-Biphenyl]-2-Carboxylic Acid (24)[1,1'-Biphenyl]-2-Ol (16)[1,1'-Biphenyl]-3-Amine (11)[1,1'-Biphenyl]-3-Carbaldehyde (20)[1,1'-Biphenyl]-3-Carboxylic Acid (40)[1,1'-Biphenyl]-3-Ol (9)[1,1'-Biphenyl]-4-Carbaldehyde (31)[1,1'-Biphenyl]-4-Carbonitrile (27)[1,1'-Biphenyl]-4-Ol (29)[1,1'-Biphenyl]-4-Ylboronic Acid (29)Benzyltriphenylphosphonium (14)Cyclopentylbenzene (28)Cyclopropanecarboxylic Acid (11)Cyclopropyl(Phenyl)Methanamine (22)Cyclopropyl(Phenyl)Methanone (13)Diphenyl Phosphonate (11)Diphenylphosphane (25)Diphenylphosphine Oxide (27)Ethane-1,1-Diyldibenzene (3)Ethene-1,1,2-Triyltribenzene (14)Ethyl Cyclopropanecarboxylate (8)Methyl 1-Naphthoate (7)Methyl 2-Naphthoate (11)Methyl [1,1'-Biphenyl]-4-Carboxylate (8)Methyl Cyclopropanecarboxylate (8)N,1-Diphenylmethanimine (15)N,N-Dimethylnaphthalen-1-Amine (4)N-((5R,6R)-7-Oxo-4-Thia-1-Azabicyclo[3.2.0]Heptan-6-Yl)-2-Phenylacetamide (5)N-Benzylbenzamide (21)N-Benzylbenzenesulfonamide (18)N-Cyclopropylbenzamide (14)N-Cyclopropylbenzenesulfonamide (10)N-Phenethylbenzamide (9)N-Phenyl-2-Naphthamide (11)Naphthalen-1-Ol (28)Naphthalen-1-Ylboronic Acid (4)Naphthalen-1-Ylmethanol (8)Naphthalen-2-Amine (12)Naphthalen-2-Ol (25)Naphthalen-2-Ylboronic Acid (8)Naphthalene-1,4-Diol (3)Naphthalene-2-Sulfonic Acid (4)Propane-2,2-Diyldibenzene (17)Tert-Butyl Cyclopropylcarbamate (7)Tetrahydro-2H-Pyran-2-Ol (12)Tetrahydro-2H-Pyran-2-Yl Acetate (12)Tetraphenylmethane (16)Triphenylene (12)Vinylcyclopropane (8)Acetylbenzonitrile (3)Aminobenzyl (6)Benzylamine (21)Benzylamino (47)Benzylhydroxylamine (25)Benzylidene (16)Benzylidene (16)Benzyloxy (1053)Benzylthio (16)Benzylthio (16)Cyanobenzyl (12)Dibenzyl (15)Dibenzylamino (7)Dichlorobenzylidene (8)Difluorobenzyl (17)Dimethoxybenzyl (20)Dimethoxybenzyl (20)Dimethylbenzonitrile (25)Dimethylbenzyl (6)Dimethylbenzyl (6)Dimethylbenzylamine (6)Ethoxybenzyl (8)Ethoxybenzyl (8)Fluorobenzyl (131)Fluorobenzylidene (2)Formylbenzonitrile (35)Iodobenzyl (9)Isopropylbenzonitrile (4)Methoxybenzyl (175)Methoxybenzylamine (7)Methoxybenzylidene (6)Methylbenzonitrile (104)Methylbenzyl (73)Methylbenzyl (73)Methylbenzylamine (8)Methylbenzylidene (1)Methylbenzylidene (1)Nitrobenzyl (31)Nitrobenzylidene (2)Phenoxybenzonitrile (5)Trifluorobenzyl (2)
Benzene Compounds (18446)
Acetamidophenoxy (1)Acetamidophenyl (5)Acetylbenzoate (7)Acetylphenyl (11)Acetylphenyl (11)Acetylphenylboronic (3)Aminobenzoyl (4)Aminobiphenyl (4)Aminonaphthalene (14)Aminophenoxy (19)Aminophenyl (233)Anisole (4)Anthracenes (143)Benzaldehyde (1587)Benzamidophenyl (1)Benzenedimethanol (4)Benzenethiol (8)Benzhydrylazetidine (10)Benzhydrylazetidine (10)Benzhydryloxy (4)Benzhydryloxy (4)Benziodoxol (2)Benzodioxole (5)Benzoquinone (6)Benzoquinone (6)Benzoylbenzoate (4)Benzoylphenyl (4)Binaphthalene (123)Binaphthalene (123)Biphenyl (1369)Boronophenyl (1)Boronophenyl (1)Bromobenzo (104)Bromobenzoyl (6)Bromodibenzo (9)Bromophenoxy (34)Bromophenyl (957)Butoxyphenyl (8)Butylaniline (7)Butylbenzene (16)Butyldiphenylsilyl (7)Butylphenol (20)Butylphenyl (32)Butylphenyl (32)Carbamoylphenoxy (12)Carbamoylphenyl (8)Carboxyphenyl (17)Carboxyphenyl (17)Chlorobenzenesulfonyl (23)Chlorobenzoyl (9)Chlorodibenzo (18)Chloronaphthalene (36)Chlorophenethyl (7)Chlorophenoxy (43)Chlorophenylacetate (4)Chlorophenylacetate Acid (9)Chlorophenylboronic (5)Cyclopropylaniline (6)Cyclopropylbenzoate (5)Cyclopropylnaphthalen (10)Cyclopropylnaphthalen (10)Cyclopropylphenyl (12)Cyclopropylphenyl (12)Diaminobenzene (7)Dibenzaldehyde (19)Dibenzene (7)Dibenzimidamide (5)Dibenzo (108)Dibenzo (108)Dibenzoate (9)Dibenzonitrile (11)Dibromobenzene (45)Dibromobenzo (14)Dibromodibenzo (8)Dibromonaphthalene (15)Dibromophenanthrene (3)Dibromophenoxy (2)Dibromophenylboronic (2)Dichlorobenzene (38)Dichlorobenzoyl (4)Dichlorophenoxy (16)Dichlorophenyl (328)Dichlorophenylboronic (5)Didehydrodibenz (2)Didehydrodibenz (2)Diethoxyphenyl (10)Diethylaniline (6)Diethylbenzamide (7)Diethylbenzene (6)Diethylbenzene (6)Difluorobenzene (57)Difluorobenzo (12)Difluorobenzoyl (9)Difluorophenoxy (9)Difluorophenyl (384)Difluorophenylboronic (11)Dihydroanthracene (1)Dihydroanthracene (1)Dihydrobenzo (188)Dihydrobenzo (188)Dihydrodibenzo (7)Dihydronaphthalene (104)Dihydronaphthalene (104)Diiodobenzene (12)Diisopropylaniline (5)Diisopropylbenzamide (8)Diisopropylphenyl (40)Diisopropylphenyl (40)Dimesityl (12)Dimesityl (12)Dimethoxyacetophenone (5)Dimethoxyaniline (20)Dimethoxybenzaldehyde (40)Dimethoxybenzamide (6)Dimethoxybenzene (47)Dimethoxybenzo (4)Dimethoxybenzo (4)Dimethoxybenzoate (20)Dimethoxybenzohydrazide (3)Dimethoxybenzonitrile (11)Dimethoxybenzoyl (4)Dimethoxybiphenyl (3)Dimethoxybiphenyl (3)Dimethoxynaphthalene (10)Dimethoxynaphthalene (10)Dimethoxyphenol (22)Dimethoxyphenyl (255)Dimethoxyphenylboronic (17)Dimethoxyphenylboronic (17)Dimethoxystyryl (4)Dimethylaminophenyl (4)Dimethylanthracene (4)Dimethylbenzaldehyde (27)Dimethylbenzamide (37)Dimethylbenzene (334)Dimethylbenzene (334)Dimethylbenzenesulfonamide (16)Dimethylbenzo (21)Dimethylbenzo (21)Dimethylbenzoate (27)Dimethylbenzoyl (3)Dimethylbenzoyl (3)Dimethylnaphthalene (6)Dimethylphenol (36)Dimethylphenoxy (13)Dimethylphenoxy (13)Dimethylphenylboronic (6)Dimethylphenylboronic (6)Dimethylphenylhydrazine (3)Dimethylphenylisothiocyanate (4)Dinaphtho (5)Dinitrobenzene (20)Diphenol (30)Diphenylamino (26)Diphenylaniline (9)Diphenylethane (10)Diphenylethanone (5)Diphenylethene (6)Diphenylethyl (4)Diphenylethyl (4)Diphenylmethyl (8)Diphenylmethyl (8)Diphenylmethylene (6)Diphenylphosphine (203)Diphenylphosphine (203)Diphenylpropane (18)Diphenylsilane (2)Diphenylurea (4)Diphenylurea (4)Ethoxyaniline (10)Ethoxybenzaldehyde (9)Ethoxybenzoate (12)Ethoxybenzonitrile (6)Ethoxyphenoxy (7)Ethoxyphenoxy (7)Ethoxyphenyl (45)Ethoxyphenyl (45)Ethoxyphenylboronic (7)Ethylaniline (8)Ethylaniline (8)Ethylbenzenesulfonamide (323)Ethylbenzo (7)Ethylbenzo (7)Ethylbenzoate (9)Ethylphenol (12)Ethylphenyl (39)Ethylphenyl (39)Ethynylaniline (7)Ethynylbenzaldehyde (17)Ethynylphenyl (35)Fluorobenzeneboronic (24)Fluoronaphthalene (34)Fluorophenoxy (46)Formylbenzoate (39)Formylphenoxy (4)Formylphenoxy (4)Formylphenyl (43)Formylphenyl (43)Formylphenylboronic (9)Formylphenylboronic (9)Hexahydrobenzo (3)Hexahydrobenzo (3)Hexylphenyl (3)Hexylphenyl (3)Homophenylalanine (16)Hydroxybenzenesulfonate (5)Hydroxybenzimidamide (11)Hydroxyisophthalaldehyde (6)Hydroxyphenethyl (2)Hydroxyphenethyl (2)Hydroxyphenyl (440)Hydroxyphenyl (440)Iodobenzo (15)Iodobenzoic (54)Iodobenzoyl (7)Iodobiphenyl (24)Iododibenzo (7)Iodonaphthalene (9)Iodophenoxy (5)Isobutoxyphenyl (15)Isobutoxyphenyl (15)Isobutylbenzene (5)Isobutylbenzene (5)Isobutylphenyl (9)Isophthalate (8)Isophthalonitrile (4)Isopropoxyaniline (10)Isopropoxyaniline (10)Isopropoxybenzaldehyde (7)Isopropoxybenzoate (7)Isopropoxyphenyl (40)Isopropoxyphenyl (40)Isopropylaniline (18)Isopropylbenzaldehyde (6)Isopropylbenzamide (9)Isopropylbenzenesulfonamide (19)Isopropylbenzoate (8)Isopropylphenyl (58)Mercaptobenzo (21)Methanobenzo (15)Methanobenzo (15)Methoxyacetophenone (12)Methoxyaniline (74)Methoxybenzaldehyde (104)Methoxybenzene (1710)Methoxybenzene (1710)Methoxybenzenesulfonamide (6)Methoxybenzenesulfonic (3)Methoxybenzenesulfonyl (13)Methoxybenzenesulfonyl (13)Methoxybenzimidamide (5)Methoxybenzo (52)Methoxybenzo (52)Methoxybenzoate (115)Methoxybenzonitrile (67)Methoxybenzoyl (12)Methoxybenzoyl (12)Methoxycarbonylphenylboronic (5)Methoxynaphthalene (54)Methoxynaphthalene (54)Methoxyphenol (72)Methoxyphenoxy (41)Methoxyphenoxy (41)Methoxyphenylacetate (4)Methoxyphenylboronic (24)Methoxyphenylboronic (24)Methoxyphenylhydrazine (4)Methoxystyryl (13)Methylacetophenone (6)Methylaniline (183)Methylanisole (3)Methylanthracene (5)Methylbenzaldehyde (101)Methylbenzamide (90)Methylbenzamido (3)Methylbenzenesulfinamide (4)Methylbenzenesulfonamide (79)Methylbenzenesulfonic (9)Methylbenzenesulfonyl (4)Methylbenzenesulfonyl (4)Methylbenzimidamide (6)Methylbenzoate (193)Methylbenzohydrazide (6)Methylbenzothioamide (6)Methylbenzothioamide (6)Methylbenzotrifluoride (5)Methylbenzoyl (24)Methylbenzoyl (24)Methylnaphthalene (34)Methylnaphthalene (34)Methylphenol (107)Methylphenoxy (18)Methylphenoxy (18)Methylphenyl (525)Methylphenylacetonitrile (45)Methylphenylboronic (24)Methylphenylhydrazine (10)Methylpropiophenone (5)Methylpropiophenone (5)Naphthaldehyde (28)Naphthalene (447)Naphthalene (447)Naphthaleneboronic (15)Naphthalenesulfonic (7)Naphthamide (15)Naphthonitrile (18)Nitrobenzenesulfonyl (8)Nitrobenzo (54)Nitrobenzoyl (13)Nitronaphthalene (24)Nitrophenoxy (43)Nitrophenyl (717)Nitrophenylboronic (14)Nitrotoluene (27)Octylbenzene (11)Octylbenzene (11)Paracyclophane (2)Paracyclophane (2)Pentafluorobenzene (13)Pentylbenzene (5)Pentylbenzene (5)Pentylbenzoate (2)Pentylphenol (4)Pentylphenyl (5)Pentylphenyl (5)Perfluorophenoxy (5)Perfluorophenyl (31)Phenoxyacetate (5)Phenoxyaniline (8)Phenoxybenzaldehyde (5)Phenoxybenzoate (5)Phenoxyethyl (6)Phenoxyethyl (6)Phenoxymethyl (7)Phenoxymethyl (7)Phenoxyphenyl (61)Phenoxyphenyl (61)Phenoxypropan (4)Phenylacetamide (34)Phenylacetonitrile (8)Phenylacetyl (1)Phenylacetyl (1)Phenylacrylamide (3)Phenylalaninol (2)Phenylamino (31)Phenylaniline (19)Phenylanthracene (16)Phenylbenzamide (23)Phenylbenzenesulfonamide (3)Phenylbutanoate (10)Phenylbutoxy (2)Phenylbutoxy (2)Phenylcarbamoyl (5)Phenylcyclohexanone (6)Phenylcyclopropanecarboxylic (6)Phenylcyclopropyl (7)Phenylcyclopropyl (7)Phenyldiazenyl (4)Phenylene (167)Phenylene (167)Phenylenediamine (13)Phenylenedimethanamine (5)Phenylethanamine (12)Phenylethane (9)Phenylethane (9)Phenylethanol (10)Phenylethanone (19)Phenylethoxy (2)Phenylethyl (66)Phenylethyl (66)Phenylethylisocyanate (2)Phenylhex (3)Phenylimino (1)Phenylisocyanate (9)Phenylisoserine (4)Phenylmethanesulfonyl (2)Phenylmethanesulfonyl (2)Phenylmethoxy (4)Phenylmethoxy (4)Phenylmethyl (8)Phenylmethyl (8)Phenylnaphthalene (6)Phenylpentan (11)Phenylpentan (11)Phenylpropanamide (11)Phenylpropyl (20)Phenylpyrazolo (8)Phenylsulfinyl (3)Phenylsulfinyl (3)Phenylsulfonyl (88)Phenylsulfonyl (88)Phenylthio (24)Phenylthio (24)Phenylthiophene (23)Phenylthiophene (23)Phenylthiourea (6)Phenylurea (3)Phenylurea (3)Phenylvinyl (9)Phthalate (23)Phthalimide (12)Phthalonitrile (8)Propoxybenzaldehyde (4)Propoxyphenyl (29)Propoxyphenyl (29)Propoxyphenylboronic (79)Propylphenyl (26)Propylphenyl (26)Pyrenes (52)Quaternaphthalene (6)Quaterphenyl (15)Quaterphenyl (15)Styryl (12)Sulfamoylphenyl (3)Sulfophenyl (4)Sulfophenyl (4)Terephthalaldehyde (11)Terephthalate (13)Terphenyl (236)Terphenyl (236)Tetraaniline (6)Tetrabenzaldehyde (7)Tetrafluorobenzene (17)Tetrahydrobenzo (70)Tetrahydrobenzo (70)Tetrahydronaphthalene (149)Tetrahydronaphthalene (149)Tetralone (12)Tetramethylbenzene (9)Tetramethylbenzene (9)Tetramethylbiphenyl (57)Tetramethylbiphenyl (57)Tetraphenyl (11)Tetraphenyl (11)Toluenesulfonyl (68)Tolyl (403)Tolyl (403)Tolylamino (3)Tolylazanediyl (4)Tolylazanediyl (4)Tolylethynyl (3)Tolyloxy (18)Tolyloxy (18)Tolylphosphine (8)Tolylphosphine (8)Tolylthio (3)Tolylthio (3)Trianiline (7)Tribenzaldehyde (5)Tribromobenzene (7)Trichlorobenzene (19)Trichlorophenoxy (3)Trichlorophenylboronic (6)Triethylbenzene (5)Trifluorophenyl (52)Trifluorophenylboronic (154)Triiodobenzene (5)Triisopropylbenzene (9)Triisopropylphenyl (6)Triisopropylphenyl (6)Trimethoxybenzene (43)Trimethoxystyryl (5)Trimethylaniline (18)Trimethylbenzene (28)Trimethylphenol (6)Trimethylphenyl (20)Trimethylphenyl (20)Trimethylpropiophenone (4)Triphenylphosphoranylidene (9)Triphenylphosphoranylidene (9)Triphenylvinyl (11)Trityl (19)Trityl (19)Tritylamino (2)Trityloxy (2)Tritylthio (3)Tritylthio (3)(((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Amino)-3-(4-Fluorophenyl)Propanoic Acid (3)(10Ar)-1,2,3,4,4A,9,10,10A-Octahydrophenanthrene (3)(1E,1'E)-N,N'-(Ethane-1,2-Diyl)Bis(1-Phenylmethanimine) (5)(1R,4R)-4-Phenyl-1,1'-Bi(Cyclohexane) (24)(9H-Fluoren-9-Yl)Methyl Benzylcarbamate (22)(9H-Fluoren-9-Yl)Methyl Piperazine-1-Carboxylate (7)(Cyclopentylmethyl)Benzene (5)(R)-3-Benzylpyrrolidine (6)(R)-3-Phenylmorpholine (7)(R)-N-(1,2-Diphenylethyl)Benzenesulfonamide (1)(S)-N-(1,2-Diphenylethyl)Benzenesulfonamide (1)1,1':2',1''-Terphenyl (4)1,1':3',1''-Terphenyl (28)1,1':4',1'':4'',1'''-Quaterphenyl (7)1,1,2,2-Tetra([1,1'-Biphenyl]-4-Yl)Ethene (10)1,1-Dimethyl-1,2,3,4-Tetrahydronaphthalene (10)1,2,3,4-Tetrahydronaphthalen-1-Amine (36)1,2,3,4-Tetrahydronaphthalen-1-Ol (5)1,2,3,4-Tetrahydronaphthalene-1-Carboxylic Acid (3)1,2,3,5,6,7-Hexahydro-S-Indacene (23)1,2,4-Trimethylbenzene (41)1,2-Dihydroacenaphthylene (6)1,2-Dinitrobenzene (6)1,3,5-Tris(Phenylethynyl)Benzene (6)1,3,6,8-Tetraphenylpyrene (10)1,3-Bis(Benzyloxy)Benzene (8)1,3-Dihydro-2H-Inden-2-One (6)1,3-Diphenoxypropane (6)1,3-Diphenyl-1H-Pyrazole (6)1,3-Diphenylpropane (6)1,3-Diphenylpropane-1,3-Dione (4)1,4-Bis(Phenylethynyl)Benzene (7)1,4-Diphenylbuta-1,3-Diyne (9)1-(2,3-Dihydro-1H-Inden-5-Yl)Ethan-1-Amine (3)1-(Anthracen-9-Yl)Ethan-1-Amine (3)1-Benzylazetidine (6)1-Bromopyrene (4)1-Cyclohexyl-4-(Phenylethynyl)Benzene (10)1-Cyclopropylnaphthalene (6)1-Phenyl-1H-Tetrazole (6)1-Phenylnaphthalene (12)1-Trityl-1H-Pyrazole (5)1H-Indene (18)1H-Indene-1,3(2H)-Dione (5)2,2 ', 3,3' - Tetrahydro-1,1 '- Spiro [Indene] (5)2,3,4,5-Tetrahydro-1H-Benzo[C]Azepine (7)2,3-Dihydro-1H-Inden-1-Amine (52)2,3-Dihydro-1H-Inden-1-Ol (13)2,3-Dihydro-1H-Inden-4-Amine (6)2,3-Dihydro-1H-Inden-4-Ol (6)2,3-Dihydro-1H-Indene-1-Carboxylic Acid (5)2,3-Dihydro-1H-Indene-4-Carbonitrile (4)2,4-Diphenyl-1,3,5-Triazine (6)2,5-Diphenyl-1,3,4-Oxadiazole (6)2,7-Bis(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)-9H-Fluorene (7)2-(2-Chlorophenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (24)2-(2-Fluorophenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (21)2-(2-Methoxyphenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (29)2-(3-Bromophenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (14)2-(3-Fluorophenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (19)2-(3-Methoxyphenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (30)2-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Aniline (17)2-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Benzaldehyde (20)2-(4-Chlorophenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (15)2-(4-Fluorophenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (16)2-Amino-2-(Anthracen-9-Yl)Ethan-1-Ol (4)2-Methyl-2,3-Dihydro-1H-Inden-1-One (9)2-Phenyl-1,3,5-Triazine (6)2-Phenyl-1,3-Dioxane (7)2-Phenyl-2H-1,2,3-Triazole (7)2-Phenylisoindoline-1,3-Dione (7)2H-Benzo[B][1,4]Thiazin-3(4H)-One (6)3,3-Dimethyl-2,3-Dihydro-1H-Inden-1-One (5)3,4-Dihydro-2H-Benzo[B][1,4]Dioxepine (7)3,5,6,7-Tetrahydro-S-Indacen-1(2H)-One (5)3-((((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Amino)-3-(2-Bromophenyl)Propanoic Acid (3)3-((((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Amino)-3-(2-Chlorophenyl)Propanoic Acid (3)3-((((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Amino)-3-(3-(Trifluoromethyl)Phenyl)Propanoic Acid (3)3-((((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Amino)-3-(3-Fluorophenyl)Propanoic Acid (3)3-((((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Amino)-3-(3-Methoxyphenyl)Propanoic Acid (4)3-((((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Amino)-3-(3-Nitrophenyl)Propanoic Acid (3)3-((((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Amino)-3-(4-Bromophenyl)Propanoic Acid (3)3-((((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Amino)-3-(4-Nitrophenyl)Propanoic Acid (3)3-((((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Amino)-3-(O-Tolyl)Propanoic Acid (3)3-((((9H-Fluoren-9-Yl)Methoxy)Carbonyl)Amino)-3-(P-Tolyl)Propanoic Acid (3)3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Aniline (25)3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Benzaldehyde (19)3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Benzoic Acid (12)3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Phenol (13)3-Methyl-2,3-Dihydro-1H-Inden-1-One (4)3-Phenyl-1H-Pyrrole (4)3-Phenyloxazolidin-2-One (4)4',5'-Diphenyl-1,1':2',1''-Terphenyl (7)4,4,5,5-Tetramethyl-2-(3-(Trifluoromethyl)Phenyl)-1,3,2-Dioxaborolane (19)4,4,5,5-Tetramethyl-2-(3-Nitrophenyl)-1,3,2-Dioxaborolane (24)4,4,5,5-Tetramethyl-2-(M-Tolyl)-1,3,2-Dioxaborolane (21)4,4,5,5-Tetramethyl-2-(O-Tolyl)-1,3,2-Dioxaborolane (24)4,5-Diphenyloxazole (7)4,7-Diphenylbenzo[C][1,2,5]Thiadiazole (6)4-(4-Cyclopropylnaphthalen-1-Yl)-4H-1,2,4-Triazole (7)4-(Phenylethynyl)-1,1'-Biphenyl (5)4-Bromo-2,3-Dihydro-1H-Inden-1-One (5)4-Bromo-2,3-Dihydro-1H-Indene (10)4-Chloro-2,3-Dihydro-1H-Inden-1-One (8)4-Chloro-2,3-Dihydro-1H-Indene (5)4-Cyclohexyl-1,1'-Biphenyl (18)4-Fluoro-2,3-Dihydro-1H-Inden-1-One (6)4-Fluoro-2,3-Dihydro-1H-Indene (14)4-Methoxy-2,3-Dihydro-1H-Inden-1-One (6)4-Methyl-2,3-Dihydro-1H-Inden-1-One (8)4-Methyl-2,3-Dihydro-1H-Indene (3)4-Phenyl-1,1'-Bi(Cyclohexane) (8)4-Phenyl-4H-1,2,4-Triazole (6)5''-([1,1'-Biphenyl]-4-Yl)-1,1':4',1'':3'',1''':4''',1''''-Quinquephenyl (5)5',5''-Diphenyl-1,1':3',1'':3'',1'''-Quaterphenyl (8)5,5',6,6',7,7',8,8'-Octahydro-1,1'-Binaphthalene (6)5,6,7,8-Tetrahydronaphthalen-2-Ol (5)5-Bromo-1,2,3,4-Tetrahydronaphthalene (6)5-Bromo-2,3-Dihydro-1H-Indene (8)5-Fluoro-2,3-Dihydro-1H-Indene (3)5-Methoxy-1,2,3,4-Tetrahydronaphthalene (3)5-Methoxy-2,3-Dihydro-1H-Indene (2)5-Methyl-3,4-Dihydronaphthalen-1(2H)-One (6)5-Phenyl-1H-Imidazole (6)5-Phenylcyclohexane-1,3-Dione (6)6,7,8,9-Tetrahydro-5H-Benzo[7]Annulen-5-One (6)6-Bromo-1,2,3,4-Tetrahydronaphthalene (9)6-Bromo-2,3-Dihydro-1H-Inden-1-One (8)6-Bromo-3,4-Dihydronaphthalen-1(2H)-One (3)6-Chloro-1,2,3,4-Tetrahydronaphthalene (3)6-Chloro-2,3-Dihydro-1H-Inden-1-One (5)6-Fluoro-1,2,3,4-Tetrahydronaphthalene (6)6-Fluoro-2,3-Dihydro-1H-Inden-1-One (4)6-Methoxy-2,3-Dihydro-1H-Inden-1-One (6)6-Methoxy-3,4-Dihydronaphthalen-1(2H)-One (5)6-Methyl-1,2,3,4-Tetrahydronaphthalene (5)6-Methyl-2,3-Dihydro-1H-Inden-1-One (3)6-Methyl-3,4-Dihydronaphthalen-1(2H)-One (2)7-Bromo-2,3-Dihydro-1H-Inden-1-One (13)7-Chloro-2,3-Dihydro-1H-Inden-1-One (9)7-Fluoro-2,3-Dihydro-1H-Inden-1-One (8)7-Hydroxy-2,3-Dihydro-1H-Inden-1-One (6)7-Methoxy-2,3-Dihydro-1H-Inden-1-One (6)7-Methoxy-3,4-Dihydronaphthalen-1(2H)-One (6)7-Methyl-2,3-Dihydro-1H-Inden-1-One (7)9,10-Bis(Phenylethynyl)Anthracene (5)9,10-Diphenylanthracene (16)9-Methylanthracene (4)9-Phenylxanthylium (7)9H-Fluoren-9-One (17)[1,1'-Binaphthalen]-2-Amine (3)[1,1':4',1''-Terphenyl]-4,4''-Dicarbaldehyde (9)Benzenesulfinic Acid (6)Benzil (17)Benzo-18-Crown-6 (7)Benzo[Lmn][3,8]Phenanthroline-1,3,6,8(2H,7H)-Tetraone (10)Benzyl ((Benzyloxy)Carbonyl)Glycinate (8)Benzyl (4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Phenyl)Carbamate (5)Benzyl (S)-Pyrrolidin-3-Ylcarbamate (7)Benzyl 3-Phenylpropanoate (1)Benzyl Piperidin-4-Ylcarbamate (7)Bicyclo[4.2.0]Octa-1,3,5-Triene (13)Cumene (181)Dibenzyl Phosphonate (6)Diphenylmethanimine (9)Ethane-1,1,2-Triyltribenzene (4)Ethylbenzene (197)Mesitylene (71)Methyl 2-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Benzoate (14)Methyl 3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Benzoate (11)N,2-Diphenylacetamide (7)N,N,N-Trimethyl-1-Phenylmethanaminium (6)N,N-Diphenyl-[1,1'-Biphenyl]-4-Amine (8)N-Benzyl-2-Phenylethan-1-Amine (1)N-Benzylcyclopropanamine (6)N-Phenylcinnamamide (16)N1,N1,N4,N4-Tetraphenylbenzene-1,4-Diamine (6)N4,N4,N4',N4'-Tetraphenyl-[1,1'-Biphenyl]-4,4'-Diamine (7)Phenanthrene (23)Phenanthrene-9,10-Dione (8)Phenethyldiphenylphosphane (6)Phenothiazin-5-Ium (6)Phenyl (1R,4R)-[1,1'-Bi(Cyclohexane)]-4-Carboxylate (6)Phenyl 4-Cyclohexylbenzoate (8)Tert-Butyl (2,3-Dihydro-1H-Inden-1-Yl)Carbamate (3)Tert-Butylbenzene (245)Triphenyl Phosphate (6)
Benzyl Bromides (1074)
Amides (6)Aryls (82)Bromides (81)Chlorides (6)Esters (14)Ethers (6)Fluorinated Building Blocks (2)Nitriles (5)Sulfamides (3)1-(Bromomethyl)-2-(Trifluoromethyl)Benzene (13)1-(Bromomethyl)-2-Chlorobenzene (28)1-(Bromomethyl)-2-Fluorobenzene (50)1-(Bromomethyl)-3-(Trifluoromethyl)Benzene (17)1-(Bromomethyl)-3-Chlorobenzene (27)1-(Bromomethyl)-3-Fluorobenzene (47)1-(Bromomethyl)-3-Methoxybenzene (19)1-Bromo-2-(Bromomethyl)Benzene (34)2-(2-(Bromomethyl)Phenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (9)2-(Bromomethyl)-1,3-Difluorobenzene (12)2-(Bromomethyl)Benzonitrile (15)Methyl 2-(Bromomethyl)Benzoate (33)
Bromides (52776)
Acyl Chlorides (5)Alcohols (147)Aldehydes (78)Aliphatic Chain Hydrocarbons (148)Aliphatic Cyclic Hydrocarbons (31)Alkenyls (45)Alkynyls (11)Amides (311)Amines (216)Anhydrides (5)Aryls (936)Azetidines (7)Benzimidazoles (8)Benzofurans (18)Benzothiazoles (11)Benzothiophenes (4)Benzoxazoles (10)Benzyl Bromides (81)Benzyl Chlorides (5)Carboxylic Acids (176)Chlorides (241)Difluoromethyls (36)Dioxolanes (6)Esters (314)Ethers (328)Fluorinated Building Blocks (305)Furans (13)Imidazoles (16)Indazoles (25)Indoles (36)Indolines (16)Iodides (54)Isoquinolines (11)Isothiazoles (4)Isoxazoles (11)Ketones (169)Morpholines (30)Naphthyridines (4)Nitriles (82)Nitroes (62)Oxadiazoles (4)Oxazoles (12)Oxetanes (9)Phenols (34)Piperazines (10)Piperidines (31)Pyrazines (9)Pyrazoles (38)Pyridazines (10)Pyridines (398)Pyrimidines (71)Pyrroles (5)Pyrrolidines (25)Pyrrolines (6)Quinazolines (13)Quinolines (106)Quinoxalines (9)Spiroes (11)Sulfamides (35)Sulfides (18)Sulfones (31)Sulfonyl Chlorides (17)Tetrahydrofurans (4)Tetrahydropyrans (6)Tetrahydroquinolines (6)Thiazoles (70)Thiophenes (66)Thiophenols (4)Trifluoromethyls (95)Other Aromatic Heterocycles (92)Bromoacetyl (14)Bromoaniline (2)Bromoanthracene (5)Bromoanthracene (5)Bromobenzaldehyde (15)Bromobenzamide (7)Bromobenzimidamide (6)Bromobenzyl (33)Bromobut (2)Bromobut (2)Bromobutoxy (4)Bromobutoxy (4)Bromobutyl (6)Bromobutyl (6)Bromochroman (26)Bromochroman (26)Bromoethoxy (22)Bromoethoxy (22)Bromoethyl (86)Bromoethyl (86)Bromoethynyl (6)Bromohexyl (11)Bromohexyl (11)Bromomethyl (759)Bromonicotinaldehyde (5)Bromonicotinamide (5)Bromooctyl (3)Bromopentyl (8)Bromopentyl (8)Bromopropane (10)Bromopropane (10)Bromopropoxy (6)Bromopropoxy (6)Bromopropyl (25)Bromopropyl (25)Bromothiochroman (4)Bromothiochroman (4)Bromovinyl (2)Dibromo (696)Dibromo (696)Dibromoanthracene (3)Dibromoanthracene (3)Dibromobenzaldehyde (8)Dibromobenzonitrile (8)Dibromomethyl (6)Dibromomethyl (6)Tetrabromo (10)Tetrabromo (10)Tribromo (9)Tribromo (9)(1-Bromoethyl)Benzene (11)(2,2-Dibromovinyl)Benzene (6)(2-(4-Bromophenyl)Ethene-1,1,2-Triyl)Tribenzene (11)(2-Bromo-6-Fluorophenyl)Methanol (4)(2-Bromoethyl)Benzene (33)(2-Bromophenyl)(Phenyl)Methanone (3)(2-Bromophenyl)Boronic Acid (39)(2-Bromophenyl)Hydrazine (8)(2-Bromophenyl)Methanamine (12)(2-Bromophenyl)Methanol (39)(3-Bromo-2-Fluorophenyl)Boronic Acid (7)(3-Bromo-2-Methoxyphenyl)Boronic Acid (6)(3-Bromophenyl)Boronic Acid (65)(3-Bromopropyl)Benzene (10)(3-Bromopyridin-2-Yl)Methanol (7)(4-Bromophenyl)(Phenyl)Methanone (9)(5-Bromopyridin-2-Yl)Methanamine (5)(5-Bromopyridin-2-Yl)Methanol (5)(Bromoethynyl)Benzene (5)(Dibromomethyl)Benzene (6)1,2,3-Tribromobenzene (5)1,2,4-Tribromobenzene (13)1,2-Dibromo-3-Chlorobenzene (3)1,2-Dibromo-3-Fluorobenzene (9)1,2-Dibromo-3-Methylbenzene (9)1,2-Dibromo-4-Fluorobenzene (11)1,2-Dibromobenzene (64)1,3-Dibromo-2,4-Difluorobenzene (5)1,3-Dibromo-2,4-Dimethylbenzene (3)1,3-Dibromo-2-Chlorobenzene (8)1,3-Dibromo-2-Fluorobenzene (15)1,3-Dibromo-2-Iodobenzene (4)1,3-Dibromo-2-Methoxybenzene (11)1,3-Dibromo-2-Methylbenzene (14)1,3-Dibromo-2-Nitrobenzene (4)1,4-Dibromo-2-Iodobenzene (3)1-(2-Bromophenyl)Ethan-1-Amine (10)1-(2-Bromophenyl)Ethan-1-One (27)1-(5-Bromopyridin-2-Yl)Piperazine (6)1-(Azetidin-1-Yl)-2-Bromoethan-1-One (3)1-(Benzyloxy)-2-Bromobenzene (12)1-(Benzyloxy)-3-Bromobenzene (12)1-(Benzyloxy)-4-Bromobenzene (8)1-(Bromomethyl)-2-Iodobenzene (9)1-(Bromomethyl)-2-Methoxybenzene (14)1-(Bromomethyl)-2-Methylbenzene (14)1-(Bromomethyl)-2-Nitrobenzene (23)1-(Bromomethyl)-3-Methylbenzene (11)1-Benzyl-3-Bromobenzene (7)1-Benzyl-4-Bromo-1H-Pyrazole (5)1-Bromo-2,3-Dichlorobenzene (18)1-Bromo-2,3-Difluorobenzene (67)1-Bromo-2,3-Dimethoxybenzene (8)1-Bromo-2,3-Dimethylbenzene (23)1-Bromo-2,4-Difluorobenzene (59)1-Bromo-2-(Bromomethyl)Benzene (34)1-Bromo-2-(Difluoromethyl)Benzene (5)1-Bromo-2-(Methylsulfonyl)Benzene (4)1-Bromo-2-(Tert-Butyl)Benzene (7)1-Bromo-2-(Trifluoromethoxy)Benzene (22)1-Bromo-2-Chloro-3-Fluorobenzene (20)1-Bromo-2-Chloro-3-Methoxybenzene (6)1-Bromo-2-Chloro-3-Methylbenzene (9)1-Bromo-2-Chloro-4-Nitrobenzene (5)1-Bromo-2-Cyclopropylbenzene (3)1-Bromo-2-Ethoxybenzene (16)1-Bromo-2-Ethylbenzene (13)1-Bromo-2-Fluoro-3-(Trifluoromethyl)Benzene (4)1-Bromo-2-Fluoro-3-Methoxybenzene (7)1-Bromo-2-Fluoro-3-Methylbenzene (11)1-Bromo-2-Fluoro-3-Nitrobenzene (11)1-Bromo-2-Fluoro-4-Nitrobenzene (13)1-Bromo-2-Iodobenzene (65)1-Bromo-2-Isopropylbenzene (10)1-Bromo-2-Phenoxybenzene (3)1-Bromo-3-(Trifluoromethyl)Benzene (100)1-Bromo-3-Chloro-2-Fluorobenzene (9)1-Bromo-3-Chloro-2-Methylbenzene (6)1-Bromo-3-Cyclopropylbenzene (6)1-Bromo-3-Fluoro-2-Iodobenzene (4)1-Bromo-3-Fluoro-2-Methoxybenzene (12)1-Bromo-3-Fluoro-2-Methylbenzene (17)1-Bromo-3-Fluoro-2-Nitrobenzene (10)1-Bromo-3-Iodobenzene (70)1-Bromo-3-Methoxybenzene (187)1-Bromo-3-Methyl-2-Nitrobenzene (3)1-Bromo-3-Nitrobenzene (235)1-Bromo-3-Phenoxybenzene (8)1-Bromo-4-(Phenylsulfonyl)Benzene (7)1-Bromo-4-Chloro-2-Fluorobenzene (22)1-Bromo-4-Fluoro-2-Methoxybenzene (11)1-Bromo-4-Fluoro-2-Methylbenzene (18)1-Bromo-4-Iodobenzene (67)1-Bromo-4-Phenoxybenzene (8)1-Bromo-4-Styrylbenzene (5)1-Bromobicyclo[1.1.1]Pentane (3)1-Bromoisoquinoline (20)1-Bromonaphthalene (4)2,3-Dibromopyridine (21)2,4-Dibromo-1-Chlorobenzene (8)2,4-Dibromo-1-Fluorobenzene (18)2,4-Dibromo-1-Methoxybenzene (6)2,4-Dibromoaniline (18)2,4-Dibromophenol (16)2,4-Dibromopyridine (5)2,4-Dibromothiazole (5)2,5-Dibromopyrazine (4)2,5-Dibromopyridine (26)2,5-Dibromothiazole (5)2,5-Dibromothiophene (18)2,6-Dibromo-3-Fluoropyridine (3)2,6-Dibromoaniline (17)2,6-Dibromobenzaldehyde (4)2,6-Dibromobenzonitrile (4)2,6-Dibromophenol (20)2,6-Dibromopyrazine (9)2,6-Dibromopyridin-4-Amine (3)2,6-Dibromopyridine (28)2-(2-Bromophenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (5)2-(2-Bromophenyl)Acetic Acid (23)2-(2-Bromophenyl)Acetonitrile (10)2-(4-Bromophenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (8)2-(Bromomethyl)Furan (3)2-(Bromomethyl)Pyridine (27)2-Amino-3-Bromobenzoic Acid (8)2-Amino-3-Bromobenzonitrile (7)2-Amino-3-Bromophenol (3)2-Amino-6-Bromophenol (3)2-Bromo-1,1'-Biphenyl (12)2-Bromo-1,3,4-Thiadiazole (11)2-Bromo-1,3,4-Trifluorobenzene (16)2-Bromo-1,3,4-Trimethylbenzene (4)2-Bromo-1,3-Dichlorobenzene (12)2-Bromo-1,3-Difluorobenzene (43)2-Bromo-1,3-Dimethoxybenzene (7)2-Bromo-1,3-Dimethylbenzene (35)2-Bromo-1,4-Dichlorobenzene (12)2-Bromo-1,4-Difluorobenzene (64)2-Bromo-1,4-Dimethoxybenzene (11)2-Bromo-1,4-Dimethylbenzene (22)2-Bromo-1-Chloro-3-Fluorobenzene (9)2-Bromo-1-Chloro-4-Fluorobenzene (20)2-Bromo-1-Chloro-4-Methylbenzene (6)2-Bromo-1-Chloro-4-Nitrobenzene (9)2-Bromo-1-Fluoro-3-Nitrobenzene (9)2-Bromo-1-Fluoro-4-(Trifluoromethyl)Benzene (4)2-Bromo-1-Fluoro-4-Methoxybenzene (12)2-Bromo-1-Fluoro-4-Methylbenzene (17)2-Bromo-1-Fluoro-4-Nitrobenzene (23)2-Bromo-1-Methoxy-3-Methylbenzene (3)2-Bromo-1-Methoxy-4-Methylbenzene (7)2-Bromo-1-Methyl-4-Nitrobenzene (12)2-Bromo-1H-Benzo[D]Imidazole (4)2-Bromo-1H-Imidazole (13)2-Bromo-1H-Pyrrole (11)2-Bromo-3-(Trifluoromethyl)Pyridine (4)2-Bromo-3-Chloroaniline (4)2-Bromo-3-Chloropyridine (25)2-Bromo-3-Fluoroaniline (13)2-Bromo-3-Fluoropyridine (26)2-Bromo-3-Hydroxybenzaldehyde (4)2-Bromo-3-Methoxybenzaldehyde (4)2-Bromo-3-Methoxypyridine (7)2-Bromo-3-Methylaniline (6)2-Bromo-3-Methylphenol (4)2-Bromo-3-Methylpyrazine (5)2-Bromo-3-Methylpyridine (25)2-Bromo-3-Nitrobenzoic Acid (4)2-Bromo-3-Nitropyridine (5)2-Bromo-4-(Trifluoromethyl)Aniline (9)2-Bromo-4-Chloro-1-Fluorobenzene (16)2-Bromo-4-Chloro-1-Methoxybenzene (7)2-Bromo-4-Chloro-1-Methylbenzene (11)2-Bromo-4-Chloro-1-Nitrobenzene (5)2-Bromo-4-Chloroaniline (15)2-Bromo-4-Chlorophenol (11)2-Bromo-4-Chloropyridine (7)2-Bromo-4-Fluoro-1-Methoxybenzene (12)2-Bromo-4-Fluoro-1-Methylbenzene (21)2-Bromo-4-Fluoro-1-Nitrobenzene (23)2-Bromo-4-Fluoroaniline (28)2-Bromo-4-Fluorobenzaldehyde (7)2-Bromo-4-Fluorobenzoic Acid (8)2-Bromo-4-Fluorobenzonitrile (7)2-Bromo-4-Fluorophenol (10)2-Bromo-4-Methoxy-1-Nitrobenzene (6)2-Bromo-4-Methoxyaniline (6)2-Bromo-4-Methoxybenzaldehyde (8)2-Bromo-4-Methoxybenzoic Acid (4)2-Bromo-4-Methylaniline (14)2-Bromo-4-Methylbenzoic Acid (5)2-Bromo-4-Methylpyridine (6)2-Bromo-4-Methylthiazole (7)2-Bromo-4-Nitroaniline (12)2-Bromo-5-(Trifluoromethyl)Pyridine (6)2-Bromo-5-Chloropyrazine (6)2-Bromo-5-Chloropyridine (22)2-Bromo-5-Fluoroaniline (18)2-Bromo-5-Fluorobenzaldehyde (7)2-Bromo-5-Fluorobenzoic Acid (11)2-Bromo-5-Fluoropyridine (33)2-Bromo-5-Methoxybenzaldehyde (6)2-Bromo-5-Methoxypyridine (11)2-Bromo-5-Methylpyridine (19)2-Bromo-5-Methylthiazole (7)2-Bromo-5-Nitropyridine (11)2-Bromo-6-(Trifluoromethyl)Aniline (5)2-Bromo-6-(Trifluoromethyl)Pyridine (6)2-Bromo-6-Chloroaniline (10)2-Bromo-6-Chlorophenol (5)2-Bromo-6-Chloropyrazine (5)2-Bromo-6-Chloropyridine (23)2-Bromo-6-Fluoroaniline (14)2-Bromo-6-Fluorobenzaldehyde (15)2-Bromo-6-Fluorobenzoic Acid (11)2-Bromo-6-Fluorobenzonitrile (7)2-Bromo-6-Fluorophenol (9)2-Bromo-6-Fluoropyridine (21)2-Bromo-6-Iodoaniline (4)2-Bromo-6-Methoxyaniline (5)2-Bromo-6-Methoxyphenol (5)2-Bromo-6-Methoxypyridine (15)2-Bromo-6-Methylaniline (11)2-Bromo-6-Methylbenzoic Acid (5)2-Bromo-6-Methylphenol (7)2-Bromo-6-Methylpyrazine (5)2-Bromo-6-Methylpyridine (17)2-Bromo-6-Nitroaniline (14)2-Bromo-6-Nitrophenol (9)2-Bromo-6-Phenylpyridine (4)2-Bromo-9,9-Diphenyl-9H-Fluorene (5)2-Bromo-9H-Fluorene (15)2-Bromo-[1,2,4]Triazolo[1,5-A]Pyridine (6)2-Bromo-N,N-Dimethylaniline (5)2-Bromo-N-Methylaniline (8)2-Bromo-N-Phenylacetamide (5)2-Bromobenzene-1,3-Diol (7)2-Bromobenzene-1,4-Diol (4)2-Bromobenzenesulfonyl Chloride (8)2-Bromobenzo[D]Thiazole (28)2-Bromodibenzo[B,D]Furan (6)2-Bromofuran (21)2-Bromoimidazo[1,2-A]Pyridine (5)2-Bromonaphthalene (4)2-Bromonicotinic Acid (5)2-Bromooxazole (7)2-Bromopyrazine (15)2-Bromopyridin-3-Amine (10)2-Bromopyridin-3-Ol (7)2-Bromopyridine (10)2-Bromopyridine 1-Oxide (6)2-Bromopyrimidine (22)2-Bromoquinazoline (4)3,4-Dibromoaniline (4)3,4-Dibromophenol (3)3,5-Dibromopyridine (12)3-(Bromomethyl)Pyridine (18)3-Bromo-1,1'-Biphenyl (12)3-Bromo-1,5-Naphthyridine (8)3-Bromo-1-Methyl-1H-Pyrazole (16)3-Bromo-1H-Pyrrole (16)3-Bromo-1H-Pyrrolo[2,3-B]Pyridine (7)3-Bromo-1H-Pyrrolo[3,2-B]Pyridine (6)3-Bromo-2-Chlorobenzaldehyde (4)3-Bromo-2-Chlorobenzoic Acid (7)3-Bromo-2-Chloropyridin-4-Amine (3)3-Bromo-2-Chloropyridine (20)3-Bromo-2-Fluoroaniline (15)3-Bromo-2-Fluorobenzaldehyde (9)3-Bromo-2-Fluorobenzoic Acid (10)3-Bromo-2-Fluorobenzonitrile (10)3-Bromo-2-Fluorophenol (5)3-Bromo-2-Fluoropyridine (21)3-Bromo-2-Hydroxybenzaldehyde (10)3-Bromo-2-Hydroxybenzoic Acid (4)3-Bromo-2-Hydroxybenzonitrile (3)3-Bromo-2-Methoxypyridine (20)3-Bromo-2-Methylpyridine (27)3-Bromo-4-Chloroaniline (13)3-Bromo-4-Chlorobenzoic Acid (6)3-Bromo-4-Chlorophenol (5)3-Bromo-4-Fluoroaniline (22)3-Bromo-4-Fluorobenzaldehyde (7)3-Bromo-4-Fluorobenzamide (3)3-Bromo-4-Fluorobenzoic Acid (10)3-Bromo-4-Fluorophenol (11)3-Bromo-4-Hydroxybenzaldehyde (9)3-Bromo-4-Hydroxybenzoic Acid (3)3-Bromo-4-Methoxyaniline (7)3-Bromo-4-Methoxybenzaldehyde (9)3-Bromo-4-Methoxybenzoic Acid (7)3-Bromo-4-Methylaniline (14)3-Bromo-4-Methylbenzoic Acid (9)3-Bromo-4-Methylpyridine (13)3-Bromo-5'-Phenyl-1,1':3',1''-Terphenyl (4)3-Bromo-5-Chloropyridine (14)3-Bromo-5-Fluoropyridine (18)3-Bromo-5-Methylpyridine (14)3-Bromo-5-Nitropyridine (10)3-Bromoaniline (187)3-Bromoazetidine (3)3-Bromobenzene-1,2-Diamine (11)3-Bromobenzene-1,2-Diol (8)3-Bromobenzo[B]Thiophene (5)3-Bromobenzoic Acid (159)3-Bromobenzonitrile (110)3-Bromofuran (13)3-Bromoimidazo[1,2-A]Pyridine (26)3-Bromoimidazo[1,2-A]Pyrimidine (6)3-Bromoisoquinoline (10)3-Bromoisoxazole (7)3-Bromopicolinic Acid (9)3-Bromopicolinonitrile (13)3-Bromopyrazolo[1,5-A]Pyrimidine (11)3-Bromopyridazine (25)3-Bromopyridin-2-Amine (30)3-Bromopyridin-2-Ol (17)3-Bromopyridine 1-Oxide (8)3-Bromopyrrolidine (6)3-Bromothiophene (6)4-(Bromomethyl)-1,1'-Biphenyl (13)4-(Bromomethyl)Pyridine (8)4-Bromo-1,5-Naphthyridine (5)4-Bromo-1-Methyl-1H-Pyrazole (14)4-Bromo-1-Phenyl-1H-Imidazole (9)4-Bromo-1-Phenyl-1H-Pyrazole (11)4-Bromo-1H-Benzo[D]Imidazole (14)4-Bromo-1H-Indole (25)4-Bromo-1H-Pyrazolo[3,4-B]Pyridine (4)4-Bromo-1H-Pyrrolo[2,3-B]Pyridine (9)4-Bromo-2-Chloro-1-Iodobenzene (4)4-Bromo-2-Chloropyridine (29)4-Bromo-2-Fluoro-1-Iodobenzene (8)4-Bromo-2-Fluoropyridine (4)4-Bromo-2-Iodoaniline (8)4-Bromo-2-Methoxypyridine (4)4-Bromo-3-(Trifluoromethyl)Aniline (7)4-Bromo-3-Chloroaniline (21)4-Bromo-3-Fluoroaniline (29)4-Bromo-3-Methylaniline (19)4-Bromo-3-Methylpyridine (5)4-Bromo-3-Nitroaniline (8)4-Bromo-N,N-Diphenylaniline (10)4-Bromo-N-Phenylaniline (6)4-Bromo-N-Phenylbenzenesulfonamide (23)4-Bromobenzo[C][1,2,5]Thiadiazole (14)4-Bromobenzo[D][1,3]Dioxole (6)4-Bromobenzo[D]Thiazole (9)4-Bromobenzofuran (3)4-Bromoindoline (4)4-Bromoisoindolin-1-One (3)4-Bromoisoquinolin-1(2H)-One (3)4-Bromoisoquinoline (33)4-Bromopyridazine (5)4-Bromopyridine (31)4-Bromopyrimidine (24)4-Bromothiazole (10)5-Bromo-1,2,3,4-Tetrahydroisoquinoline (4)5-Bromo-1-Methyl-1H-Indole (3)5-Bromo-1H-1,2,4-Triazole (6)5-Bromo-1H-Benzo[D][1,2,3]Triazole (5)5-Bromo-1H-Benzo[D]Imidazole (13)5-Bromo-1H-Indazole (14)5-Bromo-1H-Pyrazole (11)5-Bromo-1H-Pyrazolo[3,4-B]Pyridine (11)5-Bromo-1H-Pyrazolo[3,4-C]Pyridine (5)5-Bromo-1H-Pyrrolo[2,3-B]Pyridine (13)5-Bromo-2,3-Dihydro-1H-Indene (8)5-Bromo-2,3-Dihydrobenzofuran (6)5-Bromo-2-Chloro-4-Methylpyridine (5)5-Bromo-2-Chloropyridine (40)5-Bromo-2-Chloropyrimidine (6)5-Bromo-2-Fluoropyridine (22)5-Bromo-2-Methoxypyridine (29)5-Bromo-2-Methylpyridine (23)5-Bromo-2-Methylpyrimidine (5)5-Bromo-2-Phenylpyridine (3)5-Bromo-4-Methylpyridin-2-Amine (5)5-Bromo-6-Chloropyridin-2-Amine (3)5-Bromo-7H-Pyrrolo[2,3-D]Pyrimidine (3)5-Bromobenzo[B]Thiophene (3)5-Bromobenzo[D][1,3]Dioxole (12)5-Bromobenzo[D]Oxazole (3)5-Bromoimidazo[1,2-A]Pyridine (6)5-Bromoindoline (8)5-Bromoisoindolin-1-One (8)5-Bromoisoquinoline (17)5-Bromonicotinaldehyde (12)5-Bromonicotinic Acid (6)5-Bromonicotinonitrile (6)5-Bromopicolinic Acid (11)5-Bromopicolinonitrile (13)5-Bromopyrazin-2-Ol (5)5-Bromopyridin-2-Amine (35)5-Bromopyridin-2-Ol (18)5-Bromopyridin-3-Amine (10)5-Bromopyridin-3-Ol (6)5-Bromoquinoxaline (6)5-Bromothiazole (8)6-Bromo-1,2,3,4-Tetrahydroisoquinoline (4)6-Bromo-1,2,3,4-Tetrahydroquinoline (6)6-Bromo-1-Methyl-1H-Indole (3)6-Bromo-1H-Indazole (23)6-Bromo-1H-Indole (15)6-Bromo-1H-Pyrazolo[4,3-B]Pyridine (4)6-Bromo-1H-Pyrrolo[2,3-B]Pyridine (6)6-Bromo-1H-Pyrrolo[3,2-B]Pyridine (7)6-Bromo-1H-Pyrrolo[3,2-C]Pyridine (5)6-Bromo-3,4-Dihydro-2H-Benzo[B][1,4]Oxazine (5)6-Bromo-3,4-Dihydroisoquinolin-1(2H)-One (6)6-Bromo-3,4-Dihydroquinolin-2(1H)-One (4)6-Bromo-[1,2,4]Triazolo[1,5-A]Pyridine (12)6-Bromo-[1,2,4]Triazolo[4,3-A]Pyridine (8)6-Bromobenzo[B]Thiophene (4)6-Bromobenzo[D]Oxazole (3)6-Bromobenzo[D]Thiazole (7)6-Bromochroman-4-One (6)6-Bromoimidazo[1,2-A]Pyrimidine (5)6-Bromoindoline-2,3-Dione (4)6-Bromoisoindolin-1-One (4)6-Bromoisoquinoline (9)6-Bromonicotinaldehyde (10)6-Bromonicotinic Acid (9)6-Bromonicotinonitrile (7)6-Bromopicolinic Acid (5)6-Bromopyrazolo[1,5-A]Pyrimidine (7)6-Bromopyridin-2-Amine (8)6-Bromopyridin-3-Amine (12)6-Bromopyridin-3-Ol (13)6-Bromoquinazoline (7)6-Bromoquinoxaline (9)7-Bromo-1,2,3,4-Tetrahydroisoquinoline (4)7-Bromo-1H-Indazole (18)7-Bromo-1H-Indole (28)7-Bromo-1H-Pyrrolo[2,3-C]Pyridine (4)7-Bromo-2,3-Dihydrobenzofuran (6)7-Bromo-3,4-Dihydro-2H-Benzo[B][1,4]Oxazine (7)7-Bromo-3,4-Dihydroisoquinolin-1(2H)-One (6)7-Bromo-3,4-Dihydronaphthalen-1(2H)-One (8)7-Bromobenzo[B]Thiophene (5)7-Bromobenzo[D]Thiazole (8)7-Bromobenzofuran (6)7-Bromochromane (7)7-Bromoindoline (3)7-Bromoisoquinoline (6)8-Bromo-[1,2,4]Triazolo[1,5-A]Pyridine (6)8-Bromochromane (6)8-Bromoimidazo[1,2-A]Pyridine (15)8-Bromoisoquinoline (17)8-Bromoquinazoline (8)9-Bromoanthracene (5)Bromocyclohexane (7)Ethyl 2-Bromo-2-Phenylacetate (6)Methyl 2-(2-Bromophenyl)Acetate (8)Methyl 2-Bromo-2-Phenylacetate (7)Methyl 2-Bromo-3-Nitrobenzoate (3)Methyl 2-Bromo-5-Fluorobenzoate (8)Methyl 2-Bromo-6-Fluorobenzoate (7)Methyl 2-Bromo-6-Methylbenzoate (8)Methyl 2-Bromobenzoate (69)Methyl 3-(Bromomethyl)Benzoate (16)Methyl 3-Bromo-4-Chlorobenzoate (6)Methyl 3-Bromo-4-Fluorobenzoate (7)Methyl 3-Bromo-4-Methoxybenzoate (10)Methyl 3-Bromo-4-Methylbenzoate (9)Methyl 3-Bromobenzoate (153)Methyl 3-Bromopicolinate (9)Methyl 4-Amino-3-Bromobenzoate (8)Methyl 5-Amino-2-Bromobenzoate (6)Methyl 6-Bromonicotinate (6)N-(2-Benzoylphenyl)-2-Bromoacetamide (3)Tert-Butyl 2-(Bromomethyl)Morpholine-4-Carboxylate (3)Tert-Butyl 3-Bromo-1H-Indole-1-Carboxylate (9)Tert-Butyl 3-Bromopiperidine-1-Carboxylate (4)
Carboxylic Acids (58085)
Alcohols (171)Aldehydes (18)Aliphatic Chain Hydrocarbons (159)Aliphatic Cyclic Hydrocarbons (65)Alkenyls (129)Alkynyls (13)Amides (226)Amines (178)Aryls (849)Azetidines (17)Benzimidazoles (6)Benzofurans (9)Benzothiazoles (5)Benzoxazoles (4)Bromides (176)Carboxylic Acid Salts (5)Chlorides (174)Difluoromethyls (19)Dioxolanes (4)Esters (73)Ethers (291)Fluorinated Building Blocks (237)Furans (35)Imidazoles (42)Imidazolidines (3)Indazoles (19)Indoles (52)Indolines (16)Iodides (24)Isoquinolines (4)Isoxazoles (18)Ketones (138)Morpholines (34)Naphthyridines (3)Nitriles (45)Nitroes (44)Oxadiazoles (4)Oxazoles (16)Oxetanes (7)Phenols (21)Piperazines (31)Piperidines (91)Pyrans (2)Pyrazines (12)Pyrazoles (71)Pyridazines (10)Pyridines (172)Pyrimidines (46)Pyrroles (28)Pyrrolidines (26)Quinazolines (4)Quinolines (90)Quinoxalines (5)Spiroes (20)Sulfamides (28)Sulfides (57)Sulfones (15)Tetrahydrofurans (7)Tetrahydropyrans (12)Thiazoles (62)Thiazolidines (10)Thioesters (7)Thiophenes (66)Triazoles (10)Trifluoromethyls (71)Ureas (4)Other Aliphatic Heterocycles (12)Other Aromatic Heterocycles (65)Aceticacid (1245)Acetylbenzoic Acid (3)Acrylic (95)Aminobenzoic (7)Aminobutanoic (8)Aminoheptanoic (6)Aminohexanoic (10)Aminonicotinic Acid (4)Aminopentanoic (8)Aminophenylacetic Acid (22)Aminopropanoic (8)Aminoundecanoic Acid (4)Benzenedicarboxylic (6)Benzenedicarboxylic Acid (6)Benzylpropanoic Acid (71)Benzylsuccinic Acid (6)Boronobenzoic (4)Bromobutanoic (5)Bromododecanoate (3)Bromoisonicotinic Acid (4)Bromonicotinic (8)Bromonicotinic Acid (9)Bromophenylacetic Acid (15)Bromovalerate (4)Butanoic (115)Chlorobenzoic (35)Chloromandelic Acid (3)Cyanonicotinic (5)Cyanonicotinic Acid (3)Diacetic (19)Diacetic (19)Diaminobenzoic (7)Diaminobutanoic (5)Diaminohexanoic (4)Dibenzoic (40)Dibromobenzoic (10)Dibromobenzoic Acid (10)Dibromonicotinic Acid (7)Dicarboxylic (183)Dicarboxylic (183)Dichlorobenzoic (21)Dichlorobenzoic Acid (22)Dichloroisonicotinic (6)Dichloroisonicotinic Acid (6)Dichlorophenylacetic Acid (4)Dienoate (14)Dienoic (10)Difluorobenzoic (54)Difluorobutanoic (3)Difluoroisonicotinic Acid (7)Difluorophenylacetic (7)Difluorophenylacetic Acid (7)Diiodobenzoic (5)Diisophthalic (17)Diisophthalic Acid (9)Diisopropylbenzoic Acid (4)Dimethoxybenzoic (33)Dimethoxyphenylacetic Acid (3)Dimethylbenzoic (32)Dimethylbutanoic (13)Dimethylbutanoic (13)Dimethylpropanoic (5)Diphenylpropanoic (4)Diyldimethanol (17)Enedioic (8)Enoic (127)Ethoxybenzoic (9)Ethylbenzoic (11)Ethylbenzoic Acid (12)Ethynylbenzoic (6)Ethynylbenzoic Acid (5)Fluoroisophthalic Acid (4)Fluorophenylacetic (10)Fluorophenylacetic Acid (11)Fluorophthalic (5)Fluorophthalic Acid (3)Fluoropicolinic (12)Formylbenzoic (22)Formylbenzoic Acid (19)Guanidinopentanoic (8)Heptanoic (6)Heptanoic (6)Hexacarboxylic Acid (5)Hexanoic (25)Hexanoic (25)Hydrazinylbenzoic (6)Hydrazinylbenzoic Acid (5)Hydroxynicotinic (15)Isophthalic (29)Isophthalic Acid (15)Isopropoxybenzoic (8)Isopropoxybenzoic Acid (7)Isopropylbenzoic (13)Isopropylnicotinic Acid (4)Malonate (39)Mercaptobenzoic (6)Mercaptobenzoic Acid (4)Mercaptopropanoic (11)Methoxybenzoic (142)Methoxybutanoic (4)Methoxybutanoic (4)Methoxyisophthalic Acid (5)Methoxyphenylacetic (10)Methoxyphenylacetic Acid (12)Methoxypropanoic (7)Methoxypropanoic (7)Methylbenzoic (174)Methylbutanoic (32)Methylbutanoic (32)Methylcinnamic Acid (9)Methylhexanoic (8)Methylhexanoic (8)Methylisophthalic Acid (17)Methylnicotinic (46)Methyloctanoic (6)Methylpentanoic (103)Methylpentanoic (103)Methylpropanoic (57)Naphthoic (64)Nitrobenzoic (161)Nitroisonicotinic Acid (5)Nitroisophthalic Acid (3)Nitronicotinic (10)Nitropicolinic (11)Octanoic (6)Octanoic (6)Oxoacetic (22)Oxobutanoic (54)Oxobutanoic (54)Oxodecanoic (5)Oxodecanoic (5)Oxododecanoic (9)Oxododecanoic (9)Oxoheptanoic (5)Oxoheptanoic (5)Oxoheptanoic Acid (4)Oxohexadecanoic (13)Oxohexadecanoic (13)Oxohexanoic (10)Oxohexanoic (10)Oxooctadecanoic (7)Oxooctadecanoic (7)Oxopentanoic (37)Oxopentanoic (37)Oxopropanoic (8)Pentanedioic (17)Pentanedioic (17)Pentanoic (27)Phenoxybenzoic (5)Phenylacetic (35)Phenylbutanoic (41)Phenylpentanoic (11)Phenylpropanoic (26)Propanoic (996)Propenoic (6)Succinic (13)Sulfamoylbenzoic (15)Sulfobenzoic (5)Terephthalic (7)Tetraacetic (5)Tetrabenzoic (13)Tetrabenzoic Acid (14)Tetracarboxylic (13)Tribenzoic (15)Tribenzoic Acid (14)Tricarboxylic (18)Trichlorobenzoic (5)Trichlorobenzoic Acid (4)Trifluorobenzoic (12)Trifluorobenzoic Acid (11)Trifluorobutanoic (3)Trifluoromethylbenzoic (5)Trifluoromethylbenzoic Acid (4)Trifluoropentanoic (4)Trifluorophenylacetic Acid (16)Trifluoropropanoic (4)Triiodobenzoic (4)Trimethylbenzoic Acid (4)1,2,3,4-Tetrahydroisoquinoline-1-Carboxylic Acid (19)1,2,3,4-Tetrahydroisoquinoline-3-Carboxylic Acid (12)1,5-Naphthyridine-3-Carboxylic Acid (6)1-((Benzyloxy)Carbonyl)Piperidine-2-Carboxylic Acid (5)1-((Benzyloxy)Carbonyl)Piperidine-3-Carboxylic Acid (3)1-((Benzyloxy)Carbonyl)Pyrrolidine-3-Carboxylic Acid (6)1-Methyl-1H-Indole-2-Carboxylic Acid (8)1-Naphthoic Acid (23)1-Phenyl-1H-Imidazole-4-Carboxylic Acid (5)1-Phenyl-1H-Pyrazole-3-Carboxylic Acid (10)1-Phenyl-1H-Pyrazole-4-Carboxylic Acid (19)1-Phenyl-1H-Pyrazole-5-Carboxylic Acid (3)1-Phenylcyclobutane-1-Carboxylic Acid (16)1-Phenylcyclohexane-1-Carboxylic Acid (6)1-Phenylcyclopropane-1-Carboxylic Acid (23)1H-Benzo[D]Imidazole-2-Carboxylic Acid (7)1H-Benzo[D]Imidazole-4-Carboxylic Acid (6)1H-Benzo[D]Imidazole-5-Carboxylic Acid (4)1H-Imidazole-2-Carboxylic Acid (7)1H-Imidazole-4-Carboxylic Acid (4)1H-Indazole-3-Carboxylic Acid (23)1H-Indazole-4-Carboxylic Acid (9)1H-Indazole-5-Carboxylic Acid (4)1H-Indazole-7-Carboxylic Acid (6)1H-Indole-3-Carboxylic Acid (28)1H-Indole-4-Carboxylic Acid (10)1H-Indole-5-Carboxylic Acid (4)1H-Indole-6-Carboxylic Acid (5)1H-Indole-7-Carboxylic Acid (7)1H-Pyrazole-4-Carboxylic Acid (9)1H-Pyrazole-5-Carboxylic Acid (10)1H-Pyrrole-2-Carboxylic Acid (30)1H-Pyrrolo[2,3-B]Pyridine-3-Carboxylic Acid (5)1H-Pyrrolo[2,3-C]Pyridine-2-Carboxylic Acid (4)1H-Pyrrolo[3,2-B]Pyridine-3-Carboxylic Acid (6)2,3-Difluorobenzoic Acid (35)2,3-Dihydrobenzo[B][1,4]Dioxine-2-Carboxylic Acid (3)2,3-Dihydrobenzo[B][1,4]Dioxine-6-Carboxylic Acid (3)2,4-Difluorobenzoic Acid (29)2,5-Dichlorobenzoic Acid (13)2,5-Difluorobenzoic Acid (32)2,6-Dichlorobenzoic Acid (13)2,6-Difluorobenzoic Acid (33)2-(1,3-Dioxoisoindolin-2-Yl)Propanoic Acid (3)2-(1H-Indol-1-Yl)Acetic Acid (5)2-(1H-Indol-3-Yl)Acetic Acid (28)2-(2-Bromophenyl)Acetic Acid (23)2-(2-Chlorophenyl)Acetic Acid (24)2-(2-Fluorophenyl)Acetic Acid (43)2-(2-Hydroxyphenyl)Acetic Acid (8)2-(2-Methoxyphenyl)Acetic Acid (16)2-(2-Methyl-1H-Indol-3-Yl)Acetic Acid (5)2-(2-Nitrophenyl)Acetic Acid (20)2-(2-Oxo-2H-Chromen-4-Yl)Acetic Acid (4)2-(3-Fluorophenyl)Acetic Acid (50)2-(3-Hydroxyphenyl)Acetic Acid (8)2-(3-Methoxyphenyl)Acetic Acid (23)2-(4-Chlorophenoxy)Acetic Acid (3)2-(4-Fluorophenyl)Acetic Acid (30)2-(4-Methoxyphenyl)Acetic Acid (21)2-(4-Nitrophenyl)Acetic Acid (9)2-(Benzofuran-3-Yl)Acetic Acid (3)2-(M-Tolyl)Acetic Acid (16)2-(Morpholin-2-Yl)Acetic Acid (5)2-(O-Tolyl)Acetic Acid (17)2-(P-Tolyl)Acetic Acid (9)2-(Phenylamino)Benzoic Acid (11)2-(Pyridin-3-Yl)Acetic Acid (14)2-(Pyrrolidin-1-Yl)Benzoic Acid (3)2-(Pyrrolidin-2-Yl)Acetic Acid (8)2-(Pyrrolidin-3-Yl)Acetic Acid (6)2-(Thiazol-4-Yl)Acetic Acid (9)2-(Trifluoromethoxy)Benzoic Acid (8)2-(Trifluoromethyl)Benzoic Acid (33)2-Amino-2-(2-Fluorophenyl)Acetic Acid (11)2-Amino-3-Bromobenzoic Acid (8)2-Amino-3-Chlorobenzoic Acid (7)2-Amino-6-Chlorobenzoic Acid (5)2-Benzoylbenzoic Acid (10)2-Bromo-4-Fluorobenzoic Acid (8)2-Bromo-4-Methylbenzoic Acid (5)2-Bromo-6-Fluorobenzoic Acid (11)2-Bromo-6-Methylbenzoic Acid (5)2-Chloro-3-Methoxybenzoic Acid (6)2-Chloro-3-Methylbenzoic Acid (4)2-Chloro-3-Nitrobenzoic Acid (4)2-Chloro-4-Fluorobenzoic Acid (9)2-Chloro-4-Methylbenzoic Acid (3)2-Chloro-6-Fluorobenzoic Acid (8)2-Chlorobenzoic Acid (130)2-Cyclohexylacetic Acid (14)2-Fluoro-4-Methylbenzoic Acid (7)2-Fluoro-5-Methoxybenzoic Acid (9)2-Fluorobenzoic Acid (220)2-Fluorocyclopropane-1-Carboxylic Acid (7)2-Iodobenzoic Acid (47)2-Methoxyisonicotinic Acid (13)2-Methyl-2-Phenylpropanoic Acid (20)2-Methyl-3-Nitrobenzoic Acid (7)2-Naphthoic Acid (14)2-Nitrobenzoic Acid (65)2-Oxo-2-Phenylacetic Acid (17)2-Oxo-2H-Chromene-3-Carboxylic Acid (14)2-Phenoxypropanoic Acid (13)3-(1H-Indol-3-Yl)Propanoic Acid (4)3-(2-Fluorophenyl)Acrylic Acid (17)3-(3-Chlorophenyl)Acrylic Acid (10)3-(3-Chlorophenyl)Propanoic Acid (11)3-(3-Methoxyphenyl)Propanoic Acid (9)3-(Benzyloxy)Benzoic Acid (5)3-(Chlorosulfonyl)Benzoic Acid (15)3-(Methylsulfonyl)Benzoic Acid (6)3-(Phenyldiazenyl)Benzoic Acid (7)3-(Piperazin-1-Yl)Benzoic Acid (3)3-(Trifluoromethoxy)Benzoic Acid (19)3-(Trifluoromethyl)Benzoic Acid (52)3-Benzamidopropanoic Acid (3)3-Bromo-2-Chlorobenzoic Acid (7)3-Bromo-2-Fluorobenzoic Acid (10)3-Bromo-2-Hydroxybenzoic Acid (4)3-Bromo-4-Chlorobenzoic Acid (6)3-Bromo-4-Fluorobenzoic Acid (10)3-Bromo-4-Hydroxybenzoic Acid (3)3-Bromo-4-Methoxybenzoic Acid (7)3-Bromo-4-Methylbenzoic Acid (9)3-Bromobenzoic Acid (159)3-Chloro-2-Fluorobenzoic Acid (9)3-Chloro-2-Methoxybenzoic Acid (4)3-Chloro-4-Fluorobenzoic Acid (8)3-Chloro-4-Methoxybenzoic Acid (5)3-Chlorobenzoic Acid (144)3-Fluoro-4-Methoxybenzoic Acid (6)3-Fluorobenzoic Acid (206)3-Fluoropicolinic Acid (6)3-Iodobenzoic Acid (55)3-Methoxy-4-Nitrobenzoic Acid (4)3-Methylpicolinic Acid (13)3-Nitrobenzoic Acid (109)3-Phenoxybenzoic Acid (6)3-Phenoxypropanoic Acid (14)4-(1H-Pyrazol-1-Yl)Benzoic Acid (3)4-(Benzyloxy)Benzoic Acid (5)4-(Phenyldiazenyl)Benzoic Acid (4)4-(Trifluoromethyl)Nicotinic Acid (3)4-Amino-3-Chlorobenzoic Acid (6)4-Benzamidobenzoic Acid (3)4-Cyclohexylbenzoic Acid (6)4-Fluoro-3-Iodobenzoic Acid (5)4-Fluorobenzoic Acid (114)4-Methylnicotinic Acid (5)4-Morpholinobenzoic Acid (5)4-Nitrobenzoic Acid (35)4-Oxo-1,4-Dihydroquinoline-3-Carboxylic Acid (3)4-Oxo-4-(Phenylamino)Butanoic Acid (7)4-Oxo-4-Phenylbutanoic Acid (16)4-Oxopiperidine-2-Carboxylic Acid (3)4-Phenoxybenzoic Acid (4)4-Phenylbutanoic Acid (19)5'-(4-Carboxyphenyl)-[1,1':3',1''-Terphenyl]-4,4''-Dicarboxylic Acid (7)5-Oxotetrahydrofuran-2-Carboxylic Acid (4)5-Phenyl-1H-Pyrazole-3-Carboxylic Acid (15)5-Phenylfuran-2-Carboxylic Acid (6)5-Phenylisoxazole-3-Carboxylic Acid (15)6,6-Dimethylmorpholine-3-Carboxylic Acid (5)6-Methoxypicolinic Acid (9)6-Methylnicotinic Acid (6)6-Methylpicolinic Acid (12)6-Oxopiperidine-2-Carboxylic Acid (3)6-Phenylpicolinic Acid (3)7-Fluoro-1H-Indole-2-Carboxylic Acid (5)[1,1'-Biphenyl]-2-Carboxylic Acid (24)[1,1'-Biphenyl]-3-Carboxylic Acid (40)Azetidine-2-Carboxylic Acid (6)Benzo[B]Thiophene-2-Carboxylic Acid (32)Benzo[D]Thiazole-6-Carboxylic Acid (5)Benzofuran-2-Carboxylic Acid (21)Benzofuran-3-Carboxylic Acid (6)Benzoic Acid (3487)Benzoylglycine (15)Bicyclo[1.1.1]Pentane-1-Carboxylic Acid (6)Chromane-2-Carboxylic Acid (6)Chromane-3-Carboxylic Acid (4)Cyclobutanecarboxylic Acid (23)Cyclohex-1-Ene-1-Carboxylic Acid (4)Cyclohex-3-Ene-1-Carboxylic Acid (7)Cyclohexanecarboxylic Acid (83)Cyclopentanecarboxylic Acid (26)Cyclopropanecarboxylic Acid (11)Ethyl 5-Oxopyrrolidine-2-Carboxylate (3)Furan-2-Carboxylic Acid (32)Furan-3-Carboxylic Acid (11)Imidazo[1,2-A]Pyridine-2-Carboxylic Acid (21)Imidazo[1,2-A]Pyridine-3-Carboxylic Acid (14)Imidazo[1,2-A]Pyridine-5-Carboxylic Acid (3)Imidazo[1,2-A]Pyridine-7-Carboxylic Acid (5)Imidazo[1,2-A]Pyridine-8-Carboxylic Acid (6)Imidazo[1,2-A]Pyrimidine-3-Carboxylic Acid (5)Indoline-2-Carboxylic Acid (3)Isonicotinic Acid (23)Isoquinoline-1-Carboxylic Acid (10)Isoquinoline-3-Carboxylic Acid (7)Isoquinoline-4-Carboxylic Acid (12)Isoquinoline-5-Carboxylic Acid (5)Isoquinoline-6-Carboxylic Acid (3)Isoxazole-3-Carboxylic Acid (8)Isoxazole-5-Carboxylic Acid (7)Methyl Benzofuran-2-Carboxylate (6)Methyl Isoquinoline-3-Carboxylate (3)Morpholine-2-Carboxylic Acid (12)Morpholine-3-Carboxylic Acid (10)Nicotinic Acid (53)Oxazole-4-Carboxylic Acid (4)Picolinic Acid (22)Piperidine-2-Carboxylic Acid (26)Piperidine-3-Carboxylic Acid (14)Piperidine-4-Carboxylic Acid (12)Proline (13)Pyrazine-2-Carboxylic Acid (34)Pyrazolo[1,5-A]Pyrimidine-3-Carboxylic Acid (7)Pyridazine-3-Carboxylic Acid (7)Pyridine-2,6-Dicarboxylic Acid (9)Pyrimidine-2-Carboxylic Acid (11)Pyrimidine-4-Carboxylic Acid (17)Pyrimidine-5-Carboxylic Acid (7)Pyrrolidine-3-Carboxylic Acid (14)Tetrahydrofuran-2-Carboxylic Acid (4)Tetrahydrofuran-3-Carboxylic Acid (4)Thiazole-2-Carboxylic Acid (9)Thiazole-4-Carboxylic Acid (5)Thiazole-5-Carboxylic Acid (10)Thiophene-3-Carboxylic Acid (8)Tryptophan (22)
Chlorides (77789)
Acyl Chlorides (88)Alcohols (157)Aldehydes (88)Aliphatic Chain Hydrocarbons (143)Aliphatic Cyclic Hydrocarbons (19)Alkenyls (57)Alkynyls (18)Amides (372)Amidines (9)Amines (309)Anhydrides (8)Aryls (1047)Azetidines (10)Benzimidazoles (15)Benzofurans (15)Benzothiazoles (6)Benzoxazoles (11)Benzyl Bromides (6)Benzyl Chlorides (49)Bromides (241)Carboxylic Acids (174)Difluoromethyls (22)Esters (298)Ethers (285)Fluorinated Building Blocks (224)Furans (9)Hydrazides (15)Hydrazines (26)Hydrazones (4)Imidazoles (15)Indazoles (10)Indoles (27)Indolines (14)Iodides (51)Isoquinolines (14)Isoxazoles (19)Ketones (139)Morpholines (24)Nitriles (97)Nitroes (81)Oxazoles (14)Oximes (7)Phenols (26)Piperazines (22)Piperidines (68)Purines (6)Pyrazines (31)Pyrazoles (27)Pyridazines (32)Pyridines (372)Pyrimidines (209)Pyrroles (13)Pyrrolidines (49)Quinazolines (20)Quinolines (110)Quinoxalines (9)Spiroes (4)Sulfamides (51)Sulfides (50)Sulfones (29)Sulfoxides (7)Tetrahydrofurans (3)Tetrahydroisoquinolines (4)Tetrahydropyrans (5)Thiadiazoles (4)Thiazoles (29)Thiophenes (49)Triazines (13)Trifluoromethyls (61)Ureas (7)Other Aliphatic Heterocycles (3)Other Aromatic Heterocycles (149)Chloroacetamide (17)Chloroacetyl (10)Chloroaniline (18)Chlorobenzaldehyde (18)Chlorobenzamide (12)Chlorobutyl (6)Chlorobutyl (6)Chlorocarbonyl (7)Chloroethoxy (10)Chloroethoxy (10)Chloroethyl (89)Chloromethyl (478)Chloropicolinaldehyde (8)Chloropicolinamide (5)Chloropropane (18)Chloropropane (18)Chloropropoxy (2)Chloropropoxy (2)Chloropropyl (23)Dichloride (28)Dichloroaniline (9)Dichloroanthracene (3)Dichlorobenzaldehyde (13)Dichlorobenzamide (8)Dichloroisonicotinaldehyde (4)Dichloromethyl (3)Dichloromethyl (3)Dichloronicotinaldehyde (6)Dichloronicotinic (6)Dichloronicotinic (6)Dichloronicotinic Acid (5)Dichloropalladium (4)Dichloropalladium (4)Tetrachloro (2)Tetrachloro (2)Trichloro (36)Trichloro (36)Trichloroacetimidate (5)Trichloroaniline (6)Trichlorobenzaldehyde (4)Trichloromethyl (11)Trichloromethyl (11)(2,3-Dichlorophenyl)Boronic Acid (11)(2,6-Dichlorophenyl)Boronic Acid (6)(2-Chloroethoxy)Benzene (5)(2-Chlorophenyl)(Phenyl)Methanone (19)(2-Chlorophenyl)Hydrazine (16)(2-Chlorophenyl)Methanamine (28)(2-Chlorophenyl)Methanol (40)(2-Chloropyridin-4-Yl)Boronic Acid (6)(3,4-Dichlorophenyl)Boronic Acid (10)(3-Chloro-4-Fluorophenyl)Boronic Acid (8)(3-Chlorophenyl)(Phenyl)Methanone (19)(3-Chlorophenyl)Boronic Acid (121)(3-Chlorophenyl)Hydrazine (12)(3-Chlorophenyl)Methanamine (19)(3-Chlorophenyl)Methanol (38)(3-Chloropyridin-2-Yl)Methanamine (6)(4-Chlorophenyl)(Phenyl)Methanone (11)(5-Chloropyridin-2-Yl)Methanamine (5)(Chloromethanetriyl)Tribenzene (7)(Chloromethylene)Dibenzene (5)1,2,3,4-Tetrachlorobenzene (8)1,2,4-Trichlorobenzene (26)1,2-Dibromo-3-Chlorobenzene (3)1,2-Dichloro-3-Fluorobenzene (13)1,2-Dichloro-3-Methoxybenzene (7)1,2-Dichloro-3-Methylbenzene (8)1,2-Dichloro-4-Fluorobenzene (15)1,2-Dichloro-4-Methoxybenzene (11)1,2-Dichloro-4-Nitrobenzene (16)1,3-Dibromo-2-Chlorobenzene (8)1,3-Dichloro-2-Fluorobenzene (23)1,3-Dichloro-2-Iodobenzene (5)1,3-Dichloro-2-Methoxybenzene (9)1,3-Dichloro-2-Nitrobenzene (5)1,3-Dichloroisoquinoline (14)1,4-Dichloro-2-Fluorobenzene (11)1,4-Dichloro-2-Methoxybenzene (8)1-(2-Chlorophenyl)Ethan-1-One (40)1-(2-Chlorophenyl)Piperazine (4)1-(3-Chlorophenyl)-1H-Pyrazole (3)1-(3-Chlorophenyl)-N-Methylmethanamine (8)1-(3-Chlorophenyl)Ethan-1-One (38)1-(3-Chlorophenyl)Pyrrolidine (3)1-(Benzyloxy)-2-Chlorobenzene (16)1-(Benzyloxy)-3-Chlorobenzene (6)1-(Bromomethyl)-2-Chlorobenzene (28)1-(Bromomethyl)-3-Chlorobenzene (27)1-(Chloromethyl)-2-Methylbenzene (7)1-Benzyl-2-Chlorobenzene (9)1-Benzyl-3-Chlorobenzene (3)1-Benzyl-4-Chlorobenzene (6)1-Bromo-2,3-Dichlorobenzene (18)1-Bromo-2-Chloro-3-Methylbenzene (9)1-Bromo-3-Chloro-2-Fluorobenzene (9)1-Bromo-3-Chloro-2-Methylbenzene (6)1-Bromo-4-Chloro-2-Fluorobenzene (22)1-Chloro-2,3-Difluorobenzene (27)1-Chloro-2,4-Difluorobenzene (35)1-Chloro-2,4-Dimethoxybenzene (7)1-Chloro-2-(Chloromethyl)Benzene (12)1-Chloro-2-(Trifluoromethoxy)Benzene (20)1-Chloro-2-(Trifluoromethyl)Benzene (81)1-Chloro-2-Cyclopropylbenzene (7)1-Chloro-2-Ethoxybenzene (14)1-Chloro-2-Fluoro-3-Iodobenzene (5)1-Chloro-2-Fluoro-4-Nitrobenzene (9)1-Chloro-2-Iodobenzene (65)1-Chloro-2-Isocyanatobenzene (6)1-Chloro-2-Isothiocyanatobenzene (7)1-Chloro-2-Methyl-3-Nitrobenzene (6)1-Chloro-2-Nitrobenzene (138)1-Chloro-2-Phenoxybenzene (10)1-Chloro-2-Propoxybenzene (5)1-Chloro-3,5-Dimethylbenzene (8)1-Chloro-3-(Chloromethyl)Benzene (7)1-Chloro-3-(Trifluoromethoxy)Benzene (18)1-Chloro-3-(Trifluoromethyl)Benzene (101)1-Chloro-3-Fluoro-2-Methylbenzene (10)1-Chloro-3-Fluoro-2-Nitrobenzene (6)1-Chloro-3-Iodo-2-Methylbenzene (3)1-Chloro-3-Iodobenzene (78)1-Chloro-3-Methoxybenzene (125)1-Chloro-3-Nitrobenzene (161)1-Chloro-3-Phenoxybenzene (5)1-Chloro-4-(Trifluoromethoxy)Benzene (8)1-Chloro-4-Iodobenzene (49)1-Chloro-4-Methoxybenzene (98)1-Chloro-4-Phenoxybenzene (13)1-Chloroanthracene-9,10-Dione (6)1-Chloroisoquinoline (20)1-Chloronaphthalene (24)2,3,5-Trichloropyridine (13)2,3,6-Trichloropyridine (16)2,3-Dichloroaniline (18)2,3-Dichlorobenzaldehyde (7)2,3-Dichlorophenol (9)2,3-Dichloropyridine (38)2,4,5-Trichloropyrimidine (6)2,4,6-Trichloropyridine (5)2,4,6-Trichloropyrimidine (14)2,4-Dibromo-1-Chlorobenzene (8)2,4-Dichloro-1-Fluorobenzene (27)2,4-Dichloro-1-Methoxybenzene (18)2,4-Dichloro-1-Nitrobenzene (19)2,4-Dichloro-6-Methylpyridine (8)2,4-Dichloro-6-Methylpyrimidine (7)2,4-Dichlorobenzaldehyde (8)2,4-Dichlorobenzonitrile (4)2,4-Dichloropyridine (50)2,4-Dichloroquinazoline (19)2,4-Dichlorothiazole (4)2,5-Dichlorophenol (7)2,5-Dichloropyrazine (4)2,5-Dichloropyridine (32)2,6-Dichloro-3-Fluoropyridine (10)2,6-Dichloro-3-Methylpyridine (5)2,6-Dichloro-3-Nitropyridine (6)2,6-Dichloro-4-Methylpyridine (5)2,6-Dichloroaniline (27)2,6-Dichlorobenzoic Acid (13)2,6-Dichlorobenzonitrile (8)2,6-Dichloronicotinonitrile (8)2,6-Dichlorophenol (16)2,6-Dichloropyrazine (14)2,6-Dichloropyridin-3-Amine (11)2,6-Dichloropyridine (49)2-(2-Chlorophenyl)Acetic Acid (24)2-(2-Chlorophenyl)Acetonitrile (16)2-(2-Chlorophenyl)Ethan-1-Ol (8)2-(2-Chlorophenyl)Pyridine (3)2-(2-Chlorophenyl)Pyrrolidine (17)2-(3-Chlorophenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (8)2-(3-Chlorophenyl)Acetonitrile (16)2-(4-Chlorophenoxy)Acetic Acid (3)2-(Chloromethyl)-1H-Benzo[D]Imidazole (9)2-(Chloromethyl)-3-Methylpyridine (12)2-(Chloromethyl)Furan (3)2-(Chloromethyl)Pyridine (12)2-(Chloromethyl)Pyridine 1-Oxide (4)2-(Chloromethyl)Pyrimidine (5)2-(Chloromethyl)Thiazole (5)2-Amino-3-Chlorobenzoic Acid (7)2-Amino-3-Chlorobenzonitrile (3)2-Amino-6-Chlorophenol (5)2-Bromo-1,3-Dichlorobenzene (12)2-Bromo-1,4-Dichlorobenzene (12)2-Bromo-1-Chloro-4-Methylbenzene (6)2-Bromo-4-Chloro-1-Iodobenzene (6)2-Bromo-4-Chloro-1-Methoxybenzene (7)2-Bromo-4-Chloro-1-Methylbenzene (11)2-Bromo-4-Chloroaniline (15)2-Bromo-4-Chlorophenol (11)2-Bromo-6-Chloroaniline (10)2-Bromo-6-Chlorophenol (5)2-Chloro-1,1'-Biphenyl (8)2-Chloro-1,3,5-Triazine (10)2-Chloro-1,3-Difluorobenzene (27)2-Chloro-1,3-Dimethoxybenzene (3)2-Chloro-1,3-Dinitrobenzene (2)2-Chloro-1,4-Difluorobenzene (25)2-Chloro-1,4-Dimethoxybenzene (4)2-Chloro-1-Fluoro-4-Nitrobenzene (16)2-Chloro-1-Methyl-3-Nitrobenzene (4)2-Chloro-1-Phenylethan-1-One (20)2-Chloro-1H-Benzo[D]Imidazole (17)2-Chloro-3,6-Dimethylpyridine (4)2-Chloro-3-(Trifluoromethyl)Aniline (3)2-Chloro-3-(Trifluoromethyl)Pyridine (12)2-Chloro-3-Fluoropyridin-4-Amine (3)2-Chloro-3-Hydroxybenzaldehyde (4)2-Chloro-3-Methoxypyridine (12)2-Chloro-3-Methylaniline (4)2-Chloro-3-Methylbenzoic Acid (4)2-Chloro-3-Methylbenzonitrile (5)2-Chloro-3-Methylpyrazine (7)2-Chloro-3-Methylpyridine (17)2-Chloro-3-Nitropyridine (10)2-Chloro-4,6-Dimethylpyridine (7)2-Chloro-4-(Trifluoromethyl)Pyridine (9)2-Chloro-4-Ethoxypyridine (5)2-Chloro-4-Fluoro-1-Nitrobenzene (13)2-Chloro-4-Fluoroaniline (19)2-Chloro-4-Fluoropyridine (10)2-Chloro-4-Methoxypyridine (22)2-Chloro-4-Methyl-1-Nitrobenzene (6)2-Chloro-4-Methylaniline (7)2-Chloro-4-Methylbenzoic Acid (3)2-Chloro-4-Methylpyridine (31)2-Chloro-4-Methylpyrimidine (16)2-Chloro-4-Methylthiazole (5)2-Chloro-4-Phenylpyrimidine (7)2-Chloro-5-(Trifluoromethyl)Pyridine (4)2-Chloro-5-Fluoropyridin-4-Amine (3)2-Chloro-5-Fluoropyridine (16)2-Chloro-5-Methoxyaniline (8)2-Chloro-5-Methylpyridine (11)2-Chloro-5-Methylpyrimidine (5)2-Chloro-5-Methylthiazole (5)2-Chloro-5-Nitropyridine (8)2-Chloro-6-(Trifluoromethyl)Aniline (4)2-Chloro-6-(Trifluoromethyl)Pyridine (16)2-Chloro-6-Fluoroaniline (8)2-Chloro-6-Fluorophenol (5)2-Chloro-6-Fluoropyridine (14)2-Chloro-6-Iodophenol (3)2-Chloro-6-Methoxyaniline (3)2-Chloro-6-Methoxyphenol (4)2-Chloro-6-Methoxypyridine (24)2-Chloro-6-Methyl-3-Nitropyridine (4)2-Chloro-6-Methylaniline (11)2-Chloro-6-Methylnicotinonitrile (10)2-Chloro-6-Methylphenol (5)2-Chloro-6-Methylpyrazine (12)2-Chloro-6-Methylpyridine (49)2-Chloro-6-Nitroaniline (11)2-Chloro-7H-Purine (3)2-Chloro-7H-Pyrrolo[2,3-D]Pyrimidine (13)2-Chloro-9H-Purine (5)2-Chloro-N-Phenylacetamide (26)2-Chloro-N-Phenylaniline (8)2-Chloroaniline (214)2-Chlorobenzamide (16)2-Chlorobenzene-1,3-Diamine (3)2-Chlorobenzene-1,3-Diol (4)2-Chlorobenzene-1,4-Diamine (6)2-Chlorobenzenesulfonyl Chloride (24)2-Chlorobenzenethiol (7)2-Chlorobenzo[D]Oxazole (11)2-Chlorobenzo[D]Thiazole (21)2-Chlorobenzoic Acid (130)2-Chlorobenzonitrile (99)2-Chlorobenzoyl Chloride (13)2-Chlorocyclohexa-2,5-Diene-1,4-Dione (4)2-Chlorofuran (3)2-Chloroisonicotinaldehyde (19)2-Chloroisonicotinic Acid (17)2-Chloroisonicotinonitrile (7)2-Chloronaphthalene (6)2-Chloronicotinic Acid (7)2-Chloronicotinonitrile (9)2-Chlorooxazole (7)2-Chlorophenol (163)2-Chloropyrazine (24)2-Chloropyridin-3-Amine (11)2-Chloropyridin-3-Ol (9)2-Chloropyridin-4-Amine (28)2-Chloropyridine 1-Oxide (10)2-Chloroquinoxaline (39)2-Chlorothiazole (32)2-Chlorothiophene (20)3,4-Dichloroaniline (21)3,5-Dichloropyridine (16)3,6-Dichloropyridazine (20)3-(3-Chlorophenyl)Propanoic Acid (11)3-(Chloromethyl)Pyridine (22)3-Chloro-1,1'-Biphenyl (12)3-Chloro-1-Methyl-1H-Pyrazole (7)3-Chloro-1-Phenylpropan-1-One (10)3-Chloro-1H-Indazole (13)3-Chloro-2-Fluoroaniline (9)3-Chloro-2-Fluorobenzaldehyde (9)3-Chloro-2-Fluorobenzoic Acid (9)3-Chloro-2-Fluorobenzonitrile (7)3-Chloro-2-Fluoropyridine (13)3-Chloro-2-Hydroxybenzaldehyde (5)3-Chloro-2-Methoxypyridine (16)3-Chloro-2-Methylaniline (11)3-Chloro-2-Methylpyridine (4)3-Chloro-2-Nitroaniline (5)3-Chloro-4-(Trifluoromethyl)Aniline (5)3-Chloro-4-Fluoroaniline (18)3-Chloro-4-Fluorobenzoic Acid (8)3-Chloro-4-Hydroxybenzaldehyde (7)3-Chloro-4-Methylaniline (12)3-Chloro-4-Methylpyridazine (7)3-Chloro-4-Nitroaniline (5)3-Chloro-5-Fluoropyridine (10)3-Chloro-5-Methylpyridine (7)3-Chloro-5-Nitropyridine (5)3-Chloro-9H-Carbazole (3)3-Chlorobenzaldehyde (106)3-Chlorobenzene-1,2-Diamine (6)3-Chlorobenzene-1,2-Diol (6)3-Chlorobenzenesulfonyl Chloride (21)3-Chlorobenzo[B]Thiophene (6)3-Chlorobenzoic Acid (144)3-Chlorobenzonitrile (100)3-Chloroimidazo[1,2-A]Pyridine (3)3-Chloroisoquinoline (20)3-Chlorophenol (112)3-Chloropicolinic Acid (9)3-Chloropicolinonitrile (6)3-Chloropyridazine (44)3-Chloropyridin-2-Amine (17)3-Chloropyridin-2-Ol (10)3-Chlorothiophene (17)4,5-Dichloropyrimidine (7)4,6-Dichloro-2-Methylpyrimidine (7)4,6-Dichloropyridin-2-Amine (6)4,6-Dichloropyridin-3-Amine (4)4,6-Dichloropyrimidine (14)4-Chloro-1,5-Naphthyridine (6)4-Chloro-1-Phenylbutan-1-One (8)4-Chloro-1H-Indazole (18)4-Chloro-1H-Indole (21)4-Chloro-1H-Pyrazole (10)4-Chloro-1H-Pyrazolo[3,4-B]Pyridine (5)4-Chloro-1H-Pyrazolo[3,4-D]Pyrimidine (5)4-Chloro-1H-Pyrazolo[4,3-C]Pyridine (7)4-Chloro-1H-Pyrrolo[3,2-C]Pyridine (10)4-Chloro-2,6-Dimethylpyridine (4)4-Chloro-2-Methoxypyridine (6)4-Chloro-2-Phenylpyrimidine (5)4-Chloro-3-Fluoroaniline (17)4-Chloro-3-Methylaniline (11)4-Chloro-3-Methylpyridine (7)4-Chloro-5-Methylpyrimidine (8)4-Chloro-N-Phenylaniline (6)4-Chlorobenzene-1,3-Diol (6)4-Chlorobenzenesulfonic Acid (4)4-Chlorobenzenesulfonyl Chloride (12)4-Chlorobenzo[D]Thiazole (7)4-Chloroindoline-2,3-Dione (6)4-Chloroisoquinoline (11)4-Chloronicotinic Acid (4)4-Chloropyridazine (16)4-Chloropyridin-2-Amine (6)4-Chloropyridine (31)4-Chloroquinazoline (39)5,6-Dichloro-1H-Benzo[D]Imidazole (4)5,6-Dichloropyridin-2-Amine (4)5-Bromo-4-Chloropyrimidine (4)5-Chloro-1,2,3,4-Tetrahydroisoquinoline (3)5-Chloro-1H-Imidazole (2)5-Chloro-1H-Indazole (9)5-Chloro-1H-Indole (64)5-Chloro-1H-Indole-2-Carboxylic Acid (6)5-Chloro-1H-Pyrazole (8)5-Chloro-1H-Pyrazolo[4,3-B]Pyridine (6)5-Chloro-1H-Pyrrolo[2,3-B]Pyridine (13)5-Chloro-1H-Pyrrolo[3,2-B]Pyridine (6)5-Chloro-2,3-Dihydrobenzofuran (5)5-Chloro-2-Fluoropyridine (13)5-Chloro-2-Methoxypyridine (10)5-Chloro-2-Methylpyridine (10)5-Chlorobenzo[B]Thiophene (4)5-Chlorobenzo[D]Oxazole (5)5-Chlorobenzo[D]Thiazole (5)5-Chloroimidazo[1,2-A]Pyridine (3)5-Chloroindolin-2-One (3)5-Chloroisoquinoline (6)5-Chloropicolinic Acid (7)5-Chloropicolinonitrile (6)5-Chloropyrazolo[1,5-A]Pyrimidine (24)5-Chloropyridin-2-Amine (26)5-Chloropyridin-2-Ol (15)5-Chloropyridin-3-Amine (7)5-Chloropyrimidine (8)5-Chloroquinazoline (4)6-Chloro-1,2,3,4-Tetrahydroisoquinoline (3)6-Chloro-1,2,3,4-Tetrahydroquinoline (3)6-Chloro-1H-Benzo[D]Imidazole (25)6-Chloro-1H-Indazole (14)6-Chloro-1H-Indole (19)6-Chloro-1H-Pyrazolo[4,3-C]Pyridine (8)6-Chloro-1H-Pyrrolo[2,3-B]Pyridine (13)6-Chloro-1H-Pyrrolo[3,2-C]Pyridine (13)6-Chloro-2,3-Dihydrobenzofuran (3)6-Chloro-3,4-Dihydro-2H-Benzo[B][1,4]Oxazine (5)6-Chloro-3-Nitropyridin-2-Amine (4)6-Chloro-5-(Trifluoromethyl)Pyridin-2-Amine (3)6-Chlorobenzo[D]Oxazole (4)6-Chlorobenzo[D]Thiazole (6)6-Chlorochroman-4-One (6)6-Chlorochromane (6)6-Chloroindoline (3)6-Chloroindoline-2,3-Dione (5)6-Chloronicotinaldehyde (24)6-Chloronicotinic Acid (4)6-Chloropicolinic Acid (12)6-Chloropicolinonitrile (9)6-Chloropyridazin-3(2H)-One (5)6-Chloropyridin-2-Amine (21)6-Chloropyridin-2-Ol (9)6-Chloropyridin-3-Amine (4)6-Chloropyridine-3-Sulfonyl Chloride (5)6-Chloroquinoxaline (3)7-Chloro-1,2,3,4-Tetrahydroisoquinoline (5)7-Chloro-1H-Indazole (7)7-Chloro-1H-Indole (29)7-Chloro-1H-Pyrrolo[2,3-C]Pyridine (5)7-Chloro-2,3-Dihydrobenzofuran (3)7-Chloro-3,4-Dihydronaphthalen-1(2H)-One (6)7-Chlorobenzo[D]Thiazole (6)7-Chlorochromane (5)7-Chloroisoquinoline (6)7-Chloropyrazolo[1,5-A]Pyrimidine (8)8-Chlorochromane (5)8-Chloroimidazo[1,2-A]Pyridine (7)8-Chloroisoquinoline (7)8-Chloroquinazoline (5)Ethyl 4,6-Dichloronicotinate (4)Ethyl 5-Chloro-1H-Indole-2-Carboxylate (5)Ethyl 6-Chloropicolinate (7)Ethyl 7-Chloro-1H-Indole-2-Carboxylate (5)Methyl 2,6-Dichlorobenzoate (7)Methyl 2-Chloro-4-Fluorobenzoate (5)Methyl 2-Chloro-6-Fluorobenzoate (7)Methyl 2-Chlorobenzoate (79)Methyl 2-Chloroisonicotinate (11)Methyl 2-Chloronicotinate (6)Methyl 3-Chlorobenzoate (88)Methyl 3-Chloropicolinate (9)Methyl 6-Chloropicolinate (11)N-(2-Chlorophenyl)Acetamide (16)N-(3-Chlorophenyl)Benzenesulfonamide (6)N-(3-Chlorophenyl)Quinazolin-4-Amine (10)N-(4-Chlorophenyl)Acetamide (9)Phenyl Carbonochloridate (5)Pyridine-2-Sulfonyl Chloride (9)Pyridine-3-Sulfonyl Chloride (25)Tert-Butyl (2-Chloropyridin-4-Yl)Carbamate (4)
Difluoromethyls (32970)
Alcohols (10)Aliphatic Chain Hydrocarbons (13)Amines (19)Aryls (43)Bromides (36)Carboxylic Acids (19)Chlorides (22)Esters (6)Ethers (64)Fluorinated Building Blocks (124)Ketones (1)Pyrazoles (11)Pyridines (56)Pyrimidines (9)(Difluoromethyl)Benzene (62)1-(Difluoromethoxy)-2-Fluorobenzene (11)1-(Difluoromethyl)-2-Fluorobenzene (13)1-Bromo-2-(Difluoromethyl)Benzene (5)2-(Difluoromethoxy)Phenol (5)2-(Difluoromethyl)Pyridine (21)3-(Difluoromethyl)Pyridine (5)4-(Difluoromethoxy)Aniline (6)5-(Difluoromethyl)-1H-Pyrazole (6)
Esters (50749)
Acyl Chlorides (22)Alcohols (239)Aldehydes (47)Aliphatic Chain Hydrocarbons (450)Aliphatic Cyclic Hydrocarbons (109)Alkenyls (245)Alkynyls (19)Amides (300)Amines (302)Anhydrides (81)Aryls (893)Azetidines (11)Benzimidazoles (12)Benzofurans (34)Benzothiazoles (5)Benzothiophenes (8)Benzoxazoles (4)Benzyl Bromides (14)Benzyl Chlorides (10)Bromides (314)Carboxylic Acid Salts (3)Carboxylic Acids (73)Chlorides (298)Difluoromethyls (6)Dioxolanes (7)Epoxides (9)Ethers (268)Fluorinated Building Blocks (194)Furans (23)Hydrazines (7)Imidazoles (34)Indazoles (15)Indoles (61)Indolines (9)Iodides (31)Isoquinolines (7)Isoxazoles (11)Ketones (319)Morpholines (32)Nitriles (102)Nitroes (72)Oxazoles (18)Oxazolidines (7)Oxazolines (6)Oxetanes (10)Oximes (5)Phenols (30)Phosphoruses (6)Piperazines (38)Piperidines (69)Pyrans (5)Pyrazines (9)Pyrazoles (33)Pyridazines (11)Pyridines (230)Pyrimidines (63)Pyrroles (21)Pyrrolidines (32)Pyrrolines (2)Quinazolines (5)Quinolines (74)Quinuclidines (3)Spiroes (19)Sulfamides (23)Sulfides (27)Sulfones (16)Tetrahydrofurans (22)Tetrahydropyrans (25)Tetrazoles (5)Thiadiazoles (4)Thiazoles (70)Thiols (9)Thiophenes (70)Triazoles (9)Trifluoromethyls (72)Other Aliphatic Heterocycles (69)Other Aromatic Heterocycles (146)Acetoxymethyl (85)Acetoxyphenyl (1)Acetyloxy (54)Acrylate (118)Acryloyloxy (3)Adipate (17)Adipate (17)Aminobenzoate (18)Aminobenzylcarbamate (8)Aminobutanoate (14)Aminocyclobutanecarboxylic Acid (4)Aminocyclohexanecarboxylic (16)Aminocyclohexanecarboxylic Acid (11)Aminocyclopentanecarboxylic (13)Aminoheptanoate (9)Aminohexanoate (6)Aminoisonicotinate (4)Aminoisophthalate (5)Aminonicotinate (7)Aminopentanedioate (15)Aminopentanoate (7)Aminopropanoate (16)Benzoyloxy (14)Benzylcarbamate (8)Bromobenzoate (26)Bromobutanoate (9)Bromodecanoate (20)Bromoisonicotinate (8)Bromoisophthalate (3)Bromonicotinate (13)Bromopropanoate (9)Bromoundecanoate (3)Butoxycarbonyl (1449)Butoxycarbonyl (1449)Carbamate (1496)Chlorobenzoate (49)Chloroformate (4)Chloropicolinate (30)Chloropropanoate (5)Cyanoacrylate (5)Decanedioate (9)Decanedioate (9)Decanoate (7)Decanoate (7)Diacetate (55)Diacetoxy (1)Diacrylate (15)Diaminobenzoate (7)Dibromobenzoate (4)Dicarbamate (14)Dichlorobenzoate (24)Dichloroisonicotinate (9)Dimethacrylate (1)Dimethylbutanoate (14)Dimethylcarbamate (3)Dimethylnicotinate (10)Dimethylnicotinate (10)Dipropanoate (6)Dipropanoate (6)Diyldicarbamate (5)Dodecanoate (11)Enecarboxylate (9)Enoate (116)Ethoxycarbonyl (69)Ethylbutanoate (6)Ethynylbenzoate (7)Ethynylbenzylcarbamate (3)Glutarate (3)Glutarate (3)Hexanoate (14)Hexanoate (14)Iodoacrylate (6)Iodobutanoate (4)Iodopropanoate (1)Isobutyrate (18)Isobutyrate (18)Methacrylate (87)Methacryloyloxy (7)Methoxycarbonyl (168)Methoxycarbonyl (168)Methoxyisonicotinate (11)Methoxyisonicotinate (11)Methoxynicotinate (24)Methoxynicotinate (24)Methoxypropanoate (5)Methoxypropanoate (5)Methylacrylate (13)Methylbutanoate (38)Methylmalonate (6)Methylnicotinate (57)Methylpentanoate (17)Methylpropanoate (47)Nitropicolinate (13)Nonanedioate (4)Nonanedioate (4)Oxoacetate (21)Oxoacetate (21)Oxobutanoate (56)Oxobutanoate (56)Oxoheptanoate (8)Oxoheptanoate (8)Oxohexanoate (14)Oxohexanoate (14)Oxopentanoate (31)Oxopropanoate (39)Oxopropanoate (39)Pentanoate (13)Pentanoate (13)Phenylacrylate (5)Phenylpropanoate (30)Propanoate (266)Propylpentanoate (5)Propylpentanoate (5)Succinate (23)Succinate (23)Sulfamoylbenzoate (6)Tetraacetate (7)Tetraacetate (7)Tetradecanoate (10)Tetradecanoate (10)Tosylcarbamate (4)Tosyloxy (12)Triacetate (12)Triacetate (12)Vinylbenzoate (3)Vinylcyclopropanecarboxylic Acid (3)(2R,3S)-2-((Benzoyloxy)Methyl)Tetrahydrofuran-3-Yl Benzoate (8)(9H-Fluoren-9-Yl)Methyl (3-Phenylpropyl)Carbamate (24)1,3-Dioxoisoindolin-2-Yl 2-Phenylacetate (5)1-Benzyl 2-Methyl Pyrrolidine-1,2-Dicarboxylate (7)1-Benzyl 3-Methyl Piperidine-1,3-Dicarboxylate (3)1-Benzyl 4-(Tert-Butyl) Piperazine-1,4-Dicarboxylate (10)1H-Indol-3-Yl Acetate (4)2,5-Dioxopyrrolidin-1-Yl Benzoate (8)2-((Tert-Butoxycarbonyl)Amino)Benzoic Acid (11)2-(Diethylamino)Ethyl Benzoate (3)2-Ethylhexyl Benzoate (13)2-Hydroxyethyl Benzoate (3)2-Phenoxyacetyl Chloride (3)4-Cyanophenyl Benzoate (11)Allyl Benzoate (5)Azetidin-3-Yl Acetate (3)Benzoic Anhydride (6)Benzyl Acetate (11)Benzyl Benzoate (12)Benzyl Benzylcarbamate (14)Benzyl Cyclohexylcarbamate (9)Benzyl Morpholine-4-Carboxylate (5)Benzyl Phenylcarbamate (15)Benzyl Piperidine-1-Carboxylate (50)Butyl Benzoate (10)Dimethyl Isophthalate (32)Dimethyl Phthalate (14)Dimethyl Pyridine-2,6-Dicarboxylate (7)Ethane-1,2-Diyl Dibenzoate (11)Ethyl 1-Phenyl-1H-Pyrazole-3-Carboxylate (4)Ethyl 1-Phenyl-1H-Pyrazole-4-Carboxylate (13)Ethyl 1-Phenylcyclopropane-1-Carboxylate (4)Ethyl 1H-1,2,4-Triazole-3-Carboxylate (3)Ethyl 1H-Imidazole-2-Carboxylate (6)Ethyl 1H-Indole-3-Carboxylate (6)Ethyl 1H-Pyrazole-4-Carboxylate (11)Ethyl 1H-Pyrrole-2-Carboxylate (24)Ethyl 1H-Pyrrole-3-Carboxylate (8)Ethyl 2-(Imidazo[1,2-A]Pyridin-3-Yl)Acetate (3)Ethyl 2-(Pyridin-2-Yl)Acetate (15)Ethyl 2-Aminobenzoate (23)Ethyl 2-Aminothiazole-4-Carboxylate (6)Ethyl 2-Aminothiazole-5-Carboxylate (5)Ethyl 2-Bromo-2-Phenylacetate (6)Ethyl 2-Fluorobenzoate (37)Ethyl 2-Hydroxybenzoate (18)Ethyl 2-Iodobenzoate (4)Ethyl 2-Methylbenzoate (26)Ethyl 2-Methylnicotinate (6)Ethyl 2-Nitrobenzoate (9)Ethyl 2-Oxo-2-(Phenylamino)Acetate (8)Ethyl 2-Oxo-2-Phenylacetate (18)Ethyl 2-Oxo-2H-Chromene-3-Carboxylate (5)Ethyl 2-Phenoxyacetate (10)Ethyl 2-Phenylacetate (60)Ethyl 2-Phenylcyclopropane-1-Carboxylate (5)Ethyl 3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Benzoate (6)Ethyl 3-Aminobenzoate (19)Ethyl 3-Fluorobenzoate (34)Ethyl 3-Hydroxybenzoate (12)Ethyl 3-Iodobenzoate (11)Ethyl 3-Nitrobenzoate (22)Ethyl 3-Oxo-3-Phenylpropanoate (41)Ethyl 3-Phenylpropanoate (17)Ethyl 4-Aminobenzoate (23)Ethyl 4-Chloropyrimidine-5-Carboxylate (5)Ethyl 5-Fluoro-1H-Indole-2-Carboxylate (5)Ethyl 6-Chloro-1H-Indole-2-Carboxylate (5)Ethyl Benzo[B]Thiophene-2-Carboxylate (9)Ethyl Cinnamate (26)Ethyl Cyclohex-1-Ene-1-Carboxylate (5)Ethyl Cyclohexanecarboxylate (33)Ethyl Cyclopropanecarboxylate (8)Ethyl Furan-2-Carboxylate (6)Ethyl Imidazo[1,2-A]Pyridine-2-Carboxylate (25)Ethyl Imidazo[1,2-A]Pyridine-3-Carboxylate (13)Ethyl Isonicotinate (12)Ethyl Isoxazole-3-Carboxylate (8)Ethyl Isoxazole-4-Carboxylate (5)Ethyl Morpholine-2-Carboxylate (4)Ethyl Nicotinate (25)Ethyl Oxazole-4-Carboxylate (8)Ethyl Oxazole-5-Carboxylate (3)Ethyl Oxirane-2-Carboxylate (4)Ethyl Picolinate (41)Ethyl Piperidine-2-Carboxylate (9)Ethyl Piperidine-3-Carboxylate (14)Ethyl Piperidine-4-Carboxylate (10)Ethyl Pyrazine-2-Carboxylate (10)Ethyl Pyridazine-3-Carboxylate (6)Ethyl Pyrimidine-4-Carboxylate (7)Ethyl Pyrimidine-5-Carboxylate (5)Ethyl Quinazoline-2-Carboxylate (4)Ethyl Thiazole-2-Carboxylate (7)Ethyl Thiazole-4-Carboxylate (7)Ethyl Thiazole-5-Carboxylate (7)Ethyl Thiophene-2-Carboxylate (19)Ethyl Thiophene-3-Carboxylate (11)Hexyl Benzoate (5)Isobutyl Benzoate (10)Isopropyl Benzoate (10)Methyl 1-Naphthoate (7)Methyl 1H-Benzo[D]Imidazole-2-Carboxylate (3)Methyl 1H-Imidazole-2-Carboxylate (4)Methyl 1H-Imidazole-5-Carboxylate (8)Methyl 1H-Indazole-3-Carboxylate (23)Methyl 1H-Indazole-4-Carboxylate (10)Methyl 1H-Indazole-5-Carboxylate (3)Methyl 1H-Indazole-6-Carboxylate (9)Methyl 1H-Indazole-7-Carboxylate (8)Methyl 1H-Indole-3-Carboxylate (21)Methyl 1H-Indole-4-Carboxylate (9)Methyl 1H-Indole-5-Carboxylate (7)Methyl 1H-Indole-6-Carboxylate (9)Methyl 1H-Indole-7-Carboxylate (7)Methyl 1H-Pyrazole-4-Carboxylate (10)Methyl 1H-Pyrrole-2-Carboxylate (30)Methyl 1H-Pyrrole-3-Carboxylate (5)Methyl 1H-Pyrrolo[2,3-B]Pyridine-2-Carboxylate (3)Methyl 1H-Pyrrolo[2,3-B]Pyridine-3-Carboxylate (4)Methyl 1H-Pyrrolo[2,3-B]Pyridine-6-Carboxylate (3)Methyl 2,6-Dichlorobenzoate (7)Methyl 2,6-Difluorobenzoate (17)Methyl 2-(1H-Indol-3-Yl)Acetate (3)Methyl 2-(2-Bromophenyl)Acetate (8)Methyl 2-(4-Hydroxyphenyl)Acetate (10)Methyl 2-(Bromomethyl)Benzoate (33)Methyl 2-(Chlorosulfonyl)Benzoate (7)Methyl 2-(Morpholin-3-Yl)Acetate (3)Methyl 2-(Pyridin-2-Yl)Acetate (9)Methyl 2-(Trifluoromethyl)Benzoate (14)Methyl 2-Amino-2-Phenylacetate (38)Methyl 2-Aminobenzoate (95)Methyl 2-Aminothiazole-4-Carboxylate (6)Methyl 2-Bromo-2-Phenylacetate (7)Methyl 2-Bromo-3-Nitrobenzoate (3)Methyl 2-Bromo-5-Fluorobenzoate (8)Methyl 2-Bromo-6-Fluorobenzoate (7)Methyl 2-Bromo-6-Methylbenzoate (8)Methyl 2-Bromobenzoate (69)Methyl 2-Chloro-4-Fluorobenzoate (5)Methyl 2-Chloro-6-Fluorobenzoate (7)Methyl 2-Chlorobenzoate (79)Methyl 2-Cyanobenzoate (22)Methyl 2-Fluorobenzoate (132)Methyl 2-Hydroxybenzoate (74)Methyl 2-Iodobenzoate (28)Methyl 2-Methoxybenzoate (70)Methyl 2-Methyl-2H-Indazole-3-Carboxylate (3)Methyl 2-Methylbenzoate (101)Methyl 2-Naphthoate (11)Methyl 2-Nitrobenzoate (54)Methyl 2-Phenoxyacetate (12)Methyl 2-Phenoxybenzoate (4)Methyl 2-Phenylacetate (114)Methyl 3-(Benzyloxy)Benzoate (5)Methyl 3-(Bromomethyl)Benzoate (16)Methyl 3-(Trifluoromethyl)Benzoate (22)Methyl 3-Amino-3-Phenylpropanoate (27)Methyl 3-Aminobenzoate (88)Methyl 3-Bromo-4-Chlorobenzoate (6)Methyl 3-Bromo-4-Fluorobenzoate (7)Methyl 3-Bromo-4-Methoxybenzoate (10)Methyl 3-Bromo-4-Methylbenzoate (9)Methyl 3-Bromobenzoate (153)Methyl 3-Bromopyrazine-2-Carboxylate (5)Methyl 3-Chlorobenzoate (88)Methyl 3-Cyanobenzoate (29)Methyl 3-Fluorobenzoate (136)Methyl 3-Formylbenzoate (23)Methyl 3-Hydroxybenzoate (58)Methyl 3-Iodo-4-Methylbenzoate (6)Methyl 3-Iodobenzoate (47)Methyl 3-Methoxybenzoate (72)Methyl 3-Methylbenzoate (75)Methyl 3-Nitrobenzoate (109)Methyl 3-Oxo-3-Phenylpropanoate (16)Methyl 3-Phenylpropanoate (28)Methyl 4-Amino-3-Bromobenzoate (8)Methyl 4-Amino-3-Iodobenzoate (7)Methyl 4-Amino-3-Nitrobenzoate (9)Methyl 4-Aminobenzoate (71)Methyl 4-Fluorobenzoate (75)Methyl 4-Nitrobenzoate (29)Methyl 4-Oxocyclohexane-1-Carboxylate (5)Methyl 4-Oxopiperidine-2-Carboxylate (3)Methyl 4-Phenoxybenzoate (3)Methyl 5-Amino-2-Bromobenzoate (6)Methyl 5-Hydroxypicolinate (5)Methyl 5-Oxopyrrolidine-2-Carboxylate (3)Methyl 5-Phenylisoxazole-3-Carboxylate (6)Methyl 6-Bromopyrazine-2-Carboxylate (7)Methyl 6-Chloropyrazine-2-Carboxylate (5)Methyl 6-Methoxynicotinate (4)Methyl [1,1'-Biphenyl]-4-Carboxylate (8)Methyl Azetidine-2-Carboxylate (3)Methyl Benzo[B]Thiophene-2-Carboxylate (27)Methyl Benzo[D]Oxazole-2-Carboxylate (3)Methyl Benzo[D]Thiazole-6-Carboxylate (5)Methyl Bicyclo[1.1.1]Pentane-1-Carboxylate (10)Methyl Cinnamate (37)Methyl Cyclobutanecarboxylate (24)Methyl Cyclohex-3-Ene-1-Carboxylate (3)Methyl Cyclohexanecarboxylate (45)Methyl Cyclopentanecarboxylate (18)Methyl Cyclopropanecarboxylate (8)Methyl Furan-2-Carboxylate (20)Methyl Imidazo[1,2-A]Pyridine-8-Carboxylate (3)Methyl Indoline-6-Carboxylate (3)Methyl Isonicotinate (16)Methyl Isoquinoline-4-Carboxylate (6)Methyl Morpholine-2-Carboxylate (4)Methyl Morpholine-3-Carboxylate (10)Methyl Nicotinate (52)Methyl Oxazole-4-Carboxylate (9)Methyl Piperidine-2-Carboxylate (12)Methyl Piperidine-3-Carboxylate (19)Methyl Prolinate (43)Methyl Pyrazine-2-Carboxylate (19)Methyl Pyridazine-3-Carboxylate (7)Methyl Pyrimidine-2-Carboxylate (10)Methyl Pyrimidine-4-Carboxylate (11)Methyl Pyrimidine-5-Carboxylate (5)Methyl Pyrrolidine-3-Carboxylate (9)Methyl Thiazole-2-Carboxylate (6)Methyl Thiazole-4-Carboxylate (6)Methyl Thiophene-3-Carboxylate (34)Phenyl (1R,4R)-[1,1'-Bi(Cyclohexane)]-4-Carboxylate (6)Phenyl Dimethylcarbamate (5)Phenyl Ethyl(Methyl)Carbamate (5)Propyl Benzoate (11)Tert-Butyl (1-Phenylethyl)Carbamate (19)Tert-Butyl (2,3-Dihydro-1H-Inden-1-Yl)Carbamate (3)Tert-Butyl (2-Phenylcyclopropyl)Carbamate (3)Tert-Butyl (3-Oxocyclohexyl)Carbamate (3)Tert-Butyl (Morpholin-2-Ylmethyl)Carbamate (3)Tert-Butyl (Tetrahydrofuran-3-Yl)Carbamate (4)Tert-Butyl 1H-Benzo[D]Imidazole-1-Carboxylate (3)Tert-Butyl 1H-Imidazole-1-Carboxylate (6)Tert-Butyl 1H-Indazole-1-Carboxylate (29)Tert-Butyl 1H-Indole-1-Carboxylate (18)Tert-Butyl 1H-Pyrrole-1-Carboxylate (8)Tert-Butyl 1H-Pyrrole-2-Carboxylate (3)Tert-Butyl 1H-Pyrrolo[2,3-B]Pyridine-1-Carboxylate (9)Tert-Butyl 3,4-Dihydroisoquinoline-2(1H)-Carboxylate (19)Tert-Butyl 3-((Methylsulfonyl)Oxy)Pyrrolidine-1-Carboxylate (5)Tert-Butyl 3-Oxopiperazine-1-Carboxylate (7)Tert-Butyl 4-Phenylpiperazine-1-Carboxylate (23)Tert-Butyl 4-Phenylpiperidine-1-Carboxylate (16)Tert-Butyl Azetidine-1-Carboxylate (64)Tert-Butyl Benzoate (66)Tert-Butyl Benzylcarbamate (52)Tert-Butyl Bicyclo[1.1.1]Pentan-1-Ylcarbamate (5)Tert-Butyl Cyclopentylcarbamate (28)Tert-Butyl Isoindoline-2-Carboxylate (13)Tert-Butyl Methyl(Phenyl)Carbamate (9)Tert-Butyl Morpholine-4-Carboxylate (23)Tert-Butyl O-Tolylcarbamate (11)Tert-Butyl Phenethylcarbamate (1)Tert-Butyl Piperazine-1-Carboxylate (30)Tert-Butyl Pyrrolidine-1-Carboxylate (21)
Ethers (82449)
Alcohols (224)Aldehydes (130)Aliphatic Chain Hydrocarbons (199)Aliphatic Cyclic Hydrocarbons (32)Alkenyls (79)Alkynyls (29)Amides (254)Amidines (4)Amines (403)Anhydrides (8)Aryls (1461)Azetidines (18)Benzimidazoles (17)Benzofurans (2)Benzothiazoles (15)Benzoxazoles (6)Benzyl Bromides (6)Benzyl Chlorides (7)Bromides (328)Carboxylic Acid Salts (5)Carboxylic Acids (291)Chlorides (285)Difluoromethyls (64)Epoxides (13)Esters (268)Fluorinated Building Blocks (235)Furans (10)Hydrazides (23)Hydrazines (40)Imidazoles (6)Indazoles (10)Indoles (54)Indolines (5)Iodides (32)Isoquinolines (8)Isoxazoles (12)Ketones (160)Morpholines (17)Nitriles (103)Nitroes (84)Oxazoles (4)Oxetanes (6)Phenols (44)Piperazines (31)Piperidines (82)Pyrans (5)Pyrazines (19)Pyrazoles (39)Pyridazines (7)Pyridines (239)Pyrimidines (113)Pyrrolidines (56)Quinazolines (17)Quinolines (82)Quinuclidines (2)Sulfamides (23)Sulfides (29)Sulfonates (4)Sulfones (12)Sulfonyl Chlorides (19)Sulfoxides (5)Tetrahydrofurans (1)Tetrahydropyrans (24)Thiadiazoles (7)Thiazoles (16)Thiophenes (5)Thiophenols (7)Triazoles (5)Trifluoromethyls (108)Other Aliphatic Heterocycles (5)Other Aromatic Heterocycles (48)(1S,4Ar,7Ar)-1-(((S)-Tetrahydro-2H-Pyran-2-Yl)Oxy)-1,4A,5,7A-Tetrahydrocyclopenta[C]Pyran (4)(2,2,2-Trifluoroethoxy)Benzene (18)(2-(Benzyloxy)Phenyl)Boronic Acid (23)(2-(Phenoxymethyl)Phenyl)Boronic Acid (3)(2-Chloro-3-Methoxyphenyl)Boronic Acid (5)(2-Chloroethoxy)Benzene (5)(2-Methoxyethoxy)Benzene (32)(3-(Benzyloxy)Phenyl)Boronic Acid (9)(3-(Phenoxymethyl)Phenyl)Boronic Acid (3)(3-Chloro-2-Methoxyphenyl)Boronic Acid (4)(3-Phenoxyphenyl)Methanol (3)(4-(Benzyloxy)Phenyl)Boronic Acid (26)(4-(Phenoxymethyl)Phenyl)Boronic Acid (6)(4-Methoxyphenyl)(Phenyl)Methanone (7)(9H-Fluoren-9-Yl)Methyl (2-(Benzyloxy)Ethyl)Carbamate (6)(9H-Fluoren-9-Yl)Methyl (4-Methoxybenzyl)Carbamate (7)(Allyloxy)Benzene (17)(Ethoxymethyl)Benzene (7)(Hexyloxy)Benzene (7)(Methoxymethyl)Benzene (30)(Prop-2-Yn-1-Yloxy)Benzene (12)(R)-2-Phenoxytetrahydro-2H-Pyran (7)(S)-7-(((S)-Tetrahydro-2H-Pyran-2-Yl)Oxy)-7,8,9,10-Tetrahydrotetracene-5,12-Dione (5)1,2-Bis(Benzyloxy)Benzene (11)1,2-Dichloro-3-Methoxybenzene (7)1,2-Dichloro-4-Methoxybenzene (11)1,2-Difluoro-3-Methoxybenzene (28)1,2-Difluoro-4-Methoxybenzene (19)1,2-Dimethoxybenzene (224)1,2-Diphenoxyethane (11)1,3-Bis(Benzyloxy)Benzene (8)1,3-Dibromo-2-Methoxybenzene (11)1,3-Dichloro-2-Methoxybenzene (9)1,3-Diiodo-2-Phenoxybenzene (1)1,3-Dimethoxybenzene (239)1,3-Diphenoxybenzene (8)1,4-Dichloro-2-Methoxybenzene (8)1,4-Dimethoxybenzene (66)1-(2-Methoxyphenyl)Ethan-1-One (34)1-(3-(Benzyloxy)Phenyl)Ethan-1-One (3)1-(3-Methoxyphenyl)Ethan-1-One (38)1-(3-Methoxyphenyl)Piperazine (4)1-(4-(Benzyloxy)Phenyl)Ethan-1-One (4)1-(4-Methoxybenzyl)-1H-Pyrazole (5)1-(Benzyloxy)-2-Bromobenzene (12)1-(Benzyloxy)-2-Chlorobenzene (16)1-(Benzyloxy)-2-Fluorobenzene (10)1-(Benzyloxy)-2-Methoxybenzene (19)1-(Benzyloxy)-2-Methylbenzene (12)1-(Benzyloxy)-2-Nitrobenzene (3)1-(Benzyloxy)-3-Bromobenzene (12)1-(Benzyloxy)-3-Chlorobenzene (6)1-(Benzyloxy)-3-Fluorobenzene (14)1-(Benzyloxy)-3-Methylbenzene (8)1-(Benzyloxy)-3-Nitrobenzene (10)1-(Benzyloxy)-4-Bromobenzene (8)1-(Benzyloxy)-4-Iodobenzene (3)1-(Bromomethyl)-2-Methoxybenzene (14)1-(Bromomethyl)-3-Methoxybenzene (19)1-(Difluoromethoxy)-2-Fluorobenzene (11)1-Benzyl-4-Methoxybenzene (6)1-Bromo-2,3-Dimethoxybenzene (8)1-Bromo-2-Chloro-3-Methoxybenzene (6)1-Bromo-2-Ethoxybenzene (16)1-Bromo-2-Fluoro-3-Methoxybenzene (7)1-Bromo-2-Phenoxybenzene (3)1-Bromo-3-Methoxybenzene (187)1-Bromo-3-Phenoxybenzene (8)1-Bromo-4-Phenoxybenzene (8)1-Chloro-2,4-Dimethoxybenzene (7)1-Chloro-2-Ethoxybenzene (14)1-Chloro-2-Phenoxybenzene (10)1-Chloro-2-Propoxybenzene (5)1-Chloro-3-Fluoro-2-Methoxybenzene (4)1-Chloro-3-Methoxybenzene (125)1-Chloro-3-Phenoxybenzene (5)1-Chloro-4-Methoxybenzene (98)1-Chloro-4-Phenoxybenzene (13)1-Cyclopropyl-3-(4-(Quinolin-4-Yloxy)Phenyl)Urea (6)1-Ethoxy-2,3-Difluorobenzene (9)1-Ethoxy-2-Fluorobenzene (37)1-Ethoxy-3-Nitrobenzene (7)1-Fluoro-2-(Trifluoromethoxy)Benzene (20)1-Fluoro-2-Isopropoxybenzene (18)1-Fluoro-2-Phenoxybenzene (11)1-Fluoro-3-Methoxybenzene (213)1-Fluoro-3-Phenoxybenzene (8)1-Fluoro-4-(Trifluoromethoxy)Benzene (14)1-Fluoro-4-Methoxybenzene (125)1-Fluoro-4-Phenoxybenzene (8)1-Iodo-2-Methoxybenzene (50)1-Iodo-3-Methoxybenzene (48)1-Iodo-4-Methoxybenzene (34)1-Iodo-4-Phenoxybenzene (4)1-Methoxy-2-(Trifluoromethyl)Benzene (21)1-Methoxy-3-(Trifluoromethoxy)Benzene (9)1-Methoxy-3-(Trifluoromethyl)Benzene (40)1-Methoxy-3-Nitrobenzene (108)1-Methoxy-4-(Phenoxymethyl)Benzene (3)1-Methoxy-4-(Trifluoromethyl)Benzene (18)1-Methoxy-4-Nitrobenzene (90)1-Methoxynaphthalene (18)1-Methyl-3-Phenoxybenzene (9)1-Nitro-2-Phenoxybenzene (4)1-Nitro-4-Phenoxybenzene (5)1-Phenoxy-3-(Trifluoromethyl)Benzene (2)1-Phenoxy-4-(Trifluoromethyl)Benzene (2)1-Phenyl-3-(4-(Pyridin-4-Yloxy)Phenyl)Urea (8)2,3,5,6,8,9,11,12-Octahydrobenzo[B][1,4,7,10,13]Pentaoxacyclopentadecine (8)2,3-Dimethoxypyridine (7)2,4,6-Triphenoxy-1,3,5-Triazine (4)2,4-Dibromo-1-Methoxybenzene (6)2,4-Dichloro-1-Methoxybenzene (18)2,4-Difluoro-1-Methoxybenzene (25)2,4-Dimethoxy-1-Nitrobenzene (5)2,4-Dimethoxypyrimidine (13)2,6-Dimethoxypyridine (11)2-(2-Methoxyphenyl)Acetic Acid (16)2-(3-Isopropoxyphenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (5)2-(3-Methoxyphenyl)Acetic Acid (23)2-(4-(Benzyloxy)Phenyl)Acetonitrile (3)2-(4-Methoxyphenyl)Acetic Acid (21)2-(Benzyloxy)Benzaldehyde (13)2-(Benzyloxy)Naphthalene (5)2-(Benzyloxy)Phenol (5)2-(Methoxymethyl)Morpholine (4)2-(Methoxymethyl)Pyrrolidine (6)2-(Phenoxymethyl)Oxirane (13)2-(Phenoxymethyl)Pyridine (5)2-(Phenoxymethyl)Tetrahydro-2H-Pyran (5)2-Bromo-1,3-Dimethoxybenzene (7)2-Bromo-1,4-Dimethoxybenzene (11)2-Bromo-1-Fluoro-4-Methoxybenzene (12)2-Bromo-1-Methoxy-3-Methylbenzene (3)2-Bromo-1-Methoxy-4-Methylbenzene (7)2-Bromo-3-Methoxypyrazine (5)2-Bromo-4-Methoxy-1-Nitrobenzene (6)2-Bromo-4-Methoxyaniline (6)2-Bromo-4-Methoxybenzaldehyde (8)2-Bromo-4-Methoxybenzoic Acid (4)2-Bromo-5-Methoxybenzaldehyde (6)2-Bromo-6-Methoxyaniline (5)2-Bromo-6-Methoxyphenol (5)2-Chloro-1,3-Dimethoxybenzene (3)2-Chloro-1,4-Dimethoxybenzene (4)2-Chloro-3-Methoxybenzaldehyde (7)2-Chloro-3-Methoxybenzoic Acid (6)2-Chloro-4-Fluoro-1-Methoxybenzene (10)2-Chloro-4-Methoxypyrimidine (14)2-Chloro-5-Methoxyaniline (8)2-Chloro-6-Methoxyaniline (3)2-Chloro-6-Methoxyphenol (4)2-Ethoxynaphthalene (4)2-Ethoxypyridine (20)2-Ethoxypyrimidine (7)2-Fluoro-1,3-Dimethoxybenzene (8)2-Fluoro-1-Methoxy-4-Nitrobenzene (8)2-Fluoro-4-Methoxyaniline (6)2-Fluoro-5-Methoxybenzoic Acid (9)2-Fluoro-5-Methoxypyridine (4)2-Fluoro-6-Methoxypyridine (6)2-Isopropoxypyridine (16)2-Methoxy-1,1'-Biphenyl (7)2-Methoxy-1,3,5-Triazine (10)2-Methoxy-4-Methylpyridine (5)2-Methoxy-5-Methylpyridine (5)2-Methoxy-6-Methylpyridine (14)2-Methoxy-6-Nitrophenol (5)2-Methoxybenzamide (9)2-Methoxybenzenesulfonyl Chloride (15)2-Methoxynaphthalene (22)2-Methoxyphenol (201)2-Methoxypyrazine (45)2-Methoxypyridine (434)2-Methoxypyrimidin-4-Amine (6)2-Methoxypyrimidine (59)2-Methoxythiophene (7)2-Methyl-N-Phenylaniline (7)2-Phenoxyacetonitrile (11)2-Phenoxyaniline (4)2-Phenoxybenzonitrile (5)2-Phenoxyethan-1-Ol (14)2-Phenoxypropanoic Acid (13)2-Phenoxypyrimidine (8)2-Phenoxytetrahydro-2H-Pyran (6)2-Phenoxythiazole (5)2-Propoxypyridine (9)3,5-Dimethoxypyridine (4)3-(3-Methoxyphenyl)Propanoic Acid (9)3-(Benzyloxy)Aniline (12)3-(Benzyloxy)Benzaldehyde (13)3-(Benzyloxy)Benzoic Acid (5)3-(Benzyloxy)Phenol (5)3-(Methoxymethyl)Morpholine (4)3-Chloro-2-Methoxybenzoic Acid (4)3-Chloro-4-Methoxyaniline (6)3-Chloro-4-Methoxybenzaldehyde (7)3-Chloro-4-Methoxybenzoic Acid (5)3-Ethoxypyridine (12)3-Fluoro-2-Methoxypyridine (14)3-Fluoro-4-Methoxybenzoic Acid (6)3-Methoxypiperidine (7)3-Methoxypyridazine (17)3-Methoxypyridine (51)3-Methoxythiophene (9)3-Phenoxyaniline (2)3-Phenoxybenzaldehyde (7)3-Phenoxybenzoic Acid (6)3-Phenoxybenzonitrile (5)3-Phenoxypropanoic Acid (14)3-Phenoxypyrrolidine (5)4-(3-Phenoxypropyl)Morpholine (7)4-(Benzyloxy)Aniline (8)4-(Benzyloxy)Benzaldehyde (23)4-(Benzyloxy)Benzoic Acid (5)4-(Benzyloxy)Phenol (2)4-(Difluoromethoxy)Aniline (6)4-(Dimethoxymethyl)Pyrimidine (5)4-(Phenylethynyl)Phenol (7)4-Chloro-2-Methoxypyrimidine (12)4-Chloro-5-Methoxypyrimidine (7)4-Chloro-6-Methoxypyrimidine (5)4-Ethoxypyridine (7)4-Ethoxypyrimidine (6)4-Methoxy-1,1'-Biphenyl (4)4-Methoxy-1H-Indazole (7)4-Methoxy-1H-Indole (11)4-Methoxy-2-Methylpyrimidine (8)4-Methoxy-N-Phenylaniline (5)4-Methoxy-N-Phenylbenzenesulfonamide (3)4-Methoxybenzenesulfonamide (5)4-Methoxypyridazine (8)4-Methoxypyridine (136)4-Methoxypyrimidin-2-Amine (6)4-Methoxypyrimidine (102)4-Phenoxyaniline (6)4-Phenoxybenzaldehyde (12)4-Phenoxybenzoic Acid (4)4-Phenoxypyridine (7)4-Phenoxypyrimidine (5)4-Phenoxyquinoline (5)4-Phenoxytetrahydro-2H-Pyran (5)5-(Benzyloxy)Pyrimidine (5)5-Fluoro-2-Methoxypyridine (15)5-Methoxy-1H-Benzo[D]Imidazole (2)5-Methoxy-1H-Indazole (5)5-Methoxy-1H-Indole (11)5-Methoxy-2-Methyl-1H-Indole (5)5-Methoxy-2-Nitroaniline (7)5-Methoxyindoline (4)5-Methoxyisobenzofuran-1(3H)-One (5)5-Methoxyisoquinoline (6)6-Methoxy-1,2,3,4-Tetrahydroisoquinoline (3)6-Methoxy-1H-Indazole (14)6-Methoxy-1H-Indole (12)6-Methoxy-1H-Pyrrolo[2,3-B]Pyridine (3)6-Methoxy-3,4-Dihydroisoquinolin-1(2H)-One (3)6-Methoxybenzo[D]Thiazole (6)6-Methoxybenzofuran (4)6-Methoxychromane (5)6-Methoxyisoindolin-1-One (3)6-Methoxyquinoxaline (6)7-(Benzyloxy)Quinoline (6)7-Methoxy-1,2,3,4-Tetrahydroisoquinoline (6)7-Methoxy-1H-Indazole (5)7-Methoxy-1H-Indole (8)7-Methoxy-3,4-Dihydroquinolin-2(1H)-One (3)7-Methoxyisoquinoline (5)8-Methoxyisoquinoline (7)8-Methoxyquinazoline (3)Butoxybenzene (51)Ethoxybenzene (283)Ethyl 2-Phenoxyacetate (10)Isobutoxybenzene (22)Isopropoxybenzene (84)Methoxycyclohexane (7)Methyl 2-Methoxybenzoate (70)Methyl 2-Phenoxyacetate (12)Methyl 2-Phenoxybenzoate (4)Methyl 3-(Benzyloxy)Benzoate (5)Methyl 3-Methoxybenzoate (72)Methyl 4-Phenoxybenzoate (3)N,N-Dimethyl-2-Phenoxyethan-1-Amine (16)N-(2-Phenoxyphenyl)Methanesulfonamide (3)N-(4-Phenoxyphenyl)Acetamide (4)N-Phenyl-N-(4-(Quinolin-4-Yloxy)Phenyl)Cyclopropane-1,1-Dicarboxamide (5)Phenethoxybenzene (23)Propoxybenzene (63)Tert-Butoxybenzene (12)Tert-Butyl 3-Methoxypyrrolidine-1-Carboxylate (6)Tert-Butyl 4-Phenoxypiperidine-1-Carboxylate (12)Tert-Butyl 5-Methoxy-1H-Indole-1-Carboxylate (6)Butoxy-Phenyl-Boronic Acid/Ester (76)Diethoxy (14)Diethoxy (14)Diethoxyethyl (9)Diethoxyethyl (9)Diethoxymethyl (8)Diethoxymethyl (8)Dimethoxyethyl (8)Dimethoxypropane (11)Dimethoxypropane (11)Dioxane (153)Dioxepine (7)Dioxepine (7)Disulfide (38)Methoxyphenylboronic-Acid (32)
Fluorinated Building Blocks (99019)
Alcohols (169)Aldehydes (79)Aliphatic Chain Hydrocarbons (169)Aliphatic Cyclic Hydrocarbons (56)Alkenyls (42)Alkynyls (6)Amides (240)Amidines (7)Amines (284)Anhydrides (8)Aryls (868)Azetidines (20)Benzimidazoles (14)Benzisoxazoles (4)Benzofurans (11)Benzothiazoles (7)Benzothiophenes (5)Benzoxazoles (5)Benzyl Bromides (2)Benzyl Chlorides (2)Bromides (305)Carboxylic Acids (237)Chlorides (224)Difluoromethyls (124)Esters (194)Ethers (235)Furans (4)Hydrazides (6)Hydrazines (12)Imidazoles (25)Indazoles (13)Indoles (39)Indolines (11)Iodides (54)Isoquinolines (7)Isoxazoles (12)Ketones (150)Morpholines (17)Naphthyridines (4)Nitriles (76)Nitroes (75)Oxazoles (5)Oxazolidines (2)Oxetanes (5)Phenols (28)Phosphoruses (5)Piperazines (28)Piperidines (95)Pyrazines (8)Pyrazoles (53)Pyridazines (4)Pyridines (296)Pyrimidines (38)Pyrroles (8)Pyrrolidines (38)Quinazolines (5)Quinolines (50)Quinoxalines (5)Spiroes (9)Sulfamides (26)Sulfides (21)Sulfonates (23)Sulfones (14)Tetrahydropyrans (8)Tetrahydroquinolines (6)Thiazoles (9)Thioesters (4)Thiophenes (18)Thiophenols (4)Trifluoromethyls (502)Other Aliphatic Heterocycles (5)Other Aromatic Heterocycles (47)Dichlorofluorobenzene (22)Difluoro (897)Difluoroaniline (37)Difluorobenzaldehyde (33)Difluorobenzamide (6)Difluorobenzenesulfonamide (5)Difluorobenzoate (59)Difluoroethyl (26)Difluoroethyl (26)Difluoromethoxy (196)Difluoromethoxy (196)Difluoropropan (7)Difluoropropan (7)Fluindamine (32)Fluoroacetanilide (5)Fluoroacetophenone (8)Fluoroaniline (65)Fluoroanisole (9)Fluorobenzaldehyde (69)Fluorobenzamide (28)Fluorobenzenesulfonamide (7)Fluorobenzenethiol (6)Fluorobenzylamine (10)Fluorobromobenzene (3)Fluorocinnamic (10)Fluorocinnamic (10)Fluorocinnamic Acid (9)Fluorocyclopropanecarboxylic (9)Fluoroethoxy (4)Fluoroethyl (8)Fluoroethyl (8)Fluoroindan (3)Fluoroindan (3)Fluoroisonicotinaldehyde (7)Fluoroisonicotinate (8)Fluoroisonicotinic (13)Fluoroisonicotinic Acid (13)Fluoromethyl (15)Fluoromethyl (15)Fluoronicotinaldehyde (20)Fluoronicotinamide (3)Fluoronicotinic (19)Fluoronicotinic (19)Fluoronicotinic Acid (14)Fluorophenylisocyanate (3)Fluoropropan (15)Fluoropropan (15)Fluorostyryl (7)Fluorosulfonyl (12)Fluorosulfonyl (12)Hexafluoro (9)Hexafluoro (9)Hexafluoropropan (4)Hexafluoropropan (4)Nonafluoro (2)Nonafluoro (2)Octafluoro (2)Octafluoro (2)Pentafluoro (4)Pentafluorobenzoate (4)Tetrafluoro (12)Tetrafluoro (12)Tetrafluoroaniline (7)Tetrafluoroethoxy (3)Tetrafluoroethoxy (3)Trifluoro (270)Trifluoro (270)Trifluoroacetamide (8)Trifluoroacetyl (13)Trifluoroaniline (11)Trifluorobenzaldehyde (9)Trifluorobenzoate (14)Trifluorobut (36)Trifluorobut (36)Trifluoroethan (2)Trifluoroethan (2)Trifluoroethanamine (23)Trifluoroethanol (6)Trifluoroethoxy (60)Trifluoroethoxy (60)Trifluoroethyl (62)Trifluoromethanesulfonyl (22)Trifluoromethoxy (477)Trifluoromethoxy (477)Trifluoromethylthio (5)Trifluoromethylthio (5)Trifluoropropane (36)Trifluoropropanoate (5)Trifluoropropyl (9)(2,3-Difluorophenyl)Boronic Acid (33)(2,6-Difluorophenyl)Boronic Acid (28)(2-Chloro-6-Fluorophenyl)Boronic Acid (9)(2-Fluorobenzyl)Hydrazine (6)(2-Fluorophenyl)(Phenyl)Methanone (15)(2-Fluorophenyl)Boronic Acid (229)(2-Fluorophenyl)Hydrazine (22)(2-Fluorophenyl)Methanamine (49)(2-Fluorophenyl)Methanol (60)(3-Fluorophenyl)(Phenyl)Methanone (5)(3-Fluorophenyl)Boronic Acid (169)(3-Fluorophenyl)Methanol (58)(4-Fluorophenyl)(Phenyl)Methanone (8)(4-Fluorophenyl)Boronic Acid (93)(4-Fluorophenyl)Methanamine (27)(4-Fluorophenyl)Methanol (29)1,1-Difluorocyclobutane (14)1,1-Difluorocyclohexane (18)1,2,3,4,5-Pentafluorobenzene (45)1,2,3,4-Tetrafluorobenzene (68)1,2,3-Trifluorobenzene (144)1,2,4-Trifluorobenzene (197)1,2-Dibromo-3-Fluorobenzene (9)1,2-Dibromo-4-Fluorobenzene (11)1,2-Dichloro-3-Fluorobenzene (13)1,2-Dichloro-4-Fluorobenzene (15)1,2-Difluoro-3-Iodobenzene (13)1,2-Difluoro-3-Methoxybenzene (28)1,2-Difluoro-3-Methylbenzene (23)1,2-Difluoro-3-Nitrobenzene (23)1,2-Difluoro-4-Methoxybenzene (19)1,2-Difluoro-4-Nitrobenzene (43)1,2-Difluorobenzene (631)1,3-Dibromo-2,4-Difluorobenzene (5)1,3-Dibromo-2-Fluorobenzene (15)1,3-Dichloro-2-Fluorobenzene (23)1,3-Difluoro-2-Iodobenzene (12)1,3-Difluoro-2-Methylbenzene (18)1,3-Difluoro-2-Nitrobenzene (16)1,4-Dichloro-2-Fluorobenzene (11)1,4-Difluoro-2-Iodobenzene (8)1,4-Difluoro-2-Nitrobenzene (28)1,4-Difluorobenzene (343)1-(2-Fluorophenyl)Ethan-1-Amine (80)1-(2-Fluorophenyl)Ethan-1-One (58)1-(2-Fluorophenyl)Piperazine (8)1-(2-Fluorophenyl)Propan-1-Amine (8)1-(3-Fluorophenyl)Ethan-1-One (63)1-(4-Fluorophenyl)Ethan-1-One (36)1-(Benzyloxy)-2-Fluorobenzene (10)1-(Benzyloxy)-3-Fluorobenzene (14)1-(Bromomethyl)-2-Fluorobenzene (50)1-(Bromomethyl)-3-Fluorobenzene (47)1-(Chloromethyl)-2-Fluorobenzene (8)1-(Chloromethyl)-3-Fluorobenzene (11)1-Benzyl-4-Fluorobenzene (2)1-Bromo-2,3-Difluorobenzene (67)1-Bromo-2,4-Difluorobenzene (59)1-Bromo-2-Chloro-3-Fluorobenzene (20)1-Bromo-2-Fluoro-3-Methylbenzene (11)1-Bromo-3-Fluoro-2-Methoxybenzene (12)1-Bromo-3-Fluoro-2-Methylbenzene (17)1-Bromo-3-Fluoro-2-Nitrobenzene (10)1-Bromo-4-Fluoro-2-Methoxybenzene (11)1-Bromo-4-Fluoro-2-Methylbenzene (18)1-Chloro-2,3-Difluorobenzene (27)1-Chloro-2,4-Difluorobenzene (35)1-Chloro-3-Fluoro-2-Methoxybenzene (4)1-Chloro-3-Fluoro-2-Methylbenzene (10)1-Cyclohexyl-2-Fluorobenzene (4)1-Ethoxy-2,3-Difluorobenzene (9)1-Ethoxy-2-Fluorobenzene (37)1-Ethynyl-2-Fluorobenzene (10)1-Fluoro-2,3-Dimethylbenzene (8)1-Fluoro-2-(Methylsulfonyl)Benzene (5)1-Fluoro-2-Iodo-4-Methylbenzene (3)1-Fluoro-2-Iodobenzene (102)1-Fluoro-2-Isopropoxybenzene (18)1-Fluoro-2-Methylbenzene (255)1-Fluoro-2-Phenoxybenzene (11)1-Fluoro-3,5-Dimethylbenzene (12)1-Fluoro-3-(Methylsulfonyl)Benzene (10)1-Fluoro-3-Iodo-2-Methylbenzene (3)1-Fluoro-3-Iodobenzene (94)1-Fluoro-3-Methoxybenzene (213)1-Fluoro-3-Methylbenzene (254)1-Fluoro-3-Nitrobenzene (216)1-Fluoro-3-Phenoxybenzene (8)1-Fluoro-4-Iodobenzene (65)1-Fluoro-4-Methoxybenzene (125)1-Fluoro-4-Methylbenzene (140)1-Fluoro-4-Phenoxybenzene (8)1-Fluorobicyclo[1.1.1]Pentane (4)1-Fluoronaphthalene (23)2'-Fluoro-1,1':4',1''-Terphenyl (6)2,2',3,3',5,5',6,6'-Octafluoro-1,1'-Biphenyl (4)2,3-Difluoroaniline (34)2,3-Difluorobenzaldehyde (18)2,3-Difluorobenzoic Acid (35)2,3-Difluorobenzonitrile (29)2,3-Difluorophenol (36)2,3-Difluoropyridine (13)2,4-Dibromo-1-Fluorobenzene (18)2,4-Dichloro-1-Fluorobenzene (27)2,4-Difluoro-1-Iodobenzene (14)2,4-Difluoro-1-Methoxybenzene (25)2,4-Difluoro-1-Nitrobenzene (46)2,4-Difluoroaniline (44)2,4-Difluorobenzaldehyde (19)2,4-Difluorobenzoic Acid (29)2,4-Difluorobenzonitrile (21)2,4-Difluorophenol (21)2,5-Difluoroaniline (34)2,5-Difluorobenzoic Acid (32)2,5-Difluoropyridine (8)2,6-Difluoroaniline (23)2,6-Difluorobenzaldehyde (25)2,6-Difluorobenzoic Acid (33)2,6-Difluorobenzonitrile (28)2,6-Difluorophenol (23)2,6-Difluoropyridine (29)2-(2-Fluorophenyl)Acetic Acid (43)2-(2-Fluorophenyl)Acetonitrile (30)2-(2-Fluorophenyl)Pyrrolidine (16)2-(3-Fluorophenyl)Acetic Acid (50)2-(3-Fluorophenyl)Pyrrolidine (5)2-(4-Fluorophenyl)-1H-Indole (4)2-(4-Fluorophenyl)Acetic Acid (30)2-(Bromomethyl)-1,3-Difluorobenzene (12)2-Amino-2-(2-Fluorophenyl)Acetic Acid (11)2-Amino-3-Fluorobenzoic Acid (11)2-Bromo-1,3,4-Trifluorobenzene (16)2-Bromo-1,3-Difluorobenzene (43)2-Bromo-1,4-Difluorobenzene (64)2-Bromo-1-Chloro-3-Fluorobenzene (9)2-Bromo-1-Chloro-4-Fluorobenzene (20)2-Bromo-1-Fluoro-4-Methylbenzene (17)2-Bromo-4-Chloro-1-Fluorobenzene (16)2-Bromo-4-Fluoro-1-Methoxybenzene (12)2-Bromo-4-Fluoro-1-Methylbenzene (21)2-Bromo-4-Fluoroaniline (28)2-Bromo-4-Fluorobenzaldehyde (7)2-Bromo-4-Fluorobenzonitrile (7)2-Bromo-4-Fluorophenol (10)2-Bromo-5-Fluoroaniline (18)2-Bromo-5-Fluorobenzaldehyde (7)2-Bromo-5-Fluorobenzoic Acid (11)2-Bromo-6-Fluoroaniline (14)2-Bromo-6-Fluorophenol (9)2-Chloro-1,3-Difluorobenzene (27)2-Chloro-1,4-Difluorobenzene (25)2-Chloro-3-Fluoropyridine (20)2-Chloro-4-Fluoro-1-Methoxybenzene (10)2-Chloro-4-Fluoroaniline (19)2-Chloro-4-Fluorobenzaldehyde (10)2-Chloro-4-Fluorobenzoic Acid (9)2-Chloro-4-Fluorobenzonitrile (9)2-Chloro-4-Fluorophenol (9)2-Chloro-5-Fluoropyrimidine (6)2-Chloro-6-Fluoroaniline (8)2-Chloro-6-Fluorobenzoic Acid (8)2-Chloro-6-Fluorobenzonitrile (5)2-Chloro-6-Fluorophenol (5)2-Fluoro-1,3-Dimethoxybenzene (8)2-Fluoro-1,3-Dimethylbenzene (14)2-Fluoro-1-Iodo-4-Methylbenzene (5)2-Fluoro-3-Methylbenzaldehyde (8)2-Fluoro-3-Methylpyridine (14)2-Fluoro-4-Methoxyaniline (6)2-Fluoro-4-Methyl-1-Nitrobenzene (13)2-Fluoro-4-Methylbenzoic Acid (7)2-Fluoro-4-Methylpyridine (6)2-Fluoro-5-Methylpyridine (6)2-Fluoro-6-Methylpyridine (20)2-Fluoro-N-Methylbenzamide (12)2-Fluoro-N-Phenylaniline (4)2-Fluoro-N-Phenylbenzenesulfonamide (6)2-Fluoroaniline (262)2-Fluorobenzaldehyde (175)2-Fluorobenzamide (32)2-Fluorobenzenesulfonamide (12)2-Fluorobenzenesulfonyl Chloride (31)2-Fluorobenzoic Acid (220)2-Fluoropyridine (15)2-Fluoropyrimidine (9)3,3-Difluoropiperidine (29)3,3-Difluoropyrrolidine (29)3,4-Difluoroaniline (44)3,4-Difluorophenol (19)3,5-Difluoropyridine (12)3-Chloro-2-Fluoroaniline (9)3-Fluoro-1,1'-Biphenyl (16)3-Fluoro-2-Iodoaniline (8)3-Fluoro-2-Methylaniline (9)3-Fluoro-2-Methylpyridine (15)3-Fluoro-4-Nitroaniline (7)3-Fluoroaniline (288)3-Fluorobenzaldehyde (137)3-Fluorobenzamide (26)3-Fluorobenzenesulfonyl Chloride (22)3-Fluorobenzoic Acid (206)3-Fluorophenol (177)3-Fluoropiperidine (26)3-Fluoropyridin-2-Amine (12)3-Fluoropyridin-4-Amine (4)3-Fluorothiophene (6)4,4-Difluoropiperidine (5)4-(2-Fluorophenyl)Morpholine (8)4-(4-Fluorophenyl)Thiazole (9)4-Chloro-5-Fluoro-2-Methylpyrimidine (5)4-Fluoro-1-Methyl-2-Nitrobenzene (12)4-Fluoro-1H-Indazole (22)4-Fluoro-1H-Indole (24)4-Fluoro-1H-Pyrrolo[2,3-B]Pyridine (6)4-Fluoro-2-Iodoaniline (7)4-Fluoro-2-Nitroaniline (14)4-Fluoroaniline (225)4-Fluorobenzaldehyde (78)4-Fluorobenzamide (19)4-Fluorobenzenesulfonyl Chloride (26)4-Fluorobenzo[D]Thiazole (6)4-Fluorobenzoic Acid (114)4-Fluorobenzonitrile (89)4-Fluoroisoquinoline (6)4-Fluorophenol (111)4-Fluoropyrimidine (8)5-Fluoro-1,2,3,4-Tetrahydroisoquinoline (3)5-Fluoro-1H-Benzo[D][1,2,3]Triazole (4)5-Fluoro-1H-Benzo[D]Imidazole (12)5-Fluoro-1H-Indole (50)5-Fluoro-1H-Pyrrolo[2,3-B]Pyridine (9)5-Fluoro-2,3-Dihydro-1H-Indene (3)5-Fluoro-2,3-Dihydrobenzofuran (5)5-Fluoro-2-Methylpyridine (8)5-Fluoro-3,4-Dihydronaphthalen-1(2H)-One (5)5-Fluorochromane (6)5-Fluoroindoline (3)5-Fluoroisoindolin-1-One (3)5-Fluoropyridin-2-Amine (15)5-Fluoropyrimidine (11)6-Fluoro-1,2,3,4-Tetrahydroisoquinoline (3)6-Fluoro-1,2,3,4-Tetrahydroquinoline (3)6-Fluoro-1H-Indazole (15)6-Fluoro-1H-Indole (17)6-Fluoro-1H-Pyrrolo[2,3-B]Pyridine (3)6-Fluorobenzo[D]Thiazole (5)6-Fluorochromane (6)6-Fluoropyridin-2-Amine (8)6-Fluoropyridin-3-Amine (4)7-Fluoro-1,2,3,4-Tetrahydroisoquinoline (3)7-Fluoro-1,2,3,4-Tetrahydroquinoline (3)7-Fluoro-1H-Indazole (12)7-Fluoro-1H-Indole (24)7-Fluorobenzo[D]Thiazole (9)7-Fluoroindoline-2,3-Dione (6)7-Fluoroisoquinoline (3)8-Fluoro-3,4-Dihydronaphthalen-1(2H)-One (5)8-Fluorochromane (5)8-Fluoroimidazo[1,2-A]Pyridine (3)8-Fluoroisoquinoline (10)8-Fluoroquinazoline (4)Benzenesulfonyl Fluoride (50)Ethyl 2-Fluorobenzoate (37)Ethyl 3-Fluorobenzoate (34)Fluorobenzene (9797)Methyl 2,6-Difluorobenzoate (17)Methyl 2-Fluorobenzoate (132)Methyl 3-Fluorobenzoate (136)Methyl 4-Fluorobenzoate (75)Methyl 6-Fluoronicotinate (4)N-(3-Fluorophenyl)Acetamide (14)N-(4-Fluorophenyl)Benzamide (3)Pentafluoro(Phenyl)-L6-Sulfane (19)Phenyl Trifluoromethanesulfonate (27)Tert-Butyl (4-Fluorophenyl)Carbamate (9)Trifluoro(Phenyl)Borate (80)
Hydrazides (3096)
Alcohols (5)Aliphatic Chain Hydrocarbons (48)Aliphatic Cyclic Hydrocarbons (5)Amides (169)Amines (8)Aryls (84)Chlorides (15)Ethers (23)Fluorinated Building Blocks (6)Hydrazines (121)Phenols (6)Pyridines (17)Thioureas (11)Ureas (7)Acetohydrazide (17)Acetohydrazide (17)Chlorobenzhydrazide (3)Dihydrazide (5)Dihydrazide (5)Fluorobenzohydrazide (6)Pyrazine-2-Carbohydrazide (3)
Hydrazines (6273)
Alcohols (5)Aliphatic Chain Hydrocarbons (61)Aliphatic Cyclic Hydrocarbons (11)Amides (176)Amidines (2)Amines (26)Aryls (121)Chlorides (26)Esters (7)Ethers (40)Fluorinated Building Blocks (12)Hydrazides (121)Nitriles (5)Phenols (2)Pyridines (27)Pyrimidines (8)Pyrrolidines (3)Quinolines (4)Sulfamides (4)Tetrahydropyrans (4)Thioureas (8)Triazoles (3)Trifluoromethyls (4)Ureas (4)Dichlorophenylhydrazine (3)Hydrazinecarbothioamide (1)Hydrazinecarboxamide (1)Hydrazinylbenzoate (6)(2-Bromophenyl)Hydrazine (8)(2-Chlorophenyl)Hydrazine (16)(2-Fluorobenzyl)Hydrazine (6)(2-Fluorophenyl)Hydrazine (22)(3-Chlorophenyl)Hydrazine (12)2-Hydrazineylpyridine (32)2-Hydrazineylpyrimidine (6)3-Hydrazineylpyridine (8)4-Hydrazineylpiperidine (5)Isonicotinohydrazide (6)Phenylhydrazine (153)Picolinohydrazide (5)
Iodides (14105)
Alcohols (16)Aliphatic Chain Hydrocarbons (38)Aliphatic Cyclic Hydrocarbons (10)Alkenyls (6)Amides (36)Amines (36)Aryls (87)Benzofurans (4)Bromides (54)Carboxylic Acids (24)Chlorides (51)Esters (31)Ethers (32)Fluorinated Building Blocks (54)Imidazoles (12)Indazoles (12)Indoles (11)Ketones (10)Nitriles (10)Nitroes (6)Oxetanes (6)Phenols (9)Piperidines (4)Pyrazoles (18)Pyridines (78)Pyrimidines (15)Quinolines (12)Tetrahydropyrans (4)Thiazoles (4)Thiophenes (3)Trifluoromethyls (15)Other Aromatic Heterocycles (28)Diiodo (18)Diiodobenzoate (5)Iodoaniline (60)Iodoanisole (5)Iodobenzaldehyde (38)Iodobenzamide (6)Iodobutane (1)Iodobutane (1)Iodoethoxy (6)Iodoethoxy (6)Iodoethyl (5)Iodoethyl (5)Iodomethyl (26)Iodomethyl (26)Iodonicotinaldehyde (6)Iodonicotinic (14)Iodonicotinic (14)Iodopropyl (4)Iodopropyl (4)(2-Iodophenyl)(Phenyl)Methanone (4)(2-Iodophenyl)Methanol (12)(3-Iodophenyl)Boronic Acid (5)(3-Iodophenyl)Methanol (13)1,2-Benziodoxol-3-(1H)-One (6)1,2-Difluoro-3-Iodobenzene (13)1,2-Diiodobenzene (10)1,3-Dibromo-2-Iodobenzene (4)1,3-Dichloro-2-Iodobenzene (5)1,3-Difluoro-2-Iodobenzene (12)1,3-Diiodo-2-Phenoxybenzene (1)1,3-Diiodobenzene (52)1,4-Dibromo-2-Iodobenzene (3)1,4-Difluoro-2-Iodobenzene (8)1-(2-Iodophenyl)Ethan-1-One (4)1-(3-Iodophenyl)Ethan-1-One (7)1-(Benzyloxy)-4-Iodobenzene (3)1-(Bromomethyl)-2-Iodobenzene (9)1-Benzyl-4-Iodo-1H-Pyrazole (5)1-Bromo-2-Iodobenzene (65)1-Bromo-3-Fluoro-2-Iodobenzene (4)1-Bromo-3-Iodobenzene (70)1-Bromo-4-Iodobenzene (67)1-Chloro-2-Fluoro-3-Iodobenzene (5)1-Chloro-2-Iodobenzene (65)1-Chloro-3-Iodo-2-Methylbenzene (3)1-Chloro-3-Iodobenzene (78)1-Chloro-4-Iodobenzene (49)1-Ethyl-2-Iodobenzene (6)1-Fluoro-2-Iodo-4-Methylbenzene (3)1-Fluoro-2-Iodobenzene (102)1-Fluoro-3-Iodo-2-Methylbenzene (3)1-Fluoro-3-Iodobenzene (94)1-Fluoro-4-Iodobenzene (65)1-Iodo-2,3-Dimethylbenzene (8)1-Iodo-2,4-Dimethylbenzene (9)1-Iodo-2-(Trifluoromethoxy)Benzene (6)1-Iodo-2-(Trifluoromethyl)Benzene (13)1-Iodo-2-Isopropylbenzene (4)1-Iodo-2-Methoxybenzene (50)1-Iodo-2-Methylbenzene (91)1-Iodo-2-Nitrobenzene (36)1-Iodo-3-(Trifluoromethoxy)Benzene (6)1-Iodo-3-(Trifluoromethyl)Benzene (30)1-Iodo-3-Methoxybenzene (48)1-Iodo-3-Methylbenzene (60)1-Iodo-3-Nitrobenzene (45)1-Iodo-4-Methoxybenzene (34)1-Iodo-4-Phenoxybenzene (4)2,4-Difluoro-1-Iodobenzene (14)2,6-Dichloro-4-Iodopyridine (4)2,6-Diiodoaniline (8)2,6-Diiodophenol (9)2-Bromo-3-Iodopyridine (6)2-Bromo-4-Chloro-1-Iodobenzene (6)2-Bromo-6-Iodoaniline (4)2-Bromo-6-Iodopyridine (5)2-Chloro-3-Iodopyridin-4-Amine (3)2-Chloro-3-Iodopyridine (13)2-Chloro-4-Iodopyridine (19)2-Chloro-5-Iodopyridine (18)2-Chloro-6-Iodophenol (3)2-Chloro-6-Iodopyridine (9)2-Fluoro-1-Iodo-4-Methylbenzene (5)2-Fluoro-3-Iodopyridine (8)2-Fluoro-6-Iodoaniline (7)2-Hydroxy-3-Iodobenzaldehyde (3)2-Iodo-1,3-Dimethylbenzene (15)2-Iodo-1,4-Dimethylbenzene (5)2-Iodo-1-Methyl-4-Nitrobenzene (3)2-Iodo-4-Methylaniline (5)2-Iodo-6-Methylpyridine (5)2-Iodo-6-Nitroaniline (3)2-Iodo-9H-Fluorene (8)2-Iodo-N,N-Dimethylaniline (3)2-Iodoaniline (87)2-Iodobenzaldehyde (17)2-Iodobenzamide (4)2-Iodobenzene-1,3-Diol (3)2-Iodobenzoic Acid (47)2-Iodobenzonitrile (26)2-Iodonaphthalene (5)2-Iodophenol (54)2-Iodopyrazine (25)2-Iodopyridine (27)2-Iodopyrimidine (8)2-Iodothiophene (14)3-Chloro-2-Iodoaniline (3)3-Chloro-4-Iodoaniline (3)3-Fluoro-2-Iodoaniline (8)3-Iodo-1-Methyl-1H-Pyrazole (6)3-Iodo-1H-Pyrrolo[2,3-B]Pyridine (11)3-Iodo-1H-Pyrrolo[3,2-B]Pyridine (6)3-Iodo-2-Methylaniline (4)3-Iodoaniline (32)3-Iodobenzaldehyde (28)3-Iodobenzoic Acid (55)3-Iodobenzonitrile (27)3-Iodoimidazo[1,2-A]Pyridine (10)3-Iodophenol (21)3-Iodopyridazine (5)3-Iodopyridin-2-Amine (12)3-Iodopyridin-2-Ol (4)3-Iodopyridin-4-Amine (6)3-Iodopyridin-4-Ol (4)3-Iodopyridine (37)3-Iodotetrahydrofuran (3)4-Bromo-2-Chloro-1-Iodobenzene (4)4-Bromo-2-Fluoro-1-Iodobenzene (8)4-Bromo-2-Iodoaniline (8)4-Chloro-2-Iodoaniline (9)4-Chloro-3-Iodoaniline (3)4-Chloro-5-Iodopyrimidine (6)4-Fluoro-2-Iodoaniline (7)4-Fluoro-3-Iodobenzoic Acid (5)4-Iodo-1,1'-Biphenyl (9)4-Iodo-1H-Pyrazole (9)4-Iodo-N,N-Diphenylaniline (3)4-Iodopyridine (17)4-Iodopyrimidine (9)5-Bromo-2-Iodopyridine (5)5-Iodo-1H-Imidazole (4)5-Iodo-1H-Pyrazole (4)5-Iodo-7H-Pyrrolo[2,3-D]Pyrimidine (4)5-Iodopyridin-2-Amine (17)5-Iodopyridin-2-Ol (8)5-Iodopyrimidine (5)6-Chloro-5-Iodopyridin-2-Amine (3)Ethyl 2-Iodobenzoate (4)Ethyl 3-Iodobenzoate (11)Methyl 2-Iodobenzoate (28)Methyl 3-Iodo-4-Methylbenzoate (6)Methyl 3-Iodobenzoate (47)Methyl 4-Amino-3-Iodobenzoate (7)N-(2-Iodophenyl)Acetamide (8)Phenyl(O-Tolyl)Iodonium (14)Tert-Butyl 3-Iodo-1H-Indole-1-Carboxylate (7)
Ketones (39956)
Alcohols (64)Aldehydes (19)Aliphatic Chain Hydrocarbons (182)Aliphatic Cyclic Hydrocarbons (156)Alkenyls (85)Alkynyls (8)Amides (185)Amines (88)Aryls (582)Azetidines (16)Benzofurans (10)Bromides (169)Carboxylic Acid Salts (4)Carboxylic Acids (138)Chlorides (139)Difluoromethyls (1)Dioxolanes (5)Epoxides (4)Esters (319)Ethers (160)Fluorinated Building Blocks (150)Furans (10)Imidazoles (7)Indoles (20)Indolines (11)Iodides (10)Morpholines (8)Naphthyridines (4)Nitriles (57)Nitroes (28)Phenols (33)Piperidines (66)Pyrans (7)Pyrazoles (14)Pyridazines (4)Pyridines (98)Pyrimidines (8)Pyrroles (16)Pyrrolidines (23)Quinolines (50)Spiroes (47)Sulfamides (4)Sulfides (18)Sulfones (9)Sulfonic Acids (4)Tetrahydrofurans (6)Tetrahydropyrans (7)Thiazoles (19)Thiophenes (42)Trifluoromethyls (42)Other Aliphatic Heterocycles (59)Other Aromatic Heterocycles (131)Acetylacetonate (26)Bromoethanone (4)Butanedione (6)Butanedione (6)Butanone (10)Butanone (10)Carbonyl-Phenyl-Boronic Acid/Ester (184)Chloroethanone (2)Dicarbonyl (2)Dicarbonyl (2)Dichloroacetophenone (5)Dienone (2)Diethanone (5)Diethanone (5)Dione (1784)Dioxobutanoate (2)Enone (21)Ethanone (1016)Heptanedionato (7)Heptanedionato (7)Hydroxyethanone (3)Methyl-Pyrazolone (87)Oxoacetaldehyde (4)Oxoazepane (3)Oxoazepane (3)Oxochroman (5)Oxotetrahydro (11)Pyrimidinone (3)Tetraone (45)Tetraone (45)Trifluoroacetophenone (9)Trifluoroethanone (15)(1E,4E)-1,5-Diphenylpenta-1,4-Dien-3-One (7)(1E,6E)-1,7-Diphenylhepta-1,6-Diene-3,5-Dione (7)(2-Aminophenyl)(Phenyl)Methanone (30)(2-Bromophenyl)(Phenyl)Methanone (3)(2-Chlorophenyl)(Phenyl)Methanone (19)(2-Fluorophenyl)(Phenyl)Methanone (15)(2-Hydroxyphenyl)(Phenyl)Methanone (24)(2-Iodophenyl)(Phenyl)Methanone (4)(3-Chlorophenyl)(Phenyl)Methanone (19)(3-Fluorophenyl)(Phenyl)Methanone (5)(3-Nitrophenyl)(Phenyl)Methanone (5)(4-Aminophenyl)(Phenyl)Methanone (4)(4-Bromophenyl)(Phenyl)Methanone (9)(4-Chlorophenyl)(Phenyl)Methanone (11)(4-Fluorophenyl)(Phenyl)Methanone (8)(4-Hydroxyphenyl)(Phenyl)Methanone (3)(4-Methoxyphenyl)(Phenyl)Methanone (7)(4-Nitrophenyl)(Phenyl)Methanone (3)(R)-5-Thia-1-Azabicyclo[4.2.0]Oct-2-En-8-One (8)1,3-Dioxoisoindolin-2-Yl 2-Phenylacetate (5)1,3-Diphenylpropan-1-One (9)1,4-Diazepan-5-One (9)1,4-Dihydroisoquinolin-3(2H)-One (5)1-(1H-Indazol-1-Yl)Ethan-1-One (7)1-(1H-Indol-1-Yl)Ethan-1-One (9)1-(1H-Indol-3-Yl)Ethan-1-One (6)1-(1H-Pyrrolo[2,3-B]Pyridin-3-Yl)Ethan-1-One (4)1-(2-(Trifluoromethyl)Phenyl)Ethan-1-One (9)1-(2-Aminophenyl)Ethan-1-One (29)1-(2-Bromophenyl)Ethan-1-One (27)1-(2-Chlorophenyl)Ethan-1-One (40)1-(2-Fluorophenyl)Ethan-1-One (58)1-(2-Hydroxyphenyl)-2-Phenylethan-1-One (6)1-(2-Hydroxyphenyl)-3-Phenylprop-2-En-1-One (11)1-(2-Hydroxyphenyl)Ethan-1-One (74)1-(2-Iodophenyl)Ethan-1-One (4)1-(2-Methoxyphenyl)Ethan-1-One (34)1-(2-Nitrophenyl)Ethan-1-One (24)1-(3-(Benzyloxy)Phenyl)Ethan-1-One (3)1-(3-Aminophenyl)Ethan-1-One (18)1-(3-Chlorophenyl)Ethan-1-One (38)1-(3-Fluorophenyl)Ethan-1-One (63)1-(3-Hydroxyphenyl)Ethan-1-One (20)1-(3-Iodophenyl)Ethan-1-One (7)1-(3-Methoxy-2-Nitrophenyl)Ethan-1-One (4)1-(3-Methoxyphenyl)Ethan-1-One (38)1-(3-Nitrophenyl)Ethan-1-One (25)1-(3-Nitrophenyl)Propan-1-One (4)1-(4-(Benzyloxy)Phenyl)Ethan-1-One (4)1-(4-Fluorophenyl)Ethan-1-One (36)1-([1,1'-Biphenyl]-4-Yl)Ethan-1-One (5)1-(Benzofuran-2-Yl)Ethan-1-One (3)1-(Imidazo[1,2-A]Pyridin-3-Yl)Ethan-1-One (3)1-(M-Tolyl)Ethan-1-One (37)1-(Naphthalen-2-Yl)Ethan-1-One (3)1-(O-Tolyl)Ethan-1-One (26)1-(Piperidin-1-Yl)Ethan-1-One (13)1-(Pyrazin-2-Yl)Ethan-1-One (5)1-(Pyridin-2-Yl)Ethan-1-One (36)1-(Pyridin-3-Yl)Ethan-1-One (29)1-(Pyridin-4-Yl)Ethan-1-One (14)1-(Pyrimidin-2-Yl)Ethan-1-One (5)1-(Pyrimidin-4-Yl)Ethan-1-One (6)1-(Pyrrolidin-1-Yl)Ethan-1-One (9)1-(Thiazol-2-Yl)Ethan-1-One (9)1-(Thiophen-2-Yl)Ethan-1-One (17)1-Aminoanthracene-9,10-Dione (8)1-Benzylindoline-2,3-Dione (5)1-Benzylpiperidin-3-One (5)1-Chloroanthracene-9,10-Dione (6)1-Hydroxyanthracene-9,10-Dione (13)1-Methylpiperazin-2-One (6)1-Methylpyrrolidin-2-One (13)1-Phenylbutan-1-One (13)1-Phenylpentan-1-One (12)1-Phenylpiperidin-2-One (8)1-Phenylpropan-2-One (36)2,2,2-Trifluoro-1-Phenylethan-1-One (51)2,2-Dimethyl-1-Phenylpropan-1-One (8)2,2-Dimethyl-2H-Benzo[B][1,4]Oxazin-3(4H)-One (3)2,3,4,9-Tetrahydro-1H-Carbazol-1-One (7)2,3-Dihydrofuran-3-One (9)2,3-Dihydropyridin-2-One (5)2,5-Dichlorobenzoic Acid (13)2-Amino-1-(Pyrrolidin-1-Yl)Ethan-1-One (5)2-Benzalacetophenone (36)2-Benzoylbenzoic Acid (10)2-Chloro-1-Phenylethan-1-One (20)2-Chlorocyclohexa-2,5-Diene-1,4-Dione (4)2-Chloroquinazolin-4(3H)-One (4)2-Hydroxy-1,2-Diphenylethan-1-One (3)2-Hydroxy-1-Phenylethan-1-One (8)2-Hydroxycyclohexa-2,5-Diene-1,4-Dione (3)2-Hydroxyquinolin-4(1H)-One (3)2-Methoxycyclohexa-2,5-Diene-1,4-Dione (7)2-Methyl-1-Phenylpropan-1-One (20)2-Methyl-2H-Benzo[B][1,4]Oxazin-3(4H)-One (3)2-Methyl-3,4-Dihydroisoquinolin-1(2H)-One (6)2-Methylcyclohexa-2,5-Diene-1,4-Dione (5)2-Methylcyclohexan-1-One (3)2-Methylisoindolin-1-One (7)2-Methylisoindoline-1,3-Dione (5)2-Methylpyridazin-3(2H)-One (7)2-Oxo-2-Phenylacetaldehyde (12)2-Oxo-2-Phenylacetic Acid (17)3,3-Difluoropiperidin-4-One (3)3,3-Dimethylcyclohexan-1-One (4)3,3-Dimethylpiperidin-4-One (3)3,4-Dihydroquinoxalin-2(1H)-One (7)3-Acetyl-2H-Chromen-2-One (3)3-Aminopyrrolidin-2-One (5)3-Bromo-1H-Pyrrole-2,5-Dione (3)3-Chloro-1-Phenylpropan-1-One (10)3-Fluoropiperidin-4-One (3)3-Hydroxy-2-Phenyl-4H-Chromen-4-One (21)3-Hydroxypyrrolidin-2-One (4)3-Methylcyclohexan-1-One (9)3-Methylene-2,3-Dihydro-1H-Inden-1-One (5)3-Methylpiperazin-2-One (7)3-Methylpyrimidin-4(3H)-One (6)3-Oxo-3-Phenylpropanenitrile (21)3-Oxo-N-Phenylbutanamide (10)3-Phenyl-1-(Pyrrolidin-1-Yl)Propan-1-One (1)3-Phenyl-2-Thioxoimidazolidin-4-One (6)3-Phenylacrylaldehyde (29)3-Phenylimidazolidine-2,4-Dione (4)4,4,4-Trifluoro-1-Phenylbutane-1,3-Dione (10)4-(1,2,2-Triphenylvinyl)Benzaldehyde (5)4-(Hydroxymethyl)Pyrrolidin-2-One (4)4-Benzylmorpholin-3-One (6)4-Bromoisoindolin-1-One (3)4-Bromopyridazin-3(2H)-One (3)4-Chloro-1-Phenylbutan-1-One (8)4-Chloropyridazin-3(2H)-One (4)4-Fluoroindoline-2,3-Dione (3)4-Hydroxy-2H-Chromen-2-One (10)4-Hydroxydihydrofuran-2(3H)-One (14)4-Hydroxypiperidin-2-One (3)4-Hydroxypyrrolidin-2-One (7)4-Methyl-2H-Chromen-2-One (27)4-Methylisobenzofuran-1(3H)-One (3)4-Oxo-4-Phenylbutanoic Acid (16)4-Phenylbutan-2-One (15)4-Phenylcyclohexan-1-One (11)4-Phenylmorpholin-3-One (6)5,6-Dihydrobenzo[D]Thiazol-7(4H)-One (5)5-(Aminomethyl)Pyrrolidin-2-One (3)5-(Hydroxymethyl)Pyrrolidin-2-One (7)5-Bromoisoindolin-1-One (8)5-Bromoisoindoline-1,3-Dione (3)5-Bromopyrimidin-4(3H)-One (3)5-Chloroisoindoline-1,3-Dione (3)5-Chloropyrimidin-4(3H)-One (4)5-Chloropyrimidine-2,4(1H,3H)-Dione (3)5-Fluoro-3,4-Dihydronaphthalen-1(2H)-One (5)5-Fluorochroman-4-One (3)5-Fluoroindolin-2-One (4)5-Fluoroisoindolin-1-One (3)5-Fluoropyrimidin-4(3H)-One (3)5-Fluoropyrimidine-2,4(1H,3H)-Dione (3)5-Hydroxy-2-Phenyl-4H-Chromen-4-One (36)5-Hydroxypiperidin-2-One (3)5-Methoxy-2-Phenyl-4H-Chromen-4-One (9)5-Methyl-3,4-Dihydronaphthalen-1(2H)-One (6)5-Methylindolin-2-One (3)5-Oxopyrrolidine-2-Carboxylic Acid (4)6-Bromoisoindolin-1-One (4)6-Fluorochroman-4-One (5)6-Fluoroindoline-2,3-Dione (3)6-Fluoroisobenzofuran-1(3H)-One (3)6-Hydroxy-2,3-Dihydro-1H-Inden-1-One (3)6-Methoxy-2,3-Dihydro-1H-Inden-1-One (6)6-Methoxy-3,4-Dihydronaphthalen-1(2H)-One (5)6-Methoxyisoindolin-1-One (3)6-Methyl-2,3-Dihydro-1H-Inden-1-One (3)7,8-Dihydroquinolin-5(6H)-One (5)7-(Diethylamino)-2H-Chromen-2-One (9)7-Bromo-3,4-Dihydronaphthalen-1(2H)-One (8)7-Bromochroman-4-One (4)7-Bromoindoline-2,3-Dione (3)7-Bromoquinazolin-4(3H)-One (4)7-Chloro-3,4-Dihydronaphthalen-1(2H)-One (6)7-Fluorochroman-4-One (5)7-Fluoroindolin-2-One (3)7-Fluoroquinazolin-4(3H)-One (4)7-Hydroxy-2H-Chromen-2-One (23)7-Methoxy-2H-Chromen-2-One (12)7-Methoxy-3,4-Dihydronaphthalen-1(2H)-One (6)7-Methoxychroman-4-One (3)7-Methylindoline-2,3-Dione (7)8-Bromochroman-4-One (4)8-Fluoro-3,4-Dihydronaphthalen-1(2H)-One (5)8-Fluoroisoquinolin-1(2H)-One (3)8-Methylchroman-4-One (4)Acetophenone (1114)Azepan-2-One (18)Benzo[Cd]Indol-2(1H)-One (5)Benzofuran-2(3H)-One (7)Chroman-2-One (5)Cyclobutanone (24)Cyclohex-2-En-1-One (27)Cyclohexane-1,3-Dione (12)Cyclopent-2-En-1-One (21)Cyclopentane-1,3-Dione (6)Dihydro-2H-Pyran-2,6(3H)-Dione (5)Dihydrofuran-2,5-Dione (12)Dihydrofuran-3(2H)-One (8)Dihydrothiophen-3(2H)-One (4)Ethene-1,1-Diyldibenzene (10)Ethyl 2-Oxo-2-Phenylacetate (18)Ethyl 3-Oxo-3-Phenylpropanoate (41)Furan-2(5H)-One (26)Isoquinolin-1-Ol (9)Methyl 3-Oxo-3-Phenylpropanoate (16)Morpholin-3-One (11)Phenyl(3-(Trifluoromethyl)Phenyl)Methanone (3)Phenyl(M-Tolyl)Methanone (3)Phenyl(P-Tolyl)Methanone (4)Phenyl(Pyridin-3-Yl)Methanone (9)Phenyl(Thiophen-2-Yl)Methanone (6)Piperidine-2,4-Dione (5)Propiophenone (59)Pyrimidine-4,6(1H,5H)-Dione (3)Tert-Butyl (2-Oxopiperidin-3-Yl)Carbamate (4)Tert-Butyl (2-Oxopyrrolidin-3-Yl)Carbamate (3)Tert-Butyl (3-Oxocyclohexyl)Carbamate (3)Tert-Butyl (5-Oxopyrrolidin-3-Yl)Carbamate (3)Tert-Butyl (6-Oxopiperidin-3-Yl)Carbamate (3)Tert-Butyl 2-Oxopiperidine-1-Carboxylate (3)Tert-Butyl 2-Oxopyrrolidine-1-Carboxylate (15)Tert-Butyl 4-Oxopiperidine-1-Carboxylate (16)Tetrahydropyrimidin-2(1H)-One (6)
Nitriles (28592)
Alcohols (45)Aldehydes (12)Aliphatic Chain Hydrocarbons (92)Aliphatic Cyclic Hydrocarbons (29)Alkenyls (62)Amides (92)Amines (142)Aryls (353)Azetidines (5)Benzyl Bromides (5)Bromides (82)Carboxylic Acids (45)Chlorides (97)Esters (102)Ethers (103)Fluorinated Building Blocks (76)Furans (7)Guanidines (4)Hydrazines (5)Indazoles (8)Indoles (31)Indolines (5)Iodides (10)Isoquinolines (4)Ketones (57)Morpholines (5)Nitroes (26)Oxazoles (6)Phenols (5)Piperazines (17)Piperidines (54)Pyrazoles (39)Pyridazines (5)Pyridines (126)Pyrimidines (15)Pyrroles (6)Pyrrolidines (11)Quinolines (19)Spiroes (2)Sulfamides (4)Sulfides (13)Sulfones (9)Sulfonyl Chlorides (5)Tetrahydropyrans (9)Thiazoles (7)Thiophenes (15)Trifluoromethyls (13)Other Aromatic Heterocycles (31)1-Phenyl-1H-Pyrazole-4-Carbonitrile (17)1-Phenylcyclobutane-1-Carbonitrile (7)1-Phenylcyclopropane-1-Carbonitrile (19)1H-Imidazole-4-Carbonitrile (2)1H-Indazole-3-Carbonitrile (9)1H-Indole-2-Carbonitrile (9)1H-Indole-3-Carbonitrile (17)1H-Indole-5-Carbonitrile (5)1H-Indole-6-Carbonitrile (4)1H-Indole-7-Carbonitrile (6)1H-Pyrazole-4-Carbonitrile (5)1H-Pyrrole-2-Carbonitrile (7)1H-Pyrrole-3-Carbonitrile (9)2,3-Difluorobenzonitrile (29)2,4-Dichlorobenzonitrile (4)2,4-Difluorobenzonitrile (21)2,6-Dibromobenzonitrile (4)2,6-Dichlorobenzonitrile (8)2,6-Difluorobenzonitrile (28)2-(1,3,2-Dioxaborinan-2-Yl)Benzonitrile (4)2-(2-Bromophenyl)Acetonitrile (10)2-(2-Chlorophenyl)Acetonitrile (16)2-(2-Fluorophenyl)Acetonitrile (30)2-(3-Chlorophenyl)Acetonitrile (16)2-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Benzonitrile (14)2-(4-(Benzyloxy)Phenyl)Acetonitrile (3)2-(Bromomethyl)Benzonitrile (15)2-(Piperazin-1-Yl)Benzonitrile (6)2-(Pyridin-2-Yl)Acetonitrile (8)2-(Pyridin-3-Yl)Acetonitrile (6)2-(Trifluoromethoxy)Benzonitrile (7)2-(Trifluoromethyl)Benzonitrile (35)2-Amino-3-Bromobenzonitrile (7)2-Amino-3-Chlorobenzonitrile (3)2-Amino-6-Chlorobenzonitrile (3)2-Aminonicotinonitrile (6)2-Bromo-6-Fluorobenzonitrile (7)2-Chloro-3-Methylbenzonitrile (5)2-Chloro-4-Fluorobenzonitrile (9)2-Chloro-6-Fluorobenzonitrile (5)2-Chloro-N-Methylaniline (4)2-Chlorobenzonitrile (99)2-Hydroxybenzonitrile (45)2-Iodobenzonitrile (26)2-Naphthonitrile (15)2-Nitrobenzonitrile (37)2-Oxo-2H-Chromene-3-Carbonitrile (4)2-Phenoxyacetonitrile (11)2-Phenoxybenzonitrile (5)2-Phenylacetonitrile (172)3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Benzonitrile (25)3-(Trifluoromethoxy)Benzonitrile (11)3-(Trifluoromethyl)Benzonitrile (47)3-Bromo-2-Fluorobenzonitrile (10)3-Bromo-2-Hydroxybenzonitrile (3)3-Bromobenzonitrile (110)3-Chloro-2-Fluorobenzonitrile (7)3-Chlorobenzonitrile (100)3-Chloropyrazine-2-Carbonitrile (6)3-Fluoropicolinonitrile (5)3-Hydroxybenzonitrile (40)3-Iodobenzonitrile (27)3-Nitrobenzonitrile (59)3-Oxo-3-Phenylpropanenitrile (21)3-Phenoxybenzonitrile (5)4-Amino-2-Chlorobenzonitrile (8)4-Chloropyrimidine-5-Carbonitrile (6)4-Cyanophenyl Benzoate (11)4-Cyclohexylbenzonitrile (10)4-Fluorobenzonitrile (89)4-Nitrobenzonitrile (25)5-Fluoropicolinonitrile (5)6-Methylnicotinonitrile (7)6-Methylpicolinonitrile (10)[1,1'-Biphenyl]-2-Carbonitrile (12)[1,1'-Biphenyl]-4-Carbonitrile (27)Benzo[B]Thiophene-2-Carbonitrile (3)Benzo[D]Thiazole-2-Carbonitrile (11)Benzonitrile (1335)Furan-2-Carbonitrile (8)Imidazo[1,2-A]Pyridine-6-Carbonitrile (3)Isonicotinonitrile (16)Isoquinoline-1-Carbonitrile (7)Methyl 2-Cyanobenzoate (22)Methyl 3-Cyanobenzoate (29)Morpholine-2-Carbonitrile (4)Nicotinonitrile (82)Pyrazine-2-Carbonitrile (15)Pyrimidine-2-Carbonitrile (14)Pyrimidine-4-Carbonitrile (9)Pyrimidine-5-Carbonitrile (6)Pyrrolidine-2-Carbonitrile (9)Pyrrolidine-3-Carbonitrile (6)Thiazole-5-Carbonitrile (7)Thiophene-2-Carbonitrile (23)Thiophene-3-Carbonitrile (13)Acetonitrile (255)Acrylonitrile (8)Aminobutanenitrile (5)Aminocyclohexanecarbonitrile (6)Bromobenzonitrile (15)Bromonicotinonitrile (9)Bromophenylacetonitrile (2)Butanenitrile (2)Butanenitrile (2)Chlorobenzonitrile (27)Chloroisonicotinonitrile (4)Chloronicotinonitrile (40)Cyanide (432)Cyanide (432)Cyanoacetamide (4)Cyanoacetate (14)Cyanobenzaldehyde (4)Cyanobenzoate (27)Cyanobenzoic (20)Cyanoethyl (15)Cyanoethyl (15)Cyanoisonicotinate (4)Cyanomethyl (42)Cyanomethyl (42)Cyanonicotinate (8)Cyanophenoxy (8)Cyanophenyl (63)Cyanophenylboronic (5)Cyanopropan (4)Cyanopropan (4)Diacetonitrile (1)Diaminobenzonitrile (8)Dibromoisonicotinonitrile (3)Dicarbonitrile (45)Dicarbonitrile (45)Dichlorobenzonitrile (12)Dichloroisonicotinonitrile (4)Difluorobenzonitrile (41)Fluorobenzonitrile (85)Fluoroisonicotinonitrile (5)Fluoronicotinonitrile (14)Fluorophenylacetonitrile (8)Fluoropicolinonitrile (13)Formylnicotinonitrile (5)Formylnicotinonitrile (5)Hydrazinylbenzonitrile (6)Iodobenzonitrile (44)Iodonicotinonitrile (8)Malononitrile (43)Malononitrile (43)Methoxyisonicotinonitrile (7)Methoxyisonicotinonitrile (7)Methoxynicotinonitrile (18)Methoxynicotinonitrile (18)Methylnicotinonitrile (57)Methylnicotinonitrile (57)Methylpropanenitrile (9)Methylpropanenitrile (9)Nitropicolinonitrile (10)Oxopropanenitrile (8)Phenylacrylonitrile (21)Propanenitrile (41)Propanenitrile (41)Pyrazino-Nitrile (50)Tetrafluorobenzonitrile (5)Thiocyanatoaniline (3)Tricarbonitrile (5)Tricarbonitrile (5)Trifluorobenzonitrile (7)Trifluoromethylbenzonitrile (5)
Nitroes (24098)
Alcohols (42)Aldehydes (22)Aliphatic Chain Hydrocarbons (7)Alkenyls (21)Amines (114)Anhydrides (5)Aryls (356)Benzofurans (5)Benzothiazoles (5)Benzoxazoles (5)Bromides (62)Carboxylic Acids (44)Chlorides (81)Epoxides (5)Esters (72)Ethers (84)Fluorinated Building Blocks (75)Furans (8)Imidazoles (13)Indazoles (7)Indoles (10)Indolines (9)Iodides (6)Ketones (28)Morpholines (12)Nitriles (26)Oxazoles (4)Phenols (12)Piperazines (14)Piperidines (13)Pyrazoles (28)Pyridines (61)Pyrimidines (13)Pyrroles (8)Pyrrolidines (12)Quinazolines (4)Quinolines (19)Quinoxalines (7)Sulfamides (7)Sulfides (12)Sulfonates (6)Thiazoles (8)Thiophenes (7)Trifluoromethyls (5)Other Aromatic Heterocycles (22)Difluoronitrobenzene (9)Dinitro (7)Dinitroaniline (5)Nitroacetophenone (7)Nitroaniline (211)Nitrobenzaldehyde (100)Nitrobenzamide (35)Nitrobenzenesulfonamide (26)Nitrobenzonitrile (106)Nitrobenzotrifluoride (16)Nitroisophthalate (4)(2-Nitrophenyl)Boronic Acid (13)(2-Nitrophenyl)Methanol (29)(2-Nitrovinyl)Benzene (28)(3-Nitrophenyl)(Phenyl)Methanone (5)(3-Nitrophenyl)Boronic Acid (32)(3-Nitrophenyl)Methanol (31)(4-Nitrophenyl)(Phenyl)Methanone (3)(4-Nitrophenyl)Methanol (17)1,2-Dichloro-4-Nitrobenzene (16)1,2-Difluoro-3-Nitrobenzene (23)1,2-Difluoro-4-Nitrobenzene (43)1,2-Dimethyl-3-Nitrobenzene (6)1,2-Dinitrobenzene (6)1,3-Dibromo-2-Nitrobenzene (4)1,3-Dichloro-2-Nitrobenzene (5)1,3-Difluoro-2-Nitrobenzene (16)1,3-Dimethyl-2-Nitrobenzene (9)1,3-Dinitrobenzene (31)1,4-Difluoro-2-Nitrobenzene (28)1,4-Dimethyl-2-Nitrobenzene (10)1-(2-Nitrophenyl)Ethan-1-One (24)1-(3-Methoxy-2-Nitrophenyl)Ethan-1-One (4)1-(3-Nitrophenyl)Ethan-1-One (25)1-(3-Nitrophenyl)Propan-1-One (4)1-(Benzyloxy)-2-Nitrobenzene (3)1-(Benzyloxy)-3-Nitrobenzene (10)1-(Bromomethyl)-2-Nitrobenzene (23)1-(Methylsulfonyl)-3-Nitrobenzene (7)1-(Tert-Butyl)-2-Nitrobenzene (4)1-(Tert-Butyl)-3-Nitrobenzene (7)1-Bromo-2-Chloro-4-Nitrobenzene (5)1-Bromo-2-Fluoro-3-Nitrobenzene (11)1-Bromo-2-Fluoro-4-Nitrobenzene (13)1-Bromo-3-Methyl-2-Nitrobenzene (3)1-Bromo-3-Nitrobenzene (235)1-Chloro-2-Fluoro-4-Nitrobenzene (9)1-Chloro-2-Methyl-3-Nitrobenzene (6)1-Chloro-2-Nitrobenzene (138)1-Chloro-3-Fluoro-2-Nitrobenzene (6)1-Chloro-3-Nitrobenzene (161)1-Ethoxy-3-Nitrobenzene (7)1-Fluoro-3-Nitrobenzene (216)1-Iodo-2-Nitrobenzene (36)1-Iodo-3-Nitrobenzene (45)1-Methoxy-3-Nitrobenzene (108)1-Methoxy-4-Nitrobenzene (90)1-Methyl-2-Nitrobenzene (176)1-Methyl-3-Nitrobenzene (147)1-Methyl-4-Nitrobenzene (82)1-Nitro-2-(Trifluoromethoxy)Benzene (8)1-Nitro-2-(Trifluoromethyl)Benzene (32)1-Nitro-2-Phenoxybenzene (4)1-Nitro-3-(Trifluoromethoxy)Benzene (9)1-Nitro-3-(Trifluoromethyl)Benzene (82)1-Nitro-4-Phenoxybenzene (5)1-Nitronaphthalene (3)2,4-Dichloro-1-Nitrobenzene (19)2,4-Difluoro-1-Nitrobenzene (46)2,4-Dimethoxy-1-Nitrobenzene (5)2,4-Dimethyl-1-Nitrobenzene (15)2-((2-Nitrophenyl)Amino)Ethan-1-Ol (5)2-(2-Nitrophenyl)Acetic Acid (20)2-(4-Nitrophenyl)Acetic Acid (9)2-Bromo-1-Chloro-4-Nitrobenzene (9)2-Bromo-1-Fluoro-3-Nitrobenzene (9)2-Bromo-1-Fluoro-4-Nitrobenzene (23)2-Bromo-1-Methyl-4-Nitrobenzene (12)2-Bromo-3-Nitrobenzoic Acid (4)2-Bromo-4-Chloro-1-Nitrobenzene (5)2-Bromo-4-Fluoro-1-Nitrobenzene (23)2-Bromo-4-Nitroaniline (12)2-Bromo-6-Nitroaniline (14)2-Bromo-6-Nitrophenol (9)2-Chloro-1,3-Dinitrobenzene (2)2-Chloro-1-Fluoro-4-Nitrobenzene (16)2-Chloro-1-Methyl-3-Nitrobenzene (4)2-Chloro-3-Nitrobenzoic Acid (4)2-Chloro-4-Fluoro-1-Nitrobenzene (13)2-Chloro-4-Methyl-1-Nitrobenzene (6)2-Chloro-4-Nitrophenol (5)2-Chloro-5-Nitropyrimidine (5)2-Chloro-6-Nitroaniline (11)2-Chloro-6-Nitrophenol (7)2-Fluoro-1-Methoxy-4-Nitrobenzene (8)2-Fluoro-4-Methyl-1-Nitrobenzene (13)2-Iodo-1-Methyl-4-Nitrobenzene (3)2-Iodo-6-Nitroaniline (3)2-Methoxy-5-Nitropyridine (8)2-Methoxy-6-Nitrophenol (5)2-Methyl-3-Nitrobenzoic Acid (7)2-Methyl-3-Nitropyridine (9)2-Methyl-6-Nitroaniline (10)2-Nitro-1H-Pyrrole (5)2-Nitro-N-Phenylaniline (3)2-Nitrobenzaldehyde (53)2-Nitrobenzoic Acid (65)2-Nitrobenzonitrile (37)2-Nitrofuran (9)2-Nitronaphthalene (4)2-Nitropyridine (40)2-Nitrothiophene (20)3-Chloro-2-Nitroaniline (5)3-Chloro-4-Nitroaniline (5)3-Fluoro-4-Nitroaniline (7)3-Methoxy-2-Nitrobenzaldehyde (3)3-Methoxy-4-Nitrobenzoic Acid (4)3-Methyl-5-Nitropyridine (4)3-Nitroaniline (98)3-Nitrobenzaldehyde (49)3-Nitrobenzamide (17)3-Nitrobenzene-1,2-Diol (6)3-Nitrobenzenesulfonamide (8)3-Nitrobenzoic Acid (109)3-Nitrobenzonitrile (59)3-Nitrophenol (72)3-Nitropyridin-2-Ol (11)3-Nitropyridin-4-Amine (5)3-Nitropyridine (54)3-Nitrothiophene (6)4,4,5,5-Tetramethyl-2-(2-Nitrophenyl)-1,3,2-Dioxaborolane (5)4-Fluoro-1-Methyl-2-Nitrobenzene (12)4-Fluoro-2-Nitroaniline (14)4-Hydroxy-3-Nitrobenzaldehyde (5)4-Methyl-3-Nitroaniline (8)4-Methyl-3-Nitropyridine (8)4-Nitro-1H-Indazole (10)4-Nitro-1H-Indole (6)4-Nitro-1H-Pyrazole (8)4-Nitroaniline (65)4-Nitrobenzaldehyde (18)4-Nitrobenzene-1,3-Diol (5)4-Nitrobenzoic Acid (35)4-Nitrobenzonitrile (25)4-Nitrophenol (67)4-Nitropyridine (11)5-(4-Nitrophenyl)Oxazole (3)5-Methoxy-2-Nitroaniline (7)5-Methyl-2-Nitroaniline (6)5-Nitro-1H-Indazole (5)5-Nitro-1H-Indole (6)5-Nitro-1H-Pyrazole (9)5-Nitro-1H-Pyrazolo[3,4-B]Pyridine (3)5-Nitro-2,3-Dihydrobenzofuran (3)5-Nitrobenzo[D][1,3]Dioxole (4)5-Nitrobenzo[D]Thiazole (3)5-Nitroisoquinoline (7)5-Nitronicotinic Acid (5)5-Nitropyridin-2-Amine (6)6-Nitro-1,2,3,4-Tetrahydroisoquinoline (3)6-Nitro-1H-Indazole (9)6-Nitro-1H-Indole (8)6-Nitrobenzo[D]Thiazole (3)7-Nitro-1,2,3,4-Tetrahydroisoquinoline (3)7-Nitro-1H-Indazole (11)7-Nitro-1H-Indole (11)Ethyl 2-Nitrobenzoate (9)Ethyl 3-Nitrobenzoate (22)Ethyl Cinnamate (26)Methyl 2-Nitrobenzoate (54)Methyl 3-Nitrobenzoate (109)Methyl 4-Amino-3-Nitrobenzoate (9)Methyl 4-Nitrobenzoate (29)N-(2-Nitrophenyl)Acetamide (11)N-Methyl-2-Nitroaniline (20)N-Methyl-4-Nitroaniline (6)Nitrobenzene (2277)
Phenols (1870)
Aminophenol (9)Bromophenol (13)Chlorophenol (28)Dibromophenol (9)Dichlorophenol (17)Difluorophenol (36)Dihydroxybenzaldehyde (16)Dihydroxybenzamide (6)Dihydroxybenzoate (15)Dihydroxybenzoic (17)Dihydroxybenzonitrile (6)Dihydroxyphenyl (93)Dihydroxystyryl (4)Diiodophenol (6)Fluorophenol (73)Hydroxyacetophenone (16)Hydroxybenzaldehyde (88)Hydroxybenzamide (24)Hydroxybenzenesulfonic (4)Hydroxybenzenesulfonic Acid (3)Hydroxybenzoate (113)Hydroxybenzohydrazide (4)Hydroxybenzoic (89)Hydroxybenzonitrile (60)Hydroxybenzoyl (1)Hydroxyphenoxy (7)Hydroxyphenylacetic (7)Hydroxyphenylacetic Acid (7)Hydroxyphenylboronic (8)Hydroxypropiophenone (3)Iodophenol (41)Nitrophenol (134)Tetrafluorophenol (9)Trifluorophenol (5)Trihydroxybenzaldehyde (5)Trihydroxybenzoate (12)Trihydroxybenzoic (5)Trihydroxybenzoic Acid (4)Trihydroxyphenyl (10)1,2,3,4-Tetrahydroisoquinolin-5-Ol (3)1,2,3,4-Tetrahydroisoquinolin-6-Ol (6)1-(2-Hydroxyphenyl)Ethan-1-One (74)1-(3-Hydroxyphenyl)Ethan-1-One (20)1-Hydroxyanthracene-9,10-Dione (13)2,3-Dichlorophenol (9)2,3-Difluorophenol (36)2,4-Dibromophenol (16)2,4-Difluorophenol (21)2,5-Dichlorophenol (7)2,6-Dibromophenol (20)2,6-Dichlorophenol (16)2,6-Difluorophenol (23)2,6-Diiodophenol (9)2,6-Dimethylphenol (19)2-(2-Hydroxyphenyl)Acetic Acid (8)2-(3-Hydroxyphenyl)Acetic Acid (8)2-(Benzyloxy)Phenol (5)2-(Difluoromethoxy)Phenol (5)2-(Tert-Butyl)Phenol (56)2-(Trifluoromethoxy)Phenol (8)2-(Trifluoromethyl)Phenol (23)2-Amino-3-Bromophenol (3)2-Amino-3-Chlorophenol (4)2-Aminophenol (107)2-Benzylphenol (8)2-Bromo-3-Methylphenol (4)2-Bromo-6-Methylphenol (7)2-Bromobenzene-1,3-Diol (7)2-Bromobenzene-1,4-Diol (4)2-Chloro-4-Fluorophenol (9)2-Chloro-4-Nitrophenol (5)2-Chloro-6-Methylphenol (5)2-Chloro-6-Nitrophenol (7)2-Chlorobenzene-1,3-Diol (4)2-Chlorophenol (163)2-Ethylphenol (10)2-Hydroxy-N-Phenylbenzamide (3)2-Hydroxybenzamide (14)2-Hydroxybenzonitrile (45)2-Iodobenzene-1,3-Diol (3)2-Iodophenol (54)2-Isopropylphenol (28)2-Methoxyphenol (201)3,4-Dibromophenol (3)3,4-Difluorophenol (19)3-(Benzyloxy)Phenol (5)3-(Trifluoromethoxy)Phenol (12)3-(Trifluoromethyl)Phenol (43)3-Bromo-2-Fluorophenol (5)3-Bromo-4-Chlorophenol (5)3-Bromo-4-Fluorophenol (11)3-Bromobenzene-1,2-Diol (8)3-Chloro-4-Hydroxybenzaldehyde (7)3-Chlorobenzene-1,2-Diol (6)3-Chlorophenol (112)3-Fluorophenol (177)3-Hydroxybenzonitrile (40)3-Iodophenol (21)3-Nitrobenzene-1,2-Diol (6)3-Nitrophenol (72)4-(Benzyloxy)Phenol (2)4-(Trifluoromethyl)Phenol (18)4-Amino-2-Fluorophenol (9)4-Amino-3-Chlorophenol (5)4-Benzylphenol (14)4-Chlorobenzene-1,3-Diol (6)4-Fluorophenol (111)4-Hydroxybenzenesulfonic Acid (5)4-Nitrobenzene-1,3-Diol (5)4-Nitrophenol (67)6-Hydroxy-2,3-Dihydro-1H-Inden-1-One (3)Ethyl 2-Hydroxybenzoate (18)Ethyl 3-Hydroxybenzoate (12)M-Cresol (145)Methyl 2-(4-Hydroxyphenyl)Acetate (10)Methyl 2-Hydroxybenzoate (74)Methyl 3-Hydroxybenzoate (58)O-Cresol (164)P-Cresol (71)Pyrocatechol (128)Resorcinol (147)
Phosphoruses (831)
Aryls (10)Esters (6)Fluorinated Building Blocks (5)1,1,1-Trifluoro-N-(4-Oxido-2,6-Diphenyldinaphtho[2,1-D:1',2'-F][1,3,2]Dioxaphosphepin-4-Yl)Methanesulfonamide (16)Diethyl Benzylphosphonate (17)Dimethyl(Phenyl)Phosphine Oxide (20)Ethyltriphenylphosphonium (2)Methyltriphenylphosphonium (3)Phenyl Phosphate (3)Phenylphosphonic Acid (6)Diethoxyphosphoryl (5)Diisopropylphosphoramidite (4)Dimethoxyphosphoryl (4)Dimethoxyphosphoryl (4)Dimethylphosphine (28)Dioxaphosphepine (146)Diphosphonic (9)Phosphorothioate (1)
Silicon Compounds (450)
Butyldimethylsilyl (94)Butyldimethylsilyl (94)Butyldimethylsilyloxy (8)Dimethylsilane (16)Dimethylsilane (16)Dimethylsilyl (4)Tetramethyldisiloxane (3)Tetramethyldisiloxane (3)Triethoxysilyl (13)Triethoxysilyl (13)Triisopropylsilyl (22)Triisopropylsilyl (22)Trimethoxysilane (19)Trimethoxysilane (19)Trimethylsilane (155)1-(Triisopropylsilyl)-1H-Pyrrolo[2,3-B]Pyridine (8)Tert-Butyldimethyl(Phenoxy)Silane (17)Tetraphenylsilane (7)Trimethoxy(Phenyl)Silane (6)Trimethyl(Phenyl)Silane (22)Trimethyl(Phenylethynyl)Silane (23)
Sulfamides (10221)
Alcohols (28)Aldehydes (6)Aliphatic Chain Hydrocarbons (37)Aliphatic Cyclic Hydrocarbons (8)Alkenyls (12)Amides (43)Amines (50)Aryls (218)Azetidines (5)Benzothiazoles (6)Benzyl Bromides (3)Bromides (35)Carboxylic Acids (28)Chlorides (51)Esters (23)Ethers (23)Fluorinated Building Blocks (26)Hydrazines (4)Imidazoles (7)Indoles (6)Ketones (4)Morpholines (11)Nitriles (4)Nitroes (7)Piperazines (12)Piperidines (25)Pyrazoles (4)Pyridines (19)Pyrimidines (1)Pyrrolidines (10)Sulfonates (6)Thiadiazoles (4)Thiophenes (16)Trifluoromethyls (5)Ureas (3)Other Aliphatic Heterocycles (11)Other Aromatic Heterocycles (17)Dimethylsulfamoyl (5)Dimethylsulfamoyl (5)Methanesulfonamide (49)Methanesulfonamide (49)Methylmethanesulfonamide (3)Methylmethanesulfonamide (3)Methylsulfonamido (4)Methylsulfonamido (4)Sulfinamide (55)Sulfinamide (55)2-Fluoro-N-Phenylbenzenesulfonamide (6)2-Fluorobenzenesulfonamide (12)2-Methylbenzenesulfonamide (8)3-Nitrobenzenesulfonamide (8)4-Amino-N-Phenylbenzenesulfonamide (3)4-Bromo-N-Phenylbenzenesulfonamide (23)4-Chloro-N-Phenylbenzenesulfonamide (4)4-Methoxy-N-Phenylbenzenesulfonamide (3)4-Methoxybenzenesulfonamide (5)4-Methyl-N-Phenylbenzenesulfonamide (13)N,N-Diethylbenzenesulfonamide (8)N,N-Dimethylbenzenesulfonamide (23)N-(2-Phenoxyphenyl)Methanesulfonamide (3)N-(3-Chlorophenyl)Benzenesulfonamide (6)N-(O-Tolyl)Benzenesulfonamide (4)N-(Tert-Butyl)Benzenesulfonamide (14)N-Ethylbenzenesulfonamide (8)N-Isopropylbenzenesulfonamide (8)N-Methyl-N-(4-Phenylpyrimidin-2-Yl)Methanesulfonamide (7)N-Methylbenzenesulfonamide (15)N-Phenylbenzenesulfonamide (81)N-Phenylmethanesulfonamide (37)Phenylmethanesulfonamide (13)Pyridine-2-Sulfonamide (6)
Sulfides (10154)
Alcohols (26)Aldehydes (8)Aliphatic Chain Hydrocarbons (27)Amides (40)Amines (42)Aryls (143)Benzimidazoles (7)Bromides (18)Carboxylic Acids (57)Chlorides (50)Esters (27)Ethers (29)Fluorinated Building Blocks (21)Furans (6)Ketones (18)Nitriles (13)Nitroes (12)Phenols (3)Pyridines (18)Pyrimidines (53)Pyrrolidines (7)Tetrahydropyrans (3)Thiadiazoles (7)Thiazoles (8)Thiophenes (4)Triazoles (3)Other Aliphatic Heterocycles (8)Other Aromatic Heterocycles (8)Methylthio (429)(S)-2-(Phenylthio)Tetrahydro-2H-Pyran (5)1,2-Diphenyldisulfane (19)2-((Pyridin-2-Ylmethyl)Thio)-1H-Benzo[D]Imidazole (7)2-(Methylthio)Benzo[D]Thiazole (6)2-(Methylthio)Pyridine (5)2-(Methylthio)Pyrimidin-4-Amine (10)2-(Methylthio)Pyrimidine (19)2-(Methylthio)Thiazole (5)2-(Propylthio)Pyrimidine (5)4,6-Dichloro-2-(Methylthio)Pyrimidine (9)4-Chloro-2-(Methylthio)Pyrimidine (29)4-Methyl-2-(Methylthio)Pyrimidine (7)5-Bromo-2-(Methylthio)Pyrimidine (6)Benzyl(Phenyl)Sulfane (5)Ethyl(Phenyl)Sulfane (8)Pentafluoro(Phenyl)-L6-Sulfane (19)Phenyl(Trifluoromethyl)Sulfane (23)
Sulfones (3921)
Alcohols (7)Aliphatic Chain Hydrocarbons (19)Alkenyls (12)Amides (13)Amines (20)Aryls (121)Bromides (31)Carboxylic Acids (15)Chlorides (29)Esters (16)Ethers (12)Fluorinated Building Blocks (14)Ketones (9)Nitriles (9)Phenols (5)Piperazines (4)Piperidines (6)Pyridines (14)Pyrimidines (7)Thiomorpholines (8)Thiophenes (4)Other Aliphatic Heterocycles (9)Other Aromatic Heterocycles (5)(4-(Methylsulfonyl)Phenyl)Boronic Acid (6)(Ethylsulfonyl)Benzene (8)1-(Methylsulfonyl)-3-Nitrobenzene (7)1-(Methylsulfonyl)Piperazine (5)1-(Methylsulfonyl)Piperidine (6)1-(Phenylsulfonyl)-1H-1,2,4-Triazole (6)1-(Phenylsulfonyl)-1H-Imidazole (5)1-(Phenylsulfonyl)Azetidine (7)1-Bromo-2-(Methylsulfonyl)Benzene (4)1-Bromo-4-(Phenylsulfonyl)Benzene (7)1-Fluoro-2-(Methylsulfonyl)Benzene (5)1-Fluoro-3-(Methylsulfonyl)Benzene (10)1-Methyl-3-(Methylsulfonyl)Benzene (5)2-(Methylsulfonyl)Aniline (3)2-(Methylsulfonyl)Pyridine (11)2-(Methylsulfonyl)Pyrimidine (25)3-(Methylsulfonyl)Aniline (7)3-(Methylsulfonyl)Benzoic Acid (6)3-(Methylsulfonyl)Pyridine (5)Benzenesulfonyl Fluoride (50)Tetrahydro-2H-Thiopyran 1,1-Dioxide (8)Tetrahydrothiophene 1,1-Dioxide (13)
Sulfonic Acids (1258)
Aliphatic Cyclic Hydrocarbons (4)Amines (12)Aryls (23)Ketones (4)Aminobenzenesulfonic (4)Bromobenzenesulfonic Acid (4)Disulfonic (9)Disulfonic (9)Ethanesulfonic (3)Methanesulfonic (2)Methanesulfonic (2)Methylbenzenesulfonate (143)Propanesulfonic Acid (2)Trifluoromethanesulfonic (3)2-Aminobenzenesulfonic Acid (9)2-Methylbenzenesulfonic Acid (8)3-Aminobenzenesulfonic Acid (6)4-Chlorobenzenesulfonic Acid (4)4-Hydroxybenzenesulfonic Acid (5)Naphthalene-2-Sulfonic Acid (4)Pyridine-3-Sulfonic Acid (6)
Sulfonyl Chlorides (4011)
Aliphatic Chain Hydrocarbons (19)Aliphatic Cyclic Hydrocarbons (8)Aryls (57)Bromides (17)Ethers (19)Nitriles (5)Piperidines (4)Pyrazoles (4)Pyridines (5)Quinolines (6)Thiophenes (5)Fluorobenzenesulfonyl (12)1H-Pyrazole-4-Sulfonyl Chloride (8)2-Bromobenzenesulfonyl Chloride (8)2-Chlorobenzenesulfonyl Chloride (24)2-Fluorobenzenesulfonyl Chloride (31)2-Methoxybenzenesulfonyl Chloride (15)2-Methylbenzenesulfonyl Chloride (22)3-(Chlorosulfonyl)Benzoic Acid (15)3-(Trifluoromethyl)Benzenesulfonyl Chloride (13)3-Chlorobenzenesulfonyl Chloride (21)3-Fluorobenzenesulfonyl Chloride (22)4-Chlorobenzenesulfonyl Chloride (12)4-Fluorobenzenesulfonyl Chloride (26)Isoquinoline-5-Sulfonyl Chloride (3)Methyl 2-(Chlorosulfonyl)Benzoate (7)Phenylmethanesulfonyl Chloride (20)
Thiols (472)
Alcohols (5)Aliphatic Chain Hydrocarbons (27)Amides (4)Aryls (8)Esters (9)Dithiole (9)Dithiole (9)Ethanethioate (9)Ethanethioate (9)Methanethiol (6)1H-Benzo[D]Imidazole-2-Thiol (10)2-Chlorobenzenethiol (7)2-Mercaptoquinazolin-4(3H)-One (3)Benzo[D]Oxazole-2-Thiol (6)Benzo[D]Thiazole-2-Thiol (11)Phenylmethanethiol (16)Pyridine-2-Thiol (8)Pyrimidine-2-Thiol (6)
Trifluoromethyls (28935)
Alcohols (71)Aldehydes (10)Aliphatic Chain Hydrocarbons (68)Aliphatic Cyclic Hydrocarbons (19)Alkenyls (5)Amides (74)Amidines (3)Amines (95)Aryls (260)Bromides (95)Carboxylic Acids (71)Chlorides (61)Esters (72)Ethers (108)Fluorinated Building Blocks (502)Hydrazines (4)Imidazoles (12)Indoles (7)Iodides (15)Ketones (42)Morpholines (5)Nitriles (13)Nitroes (5)Phenols (2)Piperazines (7)Piperidines (8)Pyrazines (5)Pyrazoles (27)Pyridines (39)Pyrimidines (6)Pyrroles (4)Pyrrolidines (4)Quinolines (7)Sulfamides (5)Sulfonates (14)Thiophenes (1)Other Aromatic Heterocycles (19)Trifluoromethyl-Picolinamide (7)Trifluoromethylaniline (3)(2,2,2-Trifluoroethoxy)Benzene (18)(2-(Trifluoromethoxy)Phenyl)Boronic Acid (5)(2-(Trifluoromethyl)Phenyl)Boronic Acid (20)(2-(Trifluoromethyl)Phenyl)Methanol (10)(2-Fluoro-3-(Trifluoromethyl)Phenyl)Boronic Acid (6)(3-(Trifluoromethoxy)Phenyl)Boronic Acid (9)(3-(Trifluoromethoxy)Phenyl)Methanol (4)(3-(Trifluoromethyl)Phenyl)Boronic Acid (41)(3-(Trifluoromethyl)Phenyl)Methanol (22)(4-(Trifluoromethyl)Phenyl)Boronic Acid (23)(Perfluoropropane-2,2-Diyl)Dibenzene (7)(Trifluoromethyl)Benzene (1314)1,3-Bis(Trifluoromethyl)Benzene (70)1-(2-(Trifluoromethyl)Phenyl)Ethan-1-One (9)1-(3-(Trifluoromethoxy)Phenyl)Ethan-1-Amine (6)1-(3-(Trifluoromethyl)Phenyl)Ethan-1-Amine (25)1-(4-(Trifluoromethoxy)Phenyl)Ethan-1-Amine (6)1-(Bromomethyl)-2-(Trifluoromethyl)Benzene (13)1-(Bromomethyl)-3-(Trifluoromethyl)Benzene (17)1-Bromo-2-(Trifluoromethoxy)Benzene (22)1-Bromo-2-Fluoro-3-(Trifluoromethyl)Benzene (4)1-Bromo-3-(Trifluoromethyl)Benzene (100)1-Chloro-2-(Trifluoromethoxy)Benzene (20)1-Chloro-2-(Trifluoromethyl)Benzene (81)1-Chloro-3-(Trifluoromethoxy)Benzene (18)1-Chloro-3-(Trifluoromethyl)Benzene (101)1-Chloro-4-(Trifluoromethoxy)Benzene (8)1-Fluoro-2-(Trifluoromethoxy)Benzene (20)1-Fluoro-2-(Trifluoromethyl)Benzene (125)1-Fluoro-3-(Trifluoromethyl)Benzene (98)1-Fluoro-4-(Trifluoromethoxy)Benzene (14)1-Fluoro-4-(Trifluoromethyl)Benzene (53)1-Iodo-2-(Trifluoromethyl)Benzene (13)1-Iodo-3-(Trifluoromethyl)Benzene (30)1-Methoxy-2-(Trifluoromethyl)Benzene (21)1-Methoxy-3-(Trifluoromethoxy)Benzene (9)1-Methoxy-3-(Trifluoromethyl)Benzene (40)1-Methoxy-4-(Trifluoromethyl)Benzene (18)1-Methyl-2-(Trifluoromethoxy)Benzene (5)1-Methyl-2-(Trifluoromethyl)Benzene (43)1-Methyl-3-(Trifluoromethyl)Benzene (22)1-Nitro-2-(Trifluoromethoxy)Benzene (8)1-Nitro-2-(Trifluoromethyl)Benzene (32)1-Nitro-3-(Trifluoromethoxy)Benzene (9)1-Nitro-3-(Trifluoromethyl)Benzene (82)1-Phenoxy-3-(Trifluoromethyl)Benzene (2)1-Phenoxy-4-(Trifluoromethyl)Benzene (2)1-Phenyl-3-(Trifluoromethyl)-1H-Pyrazole (5)2,2,2-Trifluoro-1-Phenylethan-1-Amine (48)2,2,2-Trifluoro-1-Phenylethan-1-Ol (27)2,2,2-Trifluoro-1-Phenylethan-1-One (51)2-(2,2,2-Trifluoroethoxy)Pyridine (8)2-(Trifluoromethoxy)Aniline (18)2-(Trifluoromethoxy)Benzaldehyde (7)2-(Trifluoromethoxy)Benzoic Acid (8)2-(Trifluoromethoxy)Benzonitrile (7)2-(Trifluoromethoxy)Phenol (8)2-(Trifluoromethoxy)Pyridine (6)2-(Trifluoromethyl)-1,1'-Biphenyl (8)2-(Trifluoromethyl)Aniline (58)2-(Trifluoromethyl)Benzaldehyde (27)2-(Trifluoromethyl)Benzamide (6)2-(Trifluoromethyl)Benzoic Acid (33)2-(Trifluoromethyl)Benzonitrile (35)2-(Trifluoromethyl)Phenol (23)2-(Trifluoromethyl)Pyrazine (6)2-(Trifluoromethyl)Pyridine (6)2-(Trifluoromethyl)Pyrimidine (27)2-(Trifluoromethyl)Quinazoline (4)2-(Trifluoromethyl)Thiazole (11)2-(Trifluoromethyl)Thiophene (10)2-Bromo-1-Fluoro-4-(Trifluoromethyl)Benzene (4)2-Bromo-4-(Trifluoromethyl)Aniline (9)2-Bromo-6-(Trifluoromethyl)Aniline (5)2-Chloro-3-(Trifluoromethyl)Aniline (3)2-Chloro-4-(Trifluoromethyl)Pyrimidine (6)2-Chloro-6-(Trifluoromethyl)Aniline (4)2-Methoxy-3-(Trifluoromethyl)Pyridine (6)2-Methoxy-5-(Trifluoromethyl)Pyridine (4)2-Phenoxy-5-(Trifluoromethyl)Pyridine (7)3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)-5-(Trifluoromethyl)Pyridine (3)3-(Trifluoromethoxy)Aniline (22)3-(Trifluoromethoxy)Benzaldehyde (10)3-(Trifluoromethoxy)Benzoic Acid (19)3-(Trifluoromethoxy)Benzonitrile (11)3-(Trifluoromethoxy)Phenol (12)3-(Trifluoromethyl)-1H-Pyrrolo[2,3-B]Pyridine (3)3-(Trifluoromethyl)Aniline (102)3-(Trifluoromethyl)Benzaldehyde (31)3-(Trifluoromethyl)Benzenesulfonyl Chloride (13)3-(Trifluoromethyl)Benzoic Acid (52)3-(Trifluoromethyl)Benzonitrile (47)3-(Trifluoromethyl)Phenol (43)3-(Trifluoromethyl)Pyridazine (3)3-(Trifluoromethyl)Pyridin-2-Ol (5)3-(Trifluoromethyl)Pyridine (51)3-Chloro-4-(Trifluoromethyl)Aniline (5)4,4,4-Trifluoro-1-Phenylbutane-1,3-Dione (10)4,4,5,5-Tetramethyl-2-(2-(Trifluoromethyl)Phenyl)-1,3,2-Dioxaborolane (5)4,4,5,5-Tetramethyl-2-(3-(Trifluoromethoxy)Phenyl)-1,3,2-Dioxaborolane (5)4-(Trifluoromethoxy)Aniline (26)4-(Trifluoromethyl)-2H-Chromen-2-One (6)4-(Trifluoromethyl)Phenol (18)4-(Trifluoromethyl)Pyridine (14)4-(Trifluoromethyl)Pyrimidin-2-Amine (6)4-(Trifluoromethyl)Pyrimidine (12)4-(Trifluoromethyl)Thiazole (7)4-Bromo-3-(Trifluoromethyl)Aniline (7)5-(Trifluoromethyl)Pyridin-2-Amine (5)5-(Trifluoromethyl)Pyridin-2-Ol (8)6-(Trifluoromethyl)-1H-Indazole (6)6-(Trifluoromethyl)-1H-Indole (5)6-(Trifluoromethyl)Pyridin-2-Amine (11)6-(Trifluoromethyl)Pyridin-3-Amine (5)Methyl 2-(Trifluoromethyl)Benzoate (14)Methyl 3-(Trifluoromethyl)Benzoate (22)N-Phenyl-4-(Trifluoromethyl)Aniline (3)Phenyl(3-(Trifluoromethyl)Phenyl)Methanone (3)Phenyl(Trifluoromethyl)Sulfane (23)
Organometallic Reagents
Organoborons (34751)
Fluorene-Phenyl-Boronic Acid/Ester (4)Hydroxymethyl-Phenyl-Boronic-Acid (32)Methoxyphenylboronic-Acid (32)Phenyl-Naphthalene-Boronic Acid/Ester (9)(1-(Tert-Butoxycarbonyl)-1H-Indol-2-Yl)Boronic Acid (17)(1-Phenyl-1H-Pyrazol-4-Yl)Boronic Acid (4)(1H-Indazol-4-Yl)Boronic Acid (3)(1H-Indazol-6-Yl)Boronic Acid (4)(1H-Indol-2-Yl)Boronic Acid (3)(2,3-Dichlorophenyl)Boronic Acid (11)(2,3-Difluorophenyl)Boronic Acid (33)(2,6-Dichlorophenyl)Boronic Acid (6)(2,6-Difluorophenyl)Boronic Acid (28)(2-(Phenoxymethyl)Phenyl)Boronic Acid (3)(2-(Trifluoromethoxy)Phenyl)Boronic Acid (5)(2-(Trifluoromethyl)Phenyl)Boronic Acid (20)(2-Bromophenyl)Boronic Acid (39)(2-Chloro-3-Methoxyphenyl)Boronic Acid (5)(2-Chloro-6-Fluorophenyl)Boronic Acid (9)(2-Fluoro-3-(Trifluoromethyl)Phenyl)Boronic Acid (6)(2-Fluorophenyl)Boronic Acid (229)(2-Nitrophenyl)Boronic Acid (13)(3,4-Dichlorophenyl)Boronic Acid (10)(3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Phenyl)Methanol (7)(3-(Benzyloxy)Phenyl)Boronic Acid (9)(3-(Phenoxymethyl)Phenyl)Boronic Acid (3)(3-(Phenylcarbamoyl)Phenyl)Boronic Acid (3)(3-(Trifluoromethoxy)Phenyl)Boronic Acid (9)(3-(Trifluoromethyl)Phenyl)Boronic Acid (41)(3-Bromo-2-Fluorophenyl)Boronic Acid (7)(3-Bromo-2-Methoxyphenyl)Boronic Acid (6)(3-Bromophenyl)Boronic Acid (65)(3-Chloro-2-Methoxyphenyl)Boronic Acid (4)(3-Chloro-4-Fluorophenyl)Boronic Acid (8)(3-Chlorophenyl)Boronic Acid (121)(3-Fluorophenyl)Boronic Acid (169)(3-Iodophenyl)Boronic Acid (5)(3-Nitrophenyl)Boronic Acid (32)(4-(Benzyloxy)Phenyl)Boronic Acid (26)(4-(Methylsulfonyl)Phenyl)Boronic Acid (6)(4-(Phenoxymethyl)Phenyl)Boronic Acid (6)(4-(Phenylcarbamoyl)Phenyl)Boronic Acid (11)(4-(Trifluoromethyl)Phenyl)Boronic Acid (23)(4-Cyclohexylphenyl)Boronic Acid (7)(4-Fluorophenyl)Boronic Acid (93)(5-(Trifluoromethyl)Pyridin-3-Yl)Boronic Acid (4)(6-Phenoxypyridin-3-Yl)Boronic Acid (5)(9H-Fluoren-2-Yl)Boronic Acid (6)(E)-4,4,5,5-Tetramethyl-2-Styryl-1,3,2-Dioxaborolane (16)1,3,2-Dioxaborinane (6)1-(4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Benzyl)Piperazine (5)1-(4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Phenyl)Piperazine (10)1-(5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridin-2-Yl)Piperazine (6)1-Methyl-4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)-1H-Pyrazole (6)2,7-Bis(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)-9H-Fluorene (7)2-(1,3,2-Dioxaborinan-2-Yl)Benzonitrile (4)2-(1,3,2-Dioxaborolan-2-Yl)-1-Methyl-1H-Indole (6)2-(2-(Bromomethyl)Phenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (9)2-(2-Bromophenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (5)2-(3-Chlorophenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (8)2-(3-Cyclopropylphenyl)-1,3,2-Dioxaborolane (5)2-(3-Isopropoxyphenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (5)2-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Benzonitrile (14)2-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Phenol (7)2-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (6)2-(4-(Benzyloxy)Phenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (10)2-(4-Bromophenyl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (8)2-(4-Cyclopropylphenyl)-1,3,2-Dioxaborolane (8)2-(4-Fluorophenyl)-1,3,2-Dioxaborinane (5)2-([1,1'-Biphenyl]-4-Yl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (10)2-(Cyclohex-1-En-1-Yl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (16)2-(Furan-2-Yl)-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (6)2-(M-Tolyl)-1,3,2-Dioxaborinane (4)2-Benzyl-4,4,5,5-Tetramethyl-1,3,2-Dioxaborolane (12)2-Chloro-3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (4)2-Chloro-4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (14)2-Chloro-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (6)2-Fluoro-3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (7)2-Methoxy-3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (7)2-Methoxy-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (5)2-Methyl-3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (5)2-Methyl-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (3)3-(1,3,2-Dioxaborinan-2-Yl)Aniline (3)3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Benzonitrile (25)3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (74)3-Chloro-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (5)3-Fluoro-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (7)3-Methyl-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (6)4,4,5,5-Tetramethyl-2-(2-(Trifluoromethyl)Phenyl)-1,3,2-Dioxaborolane (5)4,4,5,5-Tetramethyl-2-(2-Nitrophenyl)-1,3,2-Dioxaborolane (5)4,4,5,5-Tetramethyl-2-(3-(Trifluoromethoxy)Phenyl)-1,3,2-Dioxaborolane (5)4,4,5,5-Tetramethyl-2-(Naphthalen-1-Yl)-1,3,2-Dioxaborolane (19)4,4,5,5-Tetramethyl-2-(Naphthalen-2-Yl)-1,3,2-Dioxaborolane (12)4,4,5,5-Tetramethyl-2-(Thiophen-2-Yl)-1,3,2-Dioxaborolane (23)4,4,5,5-Tetramethyl-2-(Thiophen-3-Yl)-1,3,2-Dioxaborolane (10)4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)-1,2,3,6-Tetrahydropyridine (11)4-Methyl-3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridine (5)5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)-1H-Indole (5)5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Indoline (5)5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Nicotinonitrile (4)5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyridin-2-Amine (9)5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Pyrimidine (27)5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Thiazole (7)[1,1'-Biphenyl]-4-Ylboronic Acid (29)Benzo[B]Thiophen-2-Ylboronic Acid (5)Benzofuran-2-Ylboronic Acid (8)Benzyl (4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Phenyl)Carbamate (5)Cyclohex-1-En-1-Ylboronic Acid (4)Ethyl 3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Benzoate (6)Isoquinolin-4-Ylboronic Acid (3)Methyl 5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Nicotinate (4)N,N-Diphenyl-4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)Aniline (6)Naphthalen-1-Ylboronic Acid (4)Naphthalen-2-Ylboronic Acid (8)Phenylboronic Acid (1287)Pyridin-2-Ylboronic Acid (9)Pyridin-3-Ylboronic Acid (49)Pyridin-4-Ylboronic Acid (21)Pyrimidin-5-Ylboronic Acid (9)Tert-Butyl 2-(1,3,2-Dioxaborolan-2-Yl)-1H-Indole-1-Carboxylate (6)Tert-Butyl 4-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-Yl)-1H-Pyrazole-1-Carboxylate (3)Tetraphenylborate (8)Thiophen-2-Ylboronic Acid (20)
Electronic Materials
Organic Light-Emitting Diode (OLED) Materials (2219)
Biphenyls & Aromatic Amines (245)Boric Acids & Organoborons (376)Carbazoles & Fluorenes (278)Halogenated Benzenes & Phenols (150)Other Structures (58)Polycyclic Aromatic Hydrocarbons (214)Polymers (10)Six-Membered Heterocycles (Nitrogen) (223)Thiophenes & Furans (147)4-(Diphenylamino)Benzaldehyde (8)4-Bromo-N,N-Bis(4-Methoxyphenyl)Aniline (3)4-Methoxy-N,N-Diphenylaniline (5)4-Methyl-N,N-Diphenylaniline (6)Electron Injection Layers (EIL) (5)Electron Transport Materials (ETM) (54)Guest Materials (75)Hole Injection Layers (HIL) (27)Hole Transport Materials (113)Host Materials (230)Other Materials (4)Blue Emitters (95)Fluorescence Materials (60)Green Emitters (75)Phosphorescence Materials (119)Red Emitters (63)Thermally Activated Delayed Fluorescence Materials (TADF) (184)Yellow Emitters (13)
Energy and Battery Materials
Material Building Blocks
Organic Frameworks and Supramolecular Materials
Covalent Organic Framework (COF) Linkers (1096)
2 Reaction Sites (31)3 Reaction Sites (14)Multiple Reaction Sites (11)2 Reaction Sites (89)3 Reaction Sites (23)Multiple Reaction Sites (24)2 Reaction Sites (70)3 Reaction Sites (25)Carboxylic Anhydride Linkers (10)Halogen Linkers (133)Hydroxyl & Quinone Linkers (26)Multiple Reaction Sites (70)Other Linkers (5)2 Reaction Sites (201)3 Reaction Sites (56)4 Reaction Sites (66)Multiple Reaction Sites (16)2 Reaction Sites (210)3 Reaction Sites (58)4 Reaction Sites (72)Multiple Reaction Sites (21)2 Reaction Sites (38)3 Reaction Sites (8)Multiple Reaction Sites (16)
Metal Organic Framework (MOF) Linkers (1701)
2 Reaction Sites (261)3 Reaction Sites (97)Multiple Reaction Sites (87)2D MOF Linkers (18)2 Reaction Sites (27)3 Reaction Sites (9)Multiple Reaction Sites (12)Other Linkers (8)Phosphoric & Sulfonic Acid Linkers (38)2 Reaction Sites (439)3 Reaction Sites (145)4 Reaction Sites (219)Multiple Reaction Sites (67)2 Reaction Sites (70)3 Reaction Sites (77)Multiple Reaction Sites (65)Porphyrins and Derivatives Linkers (77)
Polymer Additives
Polymerization Reagents (287)
Iodine Transfer Radical Polymerization Reagents (6)Nitroxide-Mediated Radical Polymerization (NMP) Reagents (7)Reversible Addition Fragmentation Chain Transfer (RAFT) Polymerization Reagents (73)Initiators (19)Ligands (19)Transition Metal Catalysts (7)Group Transfer Polymerization (GTP) Reagents (13)
Polymer Reagents
Monomers (1688)
Acrylamide Monomers (39)Acrylate Monomers (112)Acrylic & Methacrylic Monomers (14)Methacrylamide Monomers (25)Methacrylate Monomers (127)Alcohol Monomers (175)Ester Monomers (22)Isocyanate & Isothiocyanate Monomers (5)Thiol Monomers (21)Anhydride Monomers (41)Carboxylic Acid Monomers (110)Tetracarboxylic Dianhydride Monomers (37)Acyl Chloride Monomers (11)Amine & Amide Monomers (203)Maleimide Monomers (126)Sulfonyl Chloride Monomers (8)Epoxide Monomers (82)Other Monomers (64)Oxazoline Monomers (52)Cyclic Olefin Monomers (35)Linear Olefin Monomers (125)Styrene Monomers (76)
Solar Cells and Photonic Materials
Donor Intermediates & Acceptor Intermediates (1038)
Benzodiazole Derivatives (109)Isoindigo Derivatives (7)Other Structures (47)Perylenes and Analogues (13)Polycyclic Aromatic Hydrocarbons (23)Pyridines & Thiazoles (29)Pyrroles & Pyrrolidinediones (70)Thiophenes & Furans (44)Triazoles (18)Aromatic Amines (54)Benzenes & Polycyclic Aromatic Hydrocarbons (122)Fluorenes and Analogues (48)Heterocycles (Nitrogen) (72)Thiophenes & Selenophens (319)TPE Derivatives (43)
Dyes and Pigments (450)
Anthracene Dyes (5)Azobenzene Dyes (4)Diarylethene Dyes (5)Spiropyran Dyes (7)Acid Dyes (19)Direct Dyes (14)Disperse Dyes (8)Heat Sensitive & Pressure Sensitive Dyes (9)NIR Dyes (15)Other Dyes and Pigments (25)Solvent Dyes (7)Vat Dyes (6)Coumarin Dyes (38)Cyanine & Squarylium Dyes (26)Dipyrromethene Dyes (21)Other Functional Dyes (16)Perylene Dyes (23)Phthalocyanine & Porphyrin Dyes (160)Xanthene Dyes (37)
Perovskite Solar Cell (PSC) Materials (241)
Heterocycles (Nitrogen) (9)Phosphonic Acids (14)Polycyclic Aromatic Hydrocarbons & Aromatic Amines (14)Polymers (8)Bromides (47)Chlorides (39)Organometallic Salts (7)Other Onium Salts (35)Additives & Dopants (30)Electron Transport Materials (ETM) (8)Hole Transport Materials (48)Interfacial Layer Materials (9)Self-Assembled Monolayers Materials (17)Surface Passivation Reagents (10)
Inhibitors/Agonists
Anti-Infection (4704)
3CLpro (42)Antibacterial (2072)Antifungal (500)Antiparasitic (639)Antiprotozoal (3)Antiviral (49)Arenavirus (5)BVDV (3)CMV (28)Dengue Virus (36)EBV (15)Enterovirus (35)FCoV (3)Filovirus (13)Flavivirus (63)GraR (1)HBV (154)HCV (220)HHV (1)HIV (442)HIV Integrase (38)HIV Protease (70)HPV (10)HRV (3)HSV (76)Influenza Virus (250)LpxC (2)M2 proton channel (2)MERS-CoV (1)Neuraminidase (4)NNRTI (12)NRTI (22)Orthopoxvirus (24)Penicillin-Binding Protein (7)PLP (13)Prion (1)Protein Synthesis (3)Reverse Transcriptase (99)RSV (43)SAMHD1 (1)SARS-CoV (297)SQLE (6)TMV (11)Viral MTase (3)Virus Protease (60)VSV (2)β-lactamase (51)
Apoptosis (2429)
Apoptosis (2132)Apoptosis Inducer (1403)Bcl-2 (194)Bcl-6 (6)BRK (3)c-Myc (47)c-RET (64)C/EBP (2)Caspase (170)DAPK (12)Ferroptosis (111)FKBP (15)GDNF Receptor (2)IAP (47)IDO (69)MDM2 (142)MRCK (2)Necroptosis (33)p32 (1)p53 (41)PERK (35)PKD (10)Pyroptosis (12)RIP Kinase (82)Survivin (12)Thymidylate Synthase (24)TNF (22)TNF Receptor (112)TNF-alpha (28)
Cell Cycle (4584)
Antifolate (31)APC (6)Aurora Kinase (86)BUB (3)Casein Kinase (101)CDK (475)Chk (46)CRISPR/Cas9 (7)CRM1 (17)DHFR (55)DNA polymerase θ (2)DNA/RNA Synthesis (620)DOCK (3)DYRK (60)E2F (2)eIF (43)Endonuclease (3)FOXM1 (3)G-quadruplex (6)Haspin Kinase (8)HDAC (334)HGF (1)HSF (4)HSP (160)IRE1 (28)Kinesin (35)Lats kinase (1)LIMK (17)Mad2 (1)Microtubule/Tubulin (302)MKLP (2)Mps1 (21)MTH2 (1)Nucleoside Antimetabolite/Analog (1442)p21 (1)PAK (29)PDI (1)PKMYT1 (2)PLK (50)PPAR (190)PTEN (5)RAD51 (17)Rho (14)RNA polymerase 2 (1)RNA polymerase 1 (2)ROCK (87)Separase (1)Sirtuin (127)SRPK (51)STK33 (2)TACC (3)Telomerase (20)TOPK (1)Topoisomerase (271)Wee1 (16)YB-1 (5)
DNA Damage (3015)
ADAR (1)AP endonuclease (1)ATAD5 (1)ATM/ATR (67)Aurora Kinase (86)Casein Kinase (101)ClpP (5)CRISPR/Cas9 (7)DNA Alkylator/Crosslinker (76)DNA-PK (39)DNA/RNA Synthesis (620)HDR (1)HSP (160)MRE11 (1)MTH1 (8)Nucleoside Antimetabolite/Analog (1442)OGG (3)p97 (15)PARP (168)RAD51 (17)RAD52 (2)Telomerase (20)Topoisomerase (271)WRN (5)YB-1 (5)
Endocrinology/Hormones (801)
5 Alpha Reductase (2)ACE (73)Adrenergic Receptor (180)Androgen Receptor (96)Aromatase (8)ERR (213)GHSR (21)Glucocorticoid Receptor (44)GNRH Receptor (19)Mineralocorticoid Receptor (12)Opioid Receptor (47)Progesterone Receptor (9)PTHR (1)Renin (17)Sepiapterin reductase (2)SSTR (28)Taste receptor (3)Thyroid Hormone Receptor (3)TPO (4)TRHR (1)TSH Receptor (16)TTR (7)
Epigenetics (2336)
AMPK (103)Aurora Kinase (86)DNA Methyltransferase (41)Epigenetic Reader Domain (391)FTO (15)HDAC (334)HIF (111)Histone Acetyltransferase (92)Histone Demethylase (146)Histone Methyltransferase (277)JAK (244)MBD-containing protein (1)METTL3 (10)MicroRNA (16)PAD (24)PARP (168)Pim (58)PKC (157)PRMT (16)SF3B1 (3)Sirtuin (127)TET (4)
GPCR/G Protein (3432)
5-HT Receptor (392)Adenosine Receptor (169)Adenylyl Cyclase (1)Adiponectin Receptor (2)Adrenergic Receptor (180)Amylin receptor (1)Angiotensin Receptor (76)Apelin Receptor (21)Bombesin Receptor (7)Bradykinin Receptor (17)cAMP (35)Cannabinoid Receptor (62)CaSR (23)CCR (88)CGRP Receptor (12)Chemerin Receptor (2)Cholecystokinin Receptor (32)CRF Receptor (19)CXCR (112)Dopamine Receptor (166)Endothelin Receptor (48)Epac (9)G protein (2)GHSR (21)GLP Receptor (24)Glucagon Receptor (50)GNRH Receptor (19)GPCR (198)GPCR19 (2)GPR109A (1)GPR119 (2)GPR139 (3)GPR24 (1)GPR35 (1)GPR40 (7)GPR52 (4)GPR6 (3)GPR68 (2)GPR84 (1)GPR88 (8)Guanylate Cyclase (30)HCAR (2)Histamine Receptor (234)ICMT (61)Kisspeptin Receptor (2)LHCGR (39)LPA Receptor (11)LPL Receptor (113)Melanocortin Receptor (26)mGluR (228)Motilin Receptor (1)MRGPR (13)Neuropeptide Y receptor (8)Neurotensin Receptor (13)NMUR (2)NPFF receptor (3)Opioid Receptor (47)OXTR (10)P2Y Receptor (76)PACAP Receptor (3)PAFR (38)PAR (54)Prostaglandin Receptor (217)Ras (275)RGS (6)RXFP Receptor (14)S1P Receptor (5)Sigma Receptor (48)SOS (7)SRPK (51)SSTR (28)SUCNR1 (5)TAAR (7)TSH Receptor (16)Urotensin Receptor (7)Vasopressin Receptor (23)β-arrestin (17)
Immunology/Inflammation (2659)
AIM2 (5)AP-1 (16)Arginase (14)Calcineurin (8)CCR (88)CD22 (1)CD28 (2)CD28-B7 (1)CD3 (2)CD38 (9)CD47 (5)CD73 (25)CD80 (1)cGAS (8)COX (344)CRTH2 (9)CTLA-4 (1)CXCR (112)Cyclophilin (7)FAP (17)FKBP (15)FLAP (17)FPR (17)Galectin (3)Glucocorticoid Receptor (44)Glycoprotein VI (3)GM-CSF (2)Histamine Receptor (234)HMGB (1)IFNAR (22)IL (161)IL Receptor (9)Immunology & Inflammation Related (76)IRAK (71)IRF3 (1)LanC-like protein (1)Legumain (1)LTA4H (3)LTR (73)MALT1 (24)MIF (17)MyD88 (6)NADPH Oxidase (32)NFAT (11)NLR (73)NO Synthesis (6)NOS (185)NOT (1)Nrf2 (99)PD-1/PD-L1 (76)PG Synthase (31)PSMA (13)Pyroptosis (12)ROS (388)Ros1 (18)S100A9 (1)Scavenger receptor (1)SIK (15)SOD (4)SPHK (24)STING (99)TENT (1)Thrombopoietin Receptor (11)Tim3 (3)TLR (166)TSPO (2)VISTA (1)XBP1 (1)
Membrane Transporter/Ion Channel (1691)
ABC Transporter (2)AKK1 (3)ANT (1)AQP (5)AQP3 (1)ARF (2)ATP synthase (19)ATPase (42)BCRP (23)Calcium Channel (267)CFTR (61)Chloride Channel (28)CRAC Channel (13)CRM1 (17)EAAT (3)Efflux pump (1)ENT (2)Ferroportin (2)GABA Receptor (182)GLUT (32)Glutamate transporter (21)Glycine transporter (21)HCN Channel (2)IBAT (10)ITPKA (2)MAT (24)MCT (17)Na+/Ca2+ Exchanger (14)Na+/K+ ATPase (14)Na/HCO3 cotransporter (1)NKCC (1)Oct (10)OCTN (1)Organic Anion Transporter (1)P-gp (55)P2X Receptor (79)Piezo (4)Potassium Channel (281)Proton Pump (54)SEO1 (1)SGLT (44)SLC (6)Sodium Channel (194)SV2 (2)TRP Channel (179)UCP (1)URAT1 (18)Urea Transporter (2)VDAC (6)VRAC (1)
Metabolic Enzyme (3461)
15-PGDH (2)2OG Oxygenase (1)3-PGDH (2)3MST (1)6PGD (3)ACAT (37)ACC (22)ACMSD (2)Aconitate hydratase (1)ACS (3)Adenosine Deaminase (12)Adenosylhomocysteinase (8)ADP-ribosyltransferase (1)AhR (50)AKR (15)ALDH (31)Aldose Reductase (53)Aminopeptidase (25)Antifolate (31)AO (2)ApoE (1)Arginase (14)Asparagine synthetase (1)ATF/CREB proteins (2)ATP Citrate Lyase (6)ATP synthase (19)BDK (1)Beta-glucuronidase (3)Carbonic Anhydrase (123)Carboxylesterase (23)Carboxypeptidase (7)Cardiolipin Peroxidase (1)Cathepsin (93)Ceramidase (4)CETP (14)Cholesterol Transporter (1)CPA (5)CPSI (2)Creatine kinase (1)Cryptochrome (5)CSE (2)CYP51 (1)D-amino acid oxidase (3)D6D (1)DAPA synthase (1)Decarboxylase (8)Dehydrogenase (156)DGAT (62)DGK (20)Dopamine β-hydroxylase (8)E-NTPDase (1)Elastase (32)Enolase (7)Enteropeptidase (5)ERAP (3)FABP (6)Factor Xa (45)Farnesyl Transferase (4)FAS (34)FBPase (4)Ferroptosis (111)FOXO (3)FPR (17)FTO (15)Fumarate hydratase (2)FVII (2)FXR (74)G0/G1 switch protein 2 (2)Galactosidases (5)GCN (1)GCS (3)GGTase (6)Glucokinase (25)Glucosidase (133)GLUT (32)Glutaminase C (2)Glutathione Reductase (1)Glyoxalase (14)GOAT (1)GPx (24)GSNOR (3)GST (16)Heme Oxygenase (HO) (7)Hexokinase (7)HMGCR (45)HPSE (2)Hydrolase (12)Hydroxylase (20)IDH (37)IDO (69)Ketohexokinase (5)LDL (11)LDLR (1)Lipase (93)Lipoxygenase (111)LOX (4)LRH-1 (4)LXR (27)MAGL (2)MASTL (1)MDH (3)MetAP (10)MGAT (7)Mineralocorticoid Receptor (12)Mitochondrial Electron Transport Chain (3)Mitochondrial Metabolism (92)MPC (3)NAMPT (33)Neprilysin (15)NNMT (8)NPP (2)NPR (15)NQO (2)OGT (2)OPA1 (1)P448 (1)P450 (254)PAI-1 (24)PARG (12)PDE (358)PDHK (12)PEPCK (2)PGAM1 (1)PGC-1α (4)PHGDH (5)Phospholipase (108)Phosphorylase (10)PI5P4K (11)PKM1 (2)PKM2 (6)Procollagen C Proteinase (1)Proprotein convertases (5)Pyruvate Kinase (28)QR (2)RAR/RXR (87)RBP (3)REV-ERB (4)ROR (67)SCD (18)sEH (33)SGK (15)SHMT (7)Sialyltransferase (1)Sphingosine acyltransferase (2)SQOR (2)Srx (1)Steroid Sulfatase (8)TDO (3)TGH (1)Thioredoxin (12)Thrombin (41)Thymidylate Synthase (24)Transferase (79)Transketolase (7)TrxR (11)Trypanothione reductase (1)UGT (3)VD/VDR (47)Xanthine Oxidase (49)αKGD (2)
Neuronal Signaling (3786)
5-HT Receptor (392)AAK1 (17)AChE (44)AChR (346)Adenosine Kinase (6)Adrenergic Receptor (180)AMPAR (53)Amylin receptor (1)Amyloid-β (169)Calcium Channel (267)CaMK (54)Cannabinoid Receptor (62)CGRP Receptor (12)Cholecystokinin Receptor (32)Cholinesterase (235)CHT (1)COMT (17)DAT (21)Dopamine Receptor (166)Endorphin (1)Enkephalinase (3)FAAH (44)GABA Receptor (182)Glucosylceramide Synthase (13)GluR (206)GPR119 (2)GPR139 (3)GPR24 (1)Histamine Receptor (234)Huntingtin (3)Imidazoline Receptor (14)Kainate receptor (5)MAO (194)Melanocortin Receptor (26)mGluR (228)MT Receptor (11)NET (3)Neuropeptide Y receptor (8)Neurotensin Receptor (13)NGF (3)NK Receptor (56)NMDA (1)NMDAR (50)Notch (26)Nurr1 (7)OLIG2 (1)Opioid Receptor (47)OX Receptor (46)P2X Receptor (79)Proprotein convertases (5)QC (3)RAGE (3)REST (1)Ryanodine Receptor-1 (1)S100β (1)Secretase (110)SERT (28)Sigma Receptor (48)SMS (4)SSRIs (5)Substance P (2)TAAR (7)Tau Protein (28)TLX (1)Trk Receptor (80)TRP Channel (179)TTR (7)α-synuclein (1)
Protein Tyrosine Kinase/RTK (2730)
Ack1 (5)ALK (84)Bcr-Abl (105)BMX Kinase (3)BRK (3)BTK (149)c-FMS (60)c-Kit (87)c-Met/HGFR (131)c-RET (64)DDR1/DDR2 Receptor (22)DYRK (60)EGFR (401)Ephrin Receptor (16)ErbB (6)FAK (56)FGF (2)FGFR (136)FLT3 (134)GAK (623)HER2 (29)IGF-1R (27)Insulin Receptor (26)ITK (18)JAK (244)MRCK (2)PDGFR (127)PTK (7)Pyk2 (4)Ros1 (18)SHP2 (48)Src (110)Syk (40)TAM Receptor (50)Tie-2 (16)Trk Receptor (80)VEGFR (268)
Other Inhibitors/Agonists (3145)
20-HETE (2)AIMP2-DX2 (2)Amino Acids and Amino Acid Derivatives (109)Aminoacyl-tRNA Synthetase (18)Anesthetic (3)Anti-inflammation (32)Anti-Thyroid Agent (1)Anti-tumor (7)Anti-ulcer (1)Antioxidant (20)Antispasmodic Agent (2)ASGPR (11)Bcat (11)c-RET (64)Cancer Stem Cells (2)CAR (5)Cardiac transcription factor (2)CBFβ-SMMHC (1)CD73 (25)CerK (1)Chelating Agent (15)Choleretic (1)Choline Kinase (3)CNT (1)CRTH2 (9)CSE (2)DCLK (1)DcpS (1)Decarboxylase (8)Dehydrogenase (156)Diabetes (2)Diuretic (1)Drug Metabolite (283)DUE-B (1)E2F (2)ECE (2)Endogenous Metabolite (1190)ERO1 (2)FAP (17)FATP (3)Fer/FerT (1)FPP synthase (1)Fucosyltransferase (4)Fungicide (61)G protein (2)Galectin (3)Galectin-3 (12)GBF1 (1)GCase (4)Glucocorticoid Receptor (44)GRK (21)GTPCH (1)Herbicide (17)HIPK2 (3)Histidine kinase (1)HNF4α (1)HPPD (3)HRGP (1)Hydroxylase (20)IMP1 (1)Importin β (1)Insecticide (26)ITPKA (2)LAT1 (3)LDL (11)Lipase (93)Lipocalin-2 (1)LYTACs (8)MALAT1 (1)MITF (1)MLKL (4)Molecular Glues (41)MPO (6)MTTP (8)Muscle Relaxant (1)MYOF (1)NAADP (1)Neuroprotection/neurogenic Agent (4)NMD (1)NT (2)Nur77 (8)O-GlcNAcase (9)O-glycosylation (1)OSBP (1)OSBPL (1)OSC (2)Osteoclasts/Bone/Osteogenesis (16)OxPhos (4)PAM complex (1)PANK (2)PAX2 (1)PBR (4)PCA1 (1)PCSK9 (5)PDX1 (1)PEPT (1)PFKFB3 (7)Phosphatase (250)Phosphorylase (10)Photosensitizer (6)PIN1 (10)PKG (10)Platelet (5)PME-1 (2)PMO/MNA (4)PNP (1)PREP (20)Protein Synthesis (3)PXR (3)Quorum Sensing (57)RasGAP (2)Rev-ErbA (3)RUNX (7)SAP (1)SMARC (6)SQS (4)SREBP (5)SRPK (51)Sweetener (3)Tgase (2)Thioredoxin (12)TIR1 (1)TNIK (8)TNK (4)TNNI3K (1)UGT1A1 (1)Urease (5)vanin (4)VDA (1)Vitamin (64)VPS (3)XOR (1)α-amylase (16)Others (2543)
Inhibitory Monoclonal Antibody
Human Inhibitory Monoclonal Antibody (549)
ADAM9/monoclonal antibody (1)Adrenomedullin/monoclonal antibody (1)ALK/monoclonal antibody (1)AMHRII/monoclonal antibody (1)Amyloid Alpha/monoclonal antibody (1)Amyloid-β/monoclonal antibody (6)Ang2/monoclonal antibody (1)ANGPTL3/monoclonal antibody (1)Antifolate/monoclonal antibody (1)APRIL/monoclonal antibody (1)ASCT2/monoclonal antibody (1)AXL/monoclonal antibody (2)B7-H4/monoclonal antibody (1)BST2/monoclonal antibody (1)BTLA/monoclonal antibody (1)BTN1A1/monoclonal antibody (1)c-FMS/monoclonal antibody (6)c-Kit/monoclonal antibody (1)c-Met/HGFR/monoclonal antibody (3)C1q/monoclonal antibody (1)C1s/monoclonal antibody (1)C5AR/monoclonal antibody (1)CA Ⅸ/monoclonal antibody (1)CA-125/monoclonal antibody (1)CCL2/monoclonal antibody (1)CCL20/monoclonal antibody (1)CCL5/monoclonal antibody (1)CCR/monoclonal antibody (3)CD105/monoclonal antibody (1)CD11/monoclonal antibody (2)CD123/monoclonal antibody (2)CD137/monoclonal antibody (1)CD138/monoclonal antibody (1)CD14/monoclonal antibody (1)CD163/monoclonal antibody (1)CD166/monoclonal antibody (1)CD19/monoclonal antibody (5)CD2/monoclonal antibody (1)CD20/monoclonal antibody (7)CD22/monoclonal antibody (3)CD23/monoclonal antibody (1)CD248/monoclonal antibody (1)CD25/monoclonal antibody (3)CD276/B7-H3/monoclonal antibody (4)CD27L/monoclonal antibody (1)CD28/monoclonal antibody (3)CD3/monoclonal antibody (3)CD33/monoclonal antibody (2)CD37/monoclonal antibody (3)CD38/monoclonal antibody (3)CD39/monoclonal antibody (1)CD4/monoclonal antibody (5)CD40/monoclonal antibody (8)CD40L/monoclonal antibody (4)CD44v6/monoclonal antibody (1)CD45/monoclonal antibody (1)CD46/monoclonal antibody (1)CD47/monoclonal antibody (5)CD52/monoclonal antibody (2)CD56/monoclonal antibody (1)CD6/monoclonal antibody (1)CD62p/monoclonal antibody (2)CD7/monoclonal antibody (1)CD70/monoclonal antibody (1)CD73/monoclonal antibody (2)CD74/monoclonal antibody (1)CD79b/monoclonal antibody (2)CD80/monoclonal antibody (1)CD98/monoclonal antibody (2)CDH6/monoclonal antibody (1)Cdiff Toxin A/monoclonal antibody (1)CEA/monoclonal antibody (1)CEACAM1/monoclonal antibody (1)CEACAM5 /monoclonal antibody (1)CEACAM6/monoclonal antibody (1)CGRP Receptor/monoclonal antibody (4)CLDN18.2/monoclonal antibody (1)CLDN6/monoclonal antibody (2)CLEC4C/monoclonal antibody (1)clusterin/monoclonal antibody (1)Complement C3/monoclonal antibody (1)Complement System/monoclonal antibody (8)CTGF/monoclonal antibody (1)CTLA-4/monoclonal antibody (5)CX3CL1/monoclonal antibody (1)CXCL10/monoclonal antibody (1)CXCR/monoclonal antibody (2)DKK1/monoclonal antibody (2)DLL3/monoclonal antibody (1)DLL4/monoclonal antibody (1)DPEP3/monoclonal antibody (1)DPP4/monoclonal antibody (1)EGFR/monoclonal antibody (9)EpCAM/monoclonal antibody (2)EphA3/monoclonal antibody (1)ErbB/monoclonal antibody (4)Factor XI/monoclonal antibody (2)Factor XII/monoclonal antibody (1)FAP/monoclonal antibody (1)FcRn/monoclonal antibody (3)FGF23/monoclonal antibody (1)FGFR/monoclonal antibody (4)Filovirus/monoclonal antibody (7)FLT3/monoclonal antibody (1)FRα/monoclonal antibody (1)FZD/monoclonal antibody (1)FZD10/monoclonal antibody (1)GCGR/monoclonal antibody (2)GD2/monoclonal antibody (2)GD3/monoclonal antibody (1)GDF-15/monoclonal antibody (2)GFRA3/monoclonal antibody (1)GFRAL/monoclonal antibody (1)glycoprotein (GP) IIb/IIIa/monoclonal antibody (1)Glycoprotein VI/monoclonal antibody (1)GM-CSF/monoclonal antibody (3)GPA33/monoclonal antibody (1)GPC3/monoclonal antibody (1)GPNMB/monoclonal antibody (1)GPRC5D/monoclonal antibody (1)Gremlin-1/monoclonal antibody (1)GUCY2C/monoclonal antibody (1)HBEGF/monoclonal antibody (1)Hepcidin/monoclonal antibody (1)HER2/monoclonal antibody (5)HFE2/monoclonal antibody (1)HGF/monoclonal antibody (2)Histone H1/monoclonal antibody (1)HTRA1/monoclonal antibody (1)ICAM-1/monoclonal antibody (1)ICOS/monoclonal antibody (4)ICOSL/monoclonal antibody (1)IFNAR/monoclonal antibody (5)IgE/monoclonal antibody (3)IGF-1R/monoclonal antibody (5)IL-12/monoclonal antibody (1)IL-13/monoclonal antibody (5)IL-13Ra1/monoclonal antibody (1)IL-15/monoclonal antibody (1)IL-17a/monoclonal antibody (5)IL-17c/monoclonal antibody (1)IL-17R/monoclonal antibody (1)IL-18/monoclonal antibody (1)IL-1a/monoclonal antibody (1)IL-1R1/monoclonal antibody (1)IL-1RL1/monoclonal antibody (2)IL-1β/monoclonal antibody (2)IL-20/monoclonal antibody (1)IL-21/monoclonal antibody (1)IL-22/monoclonal antibody (1)IL-22Ra/monoclonal antibody (1)IL-23/monoclonal antibody (5)IL-31Ra/monoclonal antibody (1)IL-33/monoclonal antibody (2)IL-36R/monoclonal antibody (1)IL-3Rβ/monoclonal antibody (1)IL-4/monoclonal antibody (1)IL-4Ra/monoclonal antibody (3)IL-5/monoclonal antibody (2)IL-5Rα/monoclonal antibody (1)IL-6/monoclonal antibody (7)IL-6R/monoclonal antibody (1)IL-6Ra/monoclonal antibody (1)IL-7Ra/monoclonal antibody (1)IL-8/monoclonal antibody (1)IL-9/monoclonal antibody (1)IL1RAP/monoclonal antibody (1)IL22RA1 /monoclonal antibody (1)ILDR2/monoclonal antibody (1)ILT7/monoclonal antibody (1)Influenza Virus/monoclonal antibody (1)Integrin/monoclonal antibody (9)KIR/monoclonal antibody (1)KIR3DL2 /monoclonal antibody (1)Klotho β/monoclonal antibody (1)L-selectin/monoclonal antibody (1)LAG-3/monoclonal antibody (3)LDLR/monoclonal antibody (1)LEPR/monoclonal antibody (1)LIF/monoclonal antibody (1)LILRB2/monoclonal antibody (1)LINGO-1/monoclonal antibody (1)LIV-1/monoclonal antibody (1)LOXL2/monoclonal antibody (1)LRRC15/monoclonal antibody (1)LT α/monoclonal antibody (1)LYPD3/monoclonal antibody (1)MAdCAM-1/monoclonal antibody (1)MASP-2/monoclonal antibody (1)MCAM/monoclonal antibody (1)Mesothelin/monoclonal antibody (2)MIF/monoclonal antibody (1)MMP/monoclonal antibody (1)MUC16/monoclonal antibody (1)MUC5AC/monoclonal antibody (1)Mucin/monoclonal antibody (2)NaPi2b /monoclonal antibody (2)Nectin-4/monoclonal antibody (1)NeuGcGM3/monoclonal antibody (1)Neuropilin-1/monoclonal antibody (1)Neurotensin Receptor/monoclonal antibody (1)NGF/monoclonal antibody (4)NKG2A/monoclonal antibody (1)NKG2D /monoclonal antibody (1)Notch/monoclonal antibody (1)NOTCH3/monoclonal antibody (1)OSMR/monoclonal antibody (1)Osteopontin/monoclonal antibody (1)OX40/monoclonal antibody (5)OX40L /monoclonal antibody (2)P-cadherin/monoclonal antibody (1)PAC1/monoclonal antibody (1)PAR/monoclonal antibody (1)PCSK9/monoclonal antibody (4)PD-1/PD-L1/monoclonal antibody (31)PDGFR/monoclonal antibody (3)Phosphatidylserine/monoclonal antibody (1)Phosphorylcholine/monoclonal antibody (1)PRLR/monoclonal antibody (1)Prolactin Receptor/monoclonal antibody (1)Protective antigen/monoclonal antibody (1)PSMA/monoclonal antibody (1)PTK/monoclonal antibody (1)PVR/monoclonal antibody (1)PVRIG/monoclonal antibody (1)Rabies virus/monoclonal antibody (2)RAGE/monoclonal antibody (1)RANKL /monoclonal antibody (1)RG1/monoclonal antibody (1)RGMα/monoclonal antibody (1)RHD/monoclonal antibody (2)RON/monoclonal antibody (1)ROR/monoclonal antibody (1)ROR2/monoclonal antibody (1)RSPO3/monoclonal antibody (1)RSV/monoclonal antibody (4)RTN4/monoclonal antibody (1)SAA1/monoclonal antibody (1)SAP/monoclonal antibody (1)SARS-CoV/monoclonal antibody (11)Sclerostin/monoclonal antibody (2)SEMA4D/monoclonal antibody (1)Sialyl-Lewis A/monoclonal antibody (1)Siglec-15/monoclonal antibody (1)Siglec-4a/monoclonal antibody (1)SIRPα/monoclonal antibody (1)SLAMF7/monoclonal antibody (1)SLC40A1/monoclonal antibody (1)SLITRK6/monoclonal antibody (1)SpA/monoclonal antibody (1)sphingosine-1-phosphate/monoclonal antibody (1)STAB1/monoclonal antibody (1)STEAP1/monoclonal antibody (1)TAG-72/monoclonal antibody (1)Tau Protein/monoclonal antibody (2)TBFbR2/monoclonal antibody (1)TCR/monoclonal antibody (1)Tenascin-C/monoclonal antibody (1)TFPI/monoclonal antibody (1)TGF-beta/monoclonal antibody (3)TGM2/monoclonal antibody (1)Tie-2/monoclonal antibody (1)TIGIT/monoclonal antibody (5)Tim3/monoclonal antibody (3)Tissue Factor/monoclonal antibody (1)TLR/monoclonal antibody (2)TNF Receptor/monoclonal antibody (15)TNF-alpha/monoclonal antibody (5)TNF/monoclonal antibody (1)TREM2/monoclonal antibody (2)TROP2/monoclonal antibody (2)TSHR/monoclonal antibody (1)TSLP/monoclonal antibody (1)TWEAK/monoclonal antibody (1)TYRP1/monoclonal antibody (1)uPAR/monoclonal antibody (1)VAP-1/monoclonal antibody (1)VEGF/monoclonal antibody (1)VEGFC/monoclonal antibody (1)VEGFR/monoclonal antibody (5)VEGI/monoclonal antibody (2)Vimentin/monoclonal antibody (2)VISTA/monoclonal antibody (1)α-Synuclein/monoclonal antibody (2)α-toxin /monoclonal antibody (2)
Recombinant Protein
CAR-T related Proteins (141)
B Cell Maturation Antigen (BCMA) (10)B7-H3 (7)c-Met/HGFR/Recombinant Protein (4)CA-125 (3)Carbonic Anhydrase Proteins (2)CD138/Syndecan-1 (3)CD19 Proteins (2)CD20 Proteins (3)CD27 Ligand/CD70 (4)CD30 Proteins (5)CD314/NKG2D (6)CD319/SLAMF7 (5)CD38 Proteins (6)CD4 Proteins (2)CD7 Proteins (5)EGFR/Recombinant Protein (5)Epithelial Cell Adhesion Molecule (EpCAM) (5)ErbB2/HER2 (6)ErbB3/HER3 (5)FLK-1/VEGFR-2 (3)Folate Receptor alpha (FR-alpha) (6)GPC-3 (5)MUC-1/CD227 (5)Nectin-4/Recombinant Protein (9)ROR1/Recombinant Protein (7)Siglec-2/CD22 (5)Siglec-3/CD33 (6)TROP-2/Recombinant Protein (7)
CD Antigens (646)
B Cell CD Proteins (138)Dendritic Cell CD Proteins (226)Endothelial cell CD Proteins (179)Epithelial cell CD Proteins (216)Erythrocyte CD Proteins (45)Macrophage CD Proteins (318)Monocyte CD Proteins (134)NK Cell CD Proteins (176)Platelet CD Proteins (90)Stem Cell CD Proteins (165)T Cell CD Proteins (160)
Cytokines and Growth Factors (662)
Angiopoietins (14)Chemokine & Receptors (54)CSF & Receptors (35)EGF Superfamily (25)Ephrin/Eph Family (24)FGF Family (10)HGF & Receptors (4)IGF family (13)Interferon & Receptors (24)Interleukin & Receptors (208)Neurotrophic Factors (52)PDGFs & PDGFRs (4)Peptide Hormone & Neuropeptides (29)TGF-beta Superfamily (57)TNF Superfamily (146)VEGF & VEGFR (3)
Enzymes & Regulators (244)
Carbonic Anhydrase Proteins (2)Carboxypeptidase Proteins (5)Caspase Proteins (1)Cathepsin Proteins (5)Cyclin-Dependent Kinase Inhibitor Proteins (2)Cystatin Family (12)Hydrolases (EC 3) (50)Isomerases (EC 5) (4)Lyases (EC 4) (9)Matrix Metalloproteinases (3)Oxidoreductases (EC 1) (18)Phosphatase/Recombinant Protein (6)Protease Inhibitors (42)Protein Tyrosine Kinases (79)Serine/Threonine Kinase Proteins (17)Transferases (EC 2) (15)
Receptor Proteins (383)
Cell Adhesion Molecules (CAMs) (114)Cytokine Receptors (51)G-Protein-Coupled Receptors (GPCRs) (32)Killer-Cell Immunoglobulin-like Receptors (4)Leukocyte Immunoglobin-like Receptors (23)Notch family (6)Nuclear Receptor Superfamily (1)Pattern Recognition Receptors (18)Receptor Serine/Threonine Kinases (16)Receptor Tyrosine Kinases (81)Receptor Tyrosine Phosphatase (2)Siglec/Recombinant Protein (36)
Animal Modeling Reagents
Immune and Inflammatory Disease Models (29)
Arthritis Model (1)Cystitis Model (1)Dermatitis Model (1)Ear Edema Model (1)Encephalomyelitis Model (2)Foot Edema Model (2)Gastrointestinal Inflammation and Ulcer Model (8)Gouty Arthritis Model (1)Liver Inflammation Model (2)Myocarditis Model (2)Nephritis Model (1)Pancreatitis Model (5)Psoriasis Model (3)Systemic Lupus Erythematosus Model (2)
Cancer
Tumor Epigenetics and Gene Regulation (2822)
Aminoacyl-tRNA Synthetase (18)ATM/ATR (67)DNA Methyltransferase (41)DNA/RNA Synthesis (620)Epigenetic Reader Domain (391)HDAC (334)HIF (111)Histone Acetyltransferase (92)Histone Demethylase (146)Histone Methyltransferase (277)METTL3 (10)MicroRNA (16)PAD (24)PARP (168)Pim (58)PKC (157)RNA polymerase 1 (2)Sirtuin (127)TET (4)Topoisomerase (271)
Enzymology
Enzyme Inhibitors (210)
Acetaldehyde Dehydrogenase Inhibitor (2)Acyltransferase Inhibitor (4)Alkaline Phosphatase Inhibitor (2)Angiotensin Converting Enzyme Inhibitor (4)ATP Enzyme Inhibitor (8)Calcineurin Inhibitor (1)Carbonic Acid Dehydratase Inhibitor (2)Caspase Inhibitor (2)Cyclooxygenase (COX) Inhibitor (54)DNA Topoisomerase II Inhibitor (11)Hypoxia Inducible Factor (HIF) Inhibitor (3)Inosine Monophosphate (IMP) Dehydrogenase Inhibitor (10)Leukotriene A4 Hydrolase Inhibitor (3)Lipoxygenase Inhibitor (11)Monoamine Oxidase Inhibitor (4)NADH Peroxidase Inhibitor (2)Pancreatic Lipase Inhibitor (2)Phosphodiesterase Inhibitor (19)Protease Inhibitor (15)Reverse Transcriptase Inhibitor (16)Starch Glucosidase Inhibitor (3)Thromboxane Synthase Inhibitor (3)Topoisomerase I Inhibitor (8)Tyrosine / EGF Receptor Protein Kinase Inhibitor (33)
Fluorescent Probe / Staining
Cell Biology Fluorescent Dyes (98)
Apoptosis Dyes (5)BODIPY (21)Cytoplasm Dyes (5)Cytotoxicity Analysis Dyes (11)Endocytosis and pinocytosis Dyes (5)Endoplasmic Reticulum Dyes (1)Exosome Dyes (4)Lipid Droplet Dyes (3)Lysosome Dyes (4)Membrane Dyes (5)Mitochondrial Dyes (5)NOS Dyes (8)Nucleic Acid Dyes (1)Nucleus Dyes (10)PH Indicators (19)ROS Dyes (27)
Natural Products
Asteraceae (228)
Achillea (3)Ageratum (1)Ambrosia (2)Arctium (2)Arnica (1)Artemisia (24)Aster (2)Atractylodes (10)Aucklandia (5)Bellis (1)Blumea (4)Cacalia (1)Carpesium (1)Carthamus (7)Centaurea (5)Centipeda (4)Chrysactinia (4)Chrysanthemum (6)Cichorium (2)Cirsium (2)Conyza (1)Coreopsis (1)Cosmos (1)Crepis (3)Cynara (1)Dahlia (2)Dittrichia (1)Doronicum (1)Echinacea (2)Eclipta (6)Elephantopus (1)Erigeron (3)Eupatorium (20)Gnaphalium (1)Gynura (2)Helianthus (4)Heterotheca (1)Inula (8)Kalimeris (1)Laggera (13)Laurentia (1)Ligularia (4)Matricaria (2)Petasites (2)Pulicaria (4)Saussurea (3)Scorzonera (2)Senecio (9)Siegesbeckia (2)Silybum (5)Smallanthus (1)Stevia (10)Tagetes (4)Telekia (1)Tessaria (1)Tussilago (2)Verbesina (1)Vernonia (5)Wedelia (2)Xanthium (6)Zinnia (1)
Fabaceae (347)
Abrus (1)Acacia (9)Adenanthera (1)Albizia (1)Ammothamnus (1)Antheroporum (1)Astragalus (23)Butea (1)Caesalpinia (18)Cajanus (3)Campylotropis (1)Cassia (27)Cicer (1)Crotalaria (5)Cytisus (3)Dalbergia (10)Derris (13)Desmodium (3)Erythrina (13)Flemingia (3)Genista (1)Glycine (21)Glycyrrhiza (42)Griffonia (1)Hedysarum (1)Lespedeza (5)Leucaena (3)Lotus (5)Lupinus (7)Maackia (4)Medicago (1)Millettia (6)Mimosa (1)Mucuna (2)Ononis (2)Parochetus (2)Phyllodium (1)Pisum (1)Pongamia (1)Prosopis (2)Psoralea (18)Pterocarpus (1)Pueraria (25)Robinia (2)Saraca (1)Senna (1)Sophora (36)Spartium (1)Spatholobus (2)Styphnolobium (1)Trifolium (6)Trigonella (2)Vicia (2)Wisteria (2)
Rutaceae (161)
Aegle (1)Boenninghausenia (2)Cedrelopsis (1)Citrus (38)Clausena (7)Dictamnus (8)Euodia (1)Evodia (13)Flindersia (1)Fortunella (1)Glycosmis (3)Haplophyllum (3)Melicope (1)Micromelum (11)Murraya (21)Phebalium (2)Phellodendron (8)Ptaeroxylon (1)Ruta (3)Skimmia (1)Tetradium (1)Toddalia (14)Zanthoxylum (19)
Vesicle
Parkinson'S Disease and Alzheimer'S Disease (5745)
Akt (208)AMPAR (53)Apoptosis (2132)Caspase (170)CDK (475)CREB (5)Dopamine Receptor (166)Dopamine β-hydroxylase (8)ERK (167)Glucagon Receptor (50)GPCR (198)GSK-3 (125)IL (161)JAK (244)JNK (79)LRRK2 (32)MyD88 (6)NF-κB (417)Notch (26)p53 (41)PI3K (337)PKA (59)PTEN (5)RAGE (3)Ras (275)Rho (14)ROCK (87)ROS (388)Secretase (110)STAT (159)STING (99)Tau Protein (28)TLR (166)TNF (22)Tyrosinase (102)Tyrosine / EGF Receptor Protein Kinase Inhibitor (33)Wnt (104)α-synuclein (1)β-catenin (72)